

Abstract
Polyvalent antigen display is an effective strategy to enhance the immunogenicity of subunit vaccines by clustering them in an array‐like manner on a scaffold system. This strategy results in a higher local density of antigens, increased high avidity interactions with B cells and other antigen presenting cells, and therefore a more effective presentation of vaccine antigens. In this study, we used lumazine synthase (LS), an icosahedral symmetry capsid derived from Bacillus anthracis, as a scaffold to present 60 copies of a linear B cell epitope (PB10) from the ricin toxin fused to the C terminus of LS via four different linkers. We then investigated the effects of linker length, linker rigidity and formaldehyde crosslinking on the protein assembly, conformational integrity, thermal stability, in vitro antibody binding, and immunogenicity in mice. Fusion of the PB10 peptide onto LS, with varying linker lengths, did not affect protein assembly, thermal stability or exposure of the epitope, but had a minor impact on protein conformation. Formaldehyde crosslinking considerably improved protein thermal stability with only minor impact on protein conformation. All LS_PB10 constructs, when administered to mice by injection without adjuvant, elicited measurable anti‐ricin serum IgG titers, although the titers were not sufficient to confer protection against a 10× lethal dose ricin challenge. This work sheds light on the biophysical properties, immunogenicity and potential feasibility of LS from B. anthracis as a scaffold system for polyvalent antigen display.
Keywords: lumazine synthase, polyvalent antigen display, ricin, vaccine, immunogenicity, stability
Introduction
Polyvalent antigen display has become a powerful strategy to enhance the immunogenicity of subunit vaccine antigens.1 This strategy generates vaccine candidates which present multiple copies of an antigen on the surface of a scaffold and thereby mimic the surface of microbial pathogens. B cells, in particular, react strongly to exogenous antigens that have repetitive structures. At the molecular level, a high local density of structurally ordered antigens triggers activation of the B cell receptor (BCR), which promotes B cell activation and proliferation.2 In contrast to monomeric antigens, polyvalent antigen display can generate IgG secretion and long‐lived memory B cells in a more efficient manner. For example, Baschong et al. found that the presentation of antigenic systems in a repetitive array on viral surfaces elicited higher antibody level than those elicited by nonoligomeric proteins.3 This general strategy has been successfully employed with natural self‐assembling antigens to develop vaccines for prevention of hepatitis B virus and human papillomavirus and others under development.4
Many classes of scaffold nanoparticle systems have been examined for use in polyvalent antigen display including synthetic polymers, self‐assembled proteins, inorganic particles, and various others.5 Self‐assembling proteins are one of the more attractive scaffold systems because they offer precise size control, ease of production using recombinant DNA technology, and biocompatibility.6 The aim of this work is to evaluate the potential of a self‐assembling protein, lumazine synthase (LS), as a scaffold system for polyvalent antigen display. LS is an enzyme involved in the biosynthesis of riboflavin in bacteria, fungi and plants. LS synthesizes 6,7‐dimethyl‐8‐ribityllumazine, which is then catalyzed by riboflavin synthase to form riboflavin.7 LS exists as a homo‐oligomer and its oligomeric state (e.g., pentamer, decamer, or 60mer) varies depending on the species.
The structure of LS, especially the 60‐mer capsid, has inspired immunologists to explore its use as a scaffold for vaccine development.8, 9 LS consists of 60 identical monomers forming an icosahedral quaternary structure (T = 1 state) capsid with a diameter of about 16 nm. The least‐common multiple assembly unit for LS is a pentameric block, of which twelve pentamers coalesce into a quasi‐sphere, forming along the five‐fold symmetry axes of an icosahedron. The N‐ and C‐termini of all LS monomers are surface exposed and face outwards.10 Antigens of interest can be fused to either or both of the two termini without inhibiting scaffold formation. Moreover, the icosahedral LS system shows significant conformational stability.11 These features make icosahedral LS an attractive scaffold to display antigens. For example, the icosahedral LS from Aquifex aeolicus has been used for the presentation of the HIV‐1 envelope glycoproteins gp120.9 LS derived from B. anthracis is an icosahedral oligomer10 and has not been studied as a vaccine delivery system, even though it is a potentially attractive platform for subunit vaccines. Therefore, a B. anthracis variant of LS was studied here as a vaccine display platform by presenting a known linear neutralizing epitope from ricin.
Ricin is a ribosome‐inactivating protein (RIP) produced by the castor bean plant, Ricinus communis, which is grown worldwide for the production of castor oil. Ricin is toxic to humans through several routes of exposure, including injection and inhalation.12 Due to its ease of preparation and high toxicity, ricin is classified as a biothreat agent.13 There are, however, no available FDA‐approved vaccines or antidotes against ricin.14 Therefore, there is an urgent need to develop an effective ricin vaccine to protect both first responders and the public from the threat of ricin.
Several linear B‐cell epitopes have been identified on ricin, as defined by a collection of toxin‐neutralizing monoclonal antibodies (MAbs).15 The MAb PB10, for example, binds an immunodominant linear epitope on ricin's enzymatic subunit (RTA). PB10 has an estimated equilibrium dissociation constant (K D) of less than 10 pM and has been shown to passively protect mice against lethal dose ricin challenge via injection or inhalation. Kumar et al. successfully displayed on the surface of tomato bushy stunt virus a 16‐amino acid peptide that encompasses 12‐residues of PB10 epitope.16 The resulting construct elicited serum antibodies in mice that recognized RTA by Western blot analysis. However, no in vivo challenge studies were attempted to test the efficacy of the candidate vaccine. Nonetheless, the study successfully demonstrated that the PB10 epitope when conjugated onto a virus‐like particle is immunogenic in mice. For that reason we chose to display PB10 peptide as model epitope to be fused to the C terminus of LS to develop novel ricin vaccine candidates (designated here as LS_PB10).
Since the immunogenicity of epitopes displayed on a scaffold system is influenced by several factors including the length and rigidity of the linker17, 18 as well as the plasticity of the scaffold,19 LS_PB10 constructs containing four linkers with varying length and rigidity were produced and the effects of linkers on protein stability and immunogenicity were examined. The rigidity of the LS_PB10 scaffold was also enhanced by intra‐molecular crosslinking using formaldehyde which is a commonly used chemical crosslinker and has been previously used to stabilize HIV virions and induce high‐titer neutralizing antibodies against HIV in mice and rabbits.20, 21 In this study, the feasibility of LS as a potential presentation platform for polyvalent antigen display was systematically investigated.
Results
Purity and integrity of LS_PB10 samples
LS_PB10 constructs containing one of the four linkers with varying length and rigidity were designed, produced, purified and treated with formaldehyde (Fig. 1). The purity of each LS_PB10 construct was analyzed by reducing SDS‐PAGE (Fig. 2). Since the interactions between LS subunits are noncovalent, LS and noncrosslinked LS_PB10 samples were dissociated into monomers in the presence of DTT and SDS at room temperature (Fig. 2, samples 12, 34, 56, and 78). All four noncrosslinked LS_PB10 showed acceptable purity on SDS‐PAGE and had a higher monomer molecular weight than LS itself (sample LS).
Figure 1.
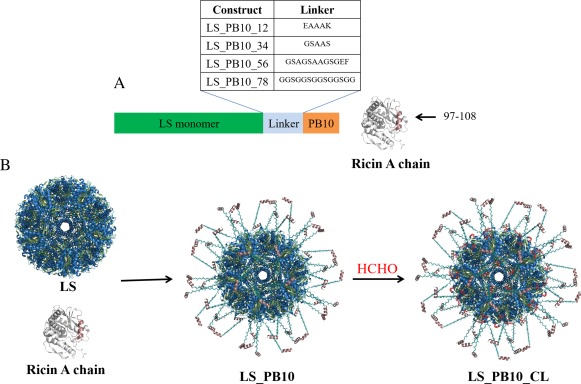
Schematic overview of design of vaccine antigens in this study. (A) LS_PB10 constructs with four different linkers and corresponding sample identification code; the sequence of PB10 peptide is “NQEDAEAITHLF” (B) Hypothetical 3‐D structure of LS_PB10 and LS_PB10_CL constructs (CL indicates formaldehyde crosslinking). The in silico model was manually constructed by combining the 60‐fold lumazine synthase structure from B. anthracis (PDBID: 4V7G), with the linear PB10 epitope from R. communis (PDBID: 3SRP), enjoined by an extended glycine‐serine linker. The PB10 epitope is shown in dirty violet color. The possible covalent bonds induced by formaldehyde crosslinking are shown in red color. The hypothetical structures were modeled using the program PyMOL v 1.7 (https://www.pymol.org/)
Figure 2.
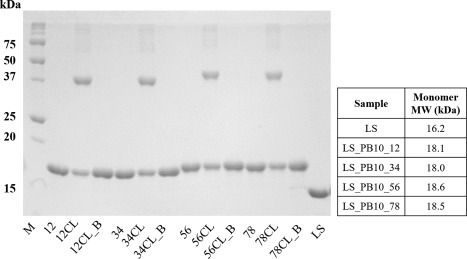
Reducing SDS‐PAGE (12%) analysis of LS and LS_PB10 capsid constructs. Two micrograms of protein were loaded in each lane. M, (molecular ladder); 12, (LS_PB10_12); 12CL, (LS_PB10_12 CL no boiling); 12CL_B, (LS_PB10_12 CL 30 min boiling); 34, (LS_PB10_34); 34CL, (LS_PB10_34 CL no boiling); 34CL_B, (LS_PB10_34 CL 30 min boiling); 56, (LS_PB10_56); 56CL, (LS_PB10_56 CL no boiling); 56CL_B, (LS_PB10_56 CL 30 min boiling); 78, (LS_PB10_78); 78CL, (LS_PB10_78 CL no boiling); 78CL_B, (LS_PB10_78 CL 30 min boiling); LS (LS no boiling). See Figure 1 for description of samples. For boiling treatment, samples were mixed with sample buffer and boiled for 30 min before sample loading, while other samples were kept at room temperature in presence of sample buffer for 30 min. Two microgram of protein was loaded in each lane. The theoretical molecular weights of the monomer for each construct are listed in the table
Formaldehyde is a reactive electrophile and primarily reacts with the amine groups of lysine residues or the N‐terminus.22, 23 Formaldehyde can covalently link two neighboring amine groups within a protein molecule to generate covalent oligomers, potentially enhancing the rigidity and stability of the protein. LS as a scaffold has several exposed neighboring lysine residues (e.g., three K28 residues along the three‐fold symmetry axis). In contrast, the PB10 epitope does not contain free amine groups and should not be chemically modified upon formaldehyde treatment. Hence, formaldehyde crosslinking was performed in this study to study its effect on the stability and immunogenicity of LS_PB10 constructs. The formaldehyde crosslinking reaction was performed under relatively mild conditions (pH 8.0, 30 min) to generate intra‐molecular covalent linkages rather than inter‐scaffold crosslinking. As shown in Figure 2 (samples 12CL, 34CL, 56CL, and 78CL), formaldehyde crosslinking induced a partial formation of oligomers (primarily dimer and slightly higher order oligomers). The disappearance of oligomeric bands observed in Figure 2 (samples 12CL_B, 34CL_B, 56CL_B, and 78CL_B) show a conversion of covalent oligomers back to monomers after 30 min of boiling, indicating that formaldehyde crosslinking is reversible at elevated temperature. This observation is consistent with the finding that formaldehyde crosslinked bonds are relatively stable at room temperature, but tend to break at high temperature.24
Protein assembly state: DLS, SEC, MALS, and TEM
The assembly states of LS_PB10 capsid were investigated by DLS, SEC, MALS and TEM. Table 1 summarizes the hydrodynamic diameter, elution volume and molecular weights (experimental and theoretical MWs) of each of the LS and LS_PB10 samples. DLS measures the hydrodynamic size of a protein sample and uses polydispersity as a measure of the size distribution of a sample. All samples showed low polydispersity (< 0.1) indicating a relatively homogenous size distribution (Table 1). The diameters of icosahedral lumazine synthases from various microorganisms have been found to be around 16 nm.10 DLS analysis shows that LS had a hydrodynamic diameter of 15.5 ± 0.19 nm in storage buffer at 25°C, consistent with the size of LS oligomers with icosahedral geometry. All four noncrosslinked LS_PB10 constructs were larger in size than LS by 2 to 3 nm. This can be attributed to the addition of the peptide linker and PB10 epitope on LS. LS_PB10 constructs with longer linkers (samples 56 and 78) had slightly larger hydrodynamic diameters than those with short linkers (samples 12 and 34). All of the crosslinked LS_PB10 constructs had similar or slightly larger diameters compared to their corresponding noncrosslinked counterparts.
Table 1.
Size Analysis of LS and LS_PB10 Capsid Constructs
| Sample | Hydrodynamic Diameter (nm) | Polydispersity | V R(mL) | Measured MW(MDa) | Theoretical MW(MDa) |
|---|---|---|---|---|---|
| LS | 15.5 ± 0.2 | 0.032 | 13.4 ± 0.1 | 0.9510.02 | 0.97 |
| LS_PB10_12 | 18.2 ± 0.2 | 0.067 | 12.5 ±0.1 | 1.0810.01 | 1.09 |
| LS_PB10_12 CL | 18.2 ± 0.5 | 0.071 | 12.8 ± 0.1 | 1.12 + 0.01 | NA |
| LS_PB10_34 | 17.6 ± 0.0 | 0.045 | 12.5 ± 0.2 | 1.07 + 0.01 | 1.08 |
| LS_PB10_34 CL | 17.6 ± 0.2 | 0.034 | 12.8 ± 0.0 | 1.11 + 0.01 | NA |
| LS_PB10_56 | 18.8 ± 0.3 | 0.031 | 12.410.1 | 1.0710.01 | 1.11 |
| LS_PB10_56 CL | 19.1 ±0.1 | 0.007 | 12.610.0 | 1.18 + 0.01 | NA |
| LS_PB10_78 | 18.4 ± 0.2 | 0.071 | 12.310.1 | 1.0910.01 | 1.11 |
| LS_PB10_78 CL | 18.6 ± 0.1 | 0.062 | 12.410.0 | 1.16 + 0.01 | NA |
Hydrodynamic diameter and polydispersity was determined by DLS, elution volume (V R) by SEC, and absolute molecular weight by MALS. Error values are standard deviations (N = 3). See Figure 1 for description of samples.
Theoretical MW was calculated by multiplying the average molecular weight of each monomer with sixty. NA indicates that it is not possible to predict the theoretical MW for crosslinked constructs.
To minimize the variation caused by varying injection volumes among samples, SEC chromatographs for LS_PB10 samples were normalized for comparison. SEC analysis shows that all LS_PB10 assemblies eluted earlier than LS indicating LS_PB10 had a larger hydrodynamic size than LS (Table 1, Supporting Information Fig. S1). LS_PB10 56 and 78 had an earlier elution volume than LS_PB10 12 and 34. These data are in good agreement with the DLS results. In contrast, all of the crosslinked LS_PB10 constructs had longer elution volumes than noncrosslinked LS_PB10. This counterintuitive phenomenon is probably attributable to the reduction of surface positive charges (primarily amine groups) induced by formaldehyde, increasing the surface hydrophobicity of LS_PB10 thereby potentially enhancing weak protein‐column interactions.
MALS analysis shows that LS_PB10 constructs had larger absolute molecular weights (MWs) than LS (Table 1). The experimental MWs of LS and noncrosslinked constructs match their theoretical MW values calculated based on a 60‐mer. All crosslinked LS_PB10 had higher MWs than the parent noncrosslinked constructs presumably due to the covalent changes induced by formaldehyde.
As an example of the microscopic morphology of the capsid constructs, TEM images (Fig. 3) of LS_PB10_78 and LS show that they share a highly similar spherical shape and hollow interior, suggesting that LS_PB10_78 successfully assembled driven by interactions between LS subunits. The size analysis shows LS and LS_PB10_78 having a size of 14.5 ± 1.0 nm and 18.5 ± 0.9 nm, respectively, which are in good agreement with DLS data. Since all of the LS_PB10 constructs shared similar hydrodynamic diameters, SEC elution times and absolute molecular weight, it seems reasonable to assume all of the other LS_PB10 constructs would show a similar morphology to LS_PB10_78, even though TEM images were not performed for the other samples.
Figure 3.
Transmission electron microscopy images of (A) LS (With kind permission from Springer Science and Business Media6) and (B) LS_PB10_78
Protein conformation: CD, intrinsic fluorescence, extrinsic fluorescence, DSC
To characterize the structural integrity of the various LS_PB10 assemblies, the secondary, tertiary, and overall conformational stability of the protein molecules within the capsid were studied by several biophysical techniques. As shown in Figure 4 and Supporting Information Figure S2, at 25°C LS and LS_PB10 constructs share similar CD spectra. In contrast to LS, however, all LS_PB10 showed slightly less negative signals at 222 nm suggesting a small loss of secondary structures [Fig. 4(B)]. The content of secondary structures for each construct was deconvoluted by BeStSel for comparison.25 The secondary structure content of LS based on its crystal structure (PDB: 1VSX) was also plotted and shows good agreement with the experimental data (Supporting Information Fig. S2). All LS_PB10 constructs showed decreased helical contents compared to LS. This reduction was relatively minor for noncrosslinked LS_PB10 constructs, but more pronounced for crosslinked constructs indicating that formaldehyde treatment slightly reduced the helical content in the LS_PB10 constructs.
Figure 4.
Comparison of the secondary structure of LS and LS_PB10 constructs in PBS buffer at 25 °C (A) Circular dichroism spectra of LS, LS_PB10_12 and LS_PB10_12CL samples; (B) Comparison of the mean residual molar ellipticity at 222 nm for LS and LS_PB10 constructs. Error bars are standard deviations (N = 3). See Figure 1 for sample composition
The tertiary structure of LS_PB10 was studied using intrinsic tryptophan (Trp) fluorescence spectroscopy. The LS_PB10 particles contain two Trp residues (Trp 51 and Trp 151) in each subunit. Neither PB10 peptide nor peptide linkers contain any Trp residue. The mean spectral center of mass (MSM) peak position was used instead of λ max to study protein tertiary structure because MSM peak position shows better sensitivity for detecting subtle conformational changes. To minimize inter‐day variations, the emission spectra for LS_PB10 samples were normalized for comparison [Fig. 5(A); Supporting Information Fig. S3]. As shown in Figure 5(A), LS and all LS_PB10 constructs showed emission λ max around 330 nm indicating that the proteins are in a folded state. Compared to LS, noncrosslinked LS_PB10 showed a minor red shift in MSM peak position (by 0.2 to 0.4 nm). This suggests that fusion of the linker and PB10 epitope produced only minor alterations of the overall tertiary structure of LS. Formaldehyde crosslinking, however, red shifted the MSM peak position by 1.1 to 1.4 nm suggesting greater alteration of the tertiary structure of the modified LS_PB10 constructs. Additionally, ratios of fluorescence intensity at 350 nm to 330 nm (F350/F330) were calculated to compare protein tertiary structures. The 330 and 350 nm values are taken to correspond to the emission maxima of Trp in buried and exposed environments, respectively. F350/F330 thus reflects the relative population of exposed Trp versus buried Trp and the overall exposure level of Trp residues in proteins. Proteins with more exposed Trp have higher F350/F330 values. The F350/F330 value for each sample manifested the same trend as the MSM peak position [Fig. 5(B)]. It should be noted that there are only two Trp residue in each LS monomer which contains 153 amino acid. Trp fluorescence may not however be able to sufficiently probe the overall protein's tertiary structure. Extrinsic fluorescence was therefore further performed to study protein tertiary structure and showed good agreement with Trp fluorescence data (see below).
Figure 5.
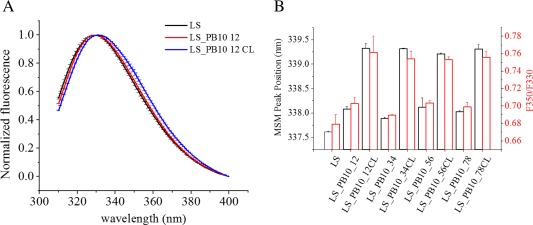
Comparison of the tertiary structure of LS and LS_PB10 constructs in PBS buffer at 25°C. (A) The emission spectra of for LS, LS_PB10_12 and LS_PB10_12CL (B) The mean spectra mass peak position and the ratio of fluorescence intensity at 350 nm to 330 nm for LS and LS_PB10 samples. Error values are standard deviations (N = 3). See Figure 1 for sample composition
The stability of the tertiary structure of LS_PB10 was investigated using extrinsic Sypro orange fluorescence. Sypro orange is a fluorescent dye commonly used to probe changes in protein tertiary structure. At 25°C, all of the crosslinked constructs showed higher initial Sypro orange fluorescent intensity than their parent noncrosslinked particles. This suggests that formaldehyde crosslinking treatment increases the exposure of hydrophobic regions of LS_PB10 constructs. Upon heating of the samples, LS showed two major thermal transitions (52.0 and 93.6°C) in PBS buffer [Fig. 6(A)]. The first thermal transition (52.0°C) results from a conformational change induced by the thermal dissociation of phosphate ions from LS/phosphate complex (Y. Wei and C.R. Middaugh, in preparation). The second transition presumably corresponds to the dissociation and unfolding of LS oligomers. All four noncrosslinked LS_PB10 constructs had T m values similar to LS, suggesting that the display of PB10 on the LS surface by four different linkers had no significant effect on the thermal stability of LS. Interestingly, the first transition (52.0°C) disappeared in all four formaldehyde crosslinked constructs. The disappearance of this transition could potentially be attributed to the change in the conformation of the scaffold induced by formaldehyde crosslinking. The T m2 value of all crosslinked constructs are above 100°C and are thus dramatically higher than those of noncrosslinked ones. The exact values of T m2 were not obtained due to the limitation of the thermal control (max 99°C) of the instrument. The significant increase in the T m2 value for crosslinked particles indicated a considerable thermal stabilization by formaldehyde on LS_PB10 constructs.
Figure 6.
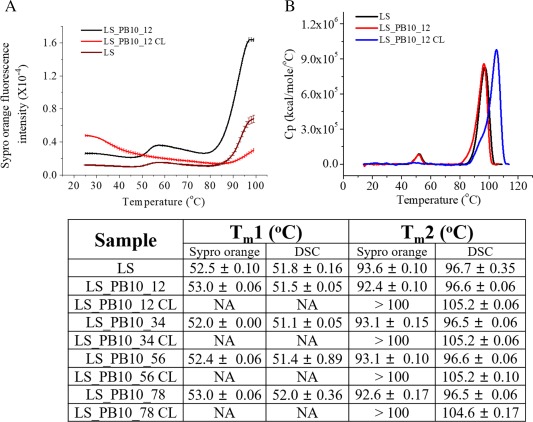
Thermal stability of LS and LS_PB10 constructs in PBS buffer. (A) Extrinsic SYPRO Orange fluorescence thermal melting curves of LS, LS_PB10_12, and LS_PB10_12 CL constructs. (B) DSC thermograms of LS, LS_PB10_12, and LS_PB10_12 CL constructs. The corresponding T m values for LS and LS_PB10 samples are displayed. Error values are standard deviations (N = 3). See Figure 1 for sample composition
The overall conformational stability of the LS_PB10 variants was studied by DSC from 10 to 110°C. Two major thermal transitions were observed for LS and noncrosslinked LS_PB10 constructs while only one major transition was observed for crosslinked LS_PB10 variants [Fig. 6(B)]. Compared to LS, there is no significant shift in the first transition for the four noncrosslinked LS_PB10 constructs, indicating that fusion of the linkers and the PB10 epitope had no significant effect on the first transition. This transition, however, was not detected in crosslinked LS_PB10 constructs and this is consistent with results observed by the extrinsic fluorescence thermal melt study described above. All four crosslinked constructs showed a T m around 105°C, which is about 9°C higher than the T m2 of the noncrosslinked particles. Therefore, formaldehyde crosslinking significantly stabilized the LS_PB10 constructs.
Storage stability: Chemical and colloidal stability
Reducing SDS‐PAGE was performed to analyze the chemical stability of the LS_PB10 constructs, including the stability of formaldehyde crosslinked bonds, proteolysis and covalent oligomerization, in storage buffer. As shown in Figure 7(B,C), all samples stored at 4°C did not produce significant degradation bands or any changes in the pattern of bands on days 15 and 30. This shows that all particles tended to be chemically stable at 4°C for at least 1 month. At 37°C, all of the noncrosslinked constructs did not manifest significant changes on the gel. However, each of crosslinked constructs stored at 37°C showed a significant decrease in the intensity of the dimer band on days 15 and 30. The probable explanation is that formaldehyde crosslinked bonds were unstable and tended to break at elevated temperature.24, 26
Figure 7.
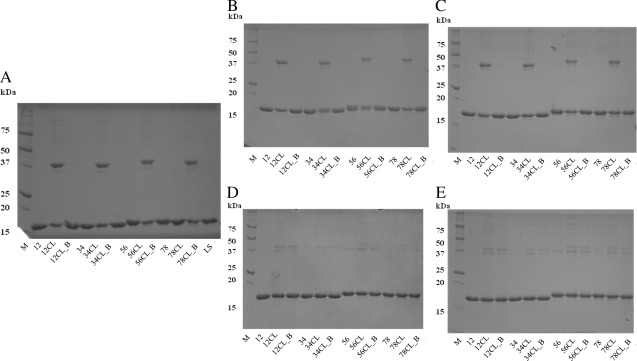
Chemical stability study of LS_PB10 constructs at 4°C and 37°C for a period of one month by SDS‐PAGE analysis. (A) Day 0; (B) Day 15 at 4°C; (C) Day 30 at 4°C; (D) Day 15 at 37°C; (E) Day 30 at 37°C. M, (molecular ladder); 12, (LS_PB10_12); 12CL, (LS_PB10_12 CL no boiling); 12CL_B, (LS_PB10_12 CL 30 min boiling); 34, (LS_PB10_34); 34CL, (LS_PB10_34 CL no boiling); 34CL_B, (LS_PB10_34 CL 30 min boiling); 56, (LS_PB10_56); 56CL, (LS_PB10_56 CL no boiling); 56CL_B, (LS_PB10_56 CL 30 min boiling); 78, (LS_PB10_78); 78CL, (LS_PB10_78 CL no boiling); 78CL_B, (LS_PB10_78 CL 30 min boiling); LS (LS no boiling). See Figure 1 for sample composition
The hydrodynamic diameter of each LS_PB10 construct was measured by DLS to monitor their colloidal stability at 4 and 37°C over 30 days (Supporting Information Fig. S6). The sizes of all constructs remain unchanged except for LS_PB10_78 and LS_PB10_78CL stored at 37°C which showed a small but statistically significant increase (P < 0.05) in the hydrodynamic diameter on day 30.
In‐vitro antibody binding
The murine MAb PB10 was used to test the accessibility of the PB10 epitope on LS_PB10 constructs using Bio‐layer interferometry (BLI). BLI is a label‐free technique for measuring biomolecular interactions between the ligand captured by the biosensor and the analyte in solution. This technique monitors the real‐time changes of the interference pattern of white light reflected from two surfaces (i.e., an internal surface layer and the ligand/analyte interaction interface) upon the analyte binding to the ligand. The readout is recorded in the form of wavelength shift (in nm) and is a direct measure of the level of binding. In the context of this study, the ligand and analyte are PB10 MAb and LS or LS_PB10 constructs, respectively. As shown in Figure 8(A) and Supporting Information Figure S7, all eight constructs (at 4.2 nM) were readily recognized by PB10 MAb. LS, at the same concentration, was not bound by PB10. These results indicate that the PB10 epitope is successfully presented on the surface of LS and readily accessible on all of the constructs. It is interesting to note that the four linkers of different length and rigidity did not influence the exposure of the epitope, because there was no significant differences in their association constant with PB10 MAb [Fig. 8(B)]. This may be due to the inherent flexibility of the C‐terminus of LS, which could diminish the effect of the linker length or rigidity. The PB10 epitope was also impervious to formaldehyde treatment, since formaldehyde crosslinking did not significantly affect the association rate of LS_PB10 constructs. Because no discernible dissociation was observed for any of the LS_PB10 constructs even though the dissociation was monitored for 30 min [only the first 7 min is shown in Fig. 8(A); Supporting Information Fig. S7], dissociation rate constant values could not be obtained (i.e., they are lower than the detection limit: 1.0−7 1/s), which is consistent with MAb PB10 having picomolar binding affinity for ricin (Y. Rong and N. Mantis, in preparation). The multivalency of the PB10 epitope displayed on the surface of LS_PB10 particles might also contribute to the tight binding to the antibody.
Figure 8.
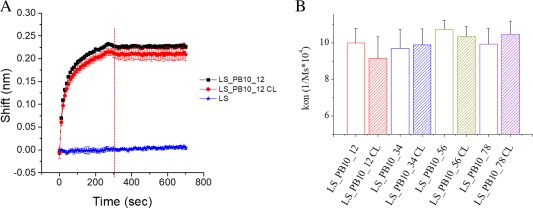
Binding kinetic of LS_PB10_12 (at 4.17 nM) with mouse PB10 IgG. (A) Sensorgram of LS_PB10_12, LS_PB10_12_CL and LS; Association phase (0 to 300 sec) and dissociation phase (300 to 700 sec) were shown. (B) The association rate constants of LS_PB10 samples with mouse PB10 IgG determined from bio‐layer interferometry. Error bars are standard deviations (N = 3). See Figure 1 for sample composition
Immunogenicity of LS_PB10 constructs
The LS_PB10 constructs were administered to groups of Swiss Webster mice three times by the intraperitoneal route. All eight LS_PB10 candidates elicited measurable levels of anti‐LS as well as anti‐ricin antibodies in mice (Table 2), as determined by ELISA. The LS_PB10 constructs stimulated detectable levels of anti‐ricin antibodies after the second immunization, with a discernible further increase in endpoint titers after a third immunization for LS_PB10_12CL, 34CL, 56, and 78. It should be noted that the levels of anti‐ricin antibodies in mouse sera following the second and third LS_PB10 construct immunizations were variable. Moreover, there was no discernible effect of linker length or formaldehyde crosslinking on the immunogenicity of LS_PB10 constructs. The LS‐PB10 mice were challenged with ricin (10 × LD50) approximately three months following the first immunization. Ultimately, all groups of ricin‐challenged mice succumbed to ricin intoxication 36–48 hours post challenge (data not shown), indicating that the LS_PB10 constructs did not confer any benefit against ricin exposure, possibly because the serum antibody titers were not sufficiently high.
Table 2.
Anti‐ricin and Anti‐lumazine Synthase Antibody Titers in Sera of Mice Immunized with LS_PB10 Constructs
| Treatment | Mouse # | Anti‐ricin end‐point titer Day 42 | Anti‐ricin end‐point titer Day 84 | Anti‐lumazine synthase end‐point titer Day 84 |
|---|---|---|---|---|
| 1 | 1600 | 800 | 100000 | |
| LS_PB10_12 | 2 | 400 | 400 | 50000 |
| 3 | 400 | 100 | NT* | |
| 1 | 50 | 100 | NT | |
| LS_PB10_12CL | 2 | 0 | 200 | 200000 |
| 3 | 100 | 3200 | 200000 | |
| 1 | 200 | 400 | 50000 | |
| LS_PB10_34 | 2 | 0 | 100 | 200000 |
| 3 | 0 | 100 | 100000 | |
| 1 | 400 | >3200 | 50000 | |
| LS_PB10_34CL | 2 | 200 | >3200 | 100000 |
| 3 | 100 | 100 | 50000 | |
| 1 | 50 | 3200 | NT | |
| LS_PB10_56 | 2 | 50 | 1600 | 50000 |
| 3 | 0 | 400 | NT | |
| 1 | 0 | 200 | 50000 | |
| LS_PB 10_56CL | 2 | 3200 | 1600 | 200000 |
| 3 | 100 | 400 | 400000 | |
| 1 | 200 | 1600 | 100000 | |
| LS_PB10_78 | 2 | 100 | 400 | 200000 |
| 3 | 100 | 800 | 100000 | |
| 1 | 0 | 200 | 50000 | |
| LS_PB10_78CL | 2 | 50 | 200 | 200000 |
| 3 | 0 | 100 | 100000 |
Groups of mice (n = 3 mice/group) were immunized with 100 µg of LS_PB10 constructs. Serum samples were collected from the mice after the second (Day 42) and third (Day 84) immunizations. NT indicates “not tested” because of insufficient sample volume for analysis. The anti‐ricin end‐point titers for the group treated with LS were not detected and therefore not shown.
Discussion
In this study, an immunodominant linear B cell epitope derived from ricin toxin's enzymatic subunit was chosen as a model epitope to evaluate the feasibility of B. anthracis LS as a platform for polyvalent antigen display. The PB10 epitope is a 12‐amino acid peptide that adopts a helical conformation within the context of ricin.27 The PB10 epitope was fused to the C terminus of the LS monomer through different peptide linkers. Four linkers of varying length and amino acid composition were chosen to study their effect on the stability and immunogenicity of LS_PB10. In addition, LS_PB10 constructs were further stabilized by formaldehyde crosslinking. The addition of PB10 epitopes with linkers onto the LS scaffold retained correct macromolecular assembly and had only a minor effect on the conformation of LS_PB10 constructs. This demonstrates the versatility of LS in presenting peptides with different sequences and lengths. LS_PB10 constructs with four different linkers showed rapid binding to a PB10 MAb, but the extremely slow off‐rates prevented accurate determination of KD values. Indeed, it is estimated that PB10's binding affinity for ricin is less than 10 pM (Y. Rong and N. Mantis, in preparation). Formaldehyde crosslinking only slightly altered the protein's overall secondary and tertiary structure, but dramatically stabilized the protein's conformation by an increase of 9°C in the T m values determined by DSC and extrinsic fluorescence. Such crosslinking did not affect the presentation of the PB10 epitope which showed no significant difference between their association rate constants with a murine PB10 antibody. After three immunizations, all LS_PB10 constructs successfully elicited detectable levels of anti‐ricin antibodies in mouse sera, as measured by ELISA. This illustrates the potential of LS as a presentation system for the generation of an immunogenic response to a short monomeric nonimmunogenic peptide via polyvalent antigen display which permits B‐cell receptor crosslinking.
The LS_PB10 constructs, however, failed to elicit a protective antibody response in mice. This suggests that protective immunity may not be achievable by a single immunogen containing just the PB10 epitope. Thus, a vaccine candidate containing multiple epitopes may be required to offer improved protection. Alternatively, the LS_PB10 constructs could be used with other ricin vaccine antigens in a prime‐boost regimen to “focus” the immune response against the PB10 epitope. Our data also suggest that antibody titers were not sufficiently robust. This issue could be overcome with the addition of an adjuvant. In fact, we are currently in the process of testing the immunogenicity of LS_PB10 constructs in the presence of Alhydrogel® and other adjuvants in mice. Several possible reasons are proposed as follows to explain the suboptimal level and specificity of antibodies elicited by these LS_PB10 constructs.
At physiological pH, the PB10 peptide fused to the C terminus of LS contains four negative charges (NQEDAEAITHLF) and negligible positive charge which could potentially make it a poor immunogen. The presentation of sixty copies of the PB10 epitope on the LS scaffold gives the surface of LS_PB10 a high density of negative charge. It has been shown that extensive negative charge on the surface of an immunogen could significantly inhibit and even completely shut down its immunogenicity.28
The X‐ray crystal structure of ricin shows that the helical PB10 epitope interacts with several surrounding positively charged residues (e.g., R48, R56, and R114).27 These interactions reduce the negative charge density on the PB10 epitope and may have a stabilizing effect on its helical structure. When this peptide was displayed on LS, the absence of stabilizing interactions may alter the conformation of the PB10 peptide. High charge density has been shown to destabilize helicity because of increased side chain repulsion and disruption of intramolecular hydrogen bonding.29 In fact, deconvolution of the far‐UV circular dichroism spectrum of the PB10 peptide (synthesized by Genscript®, Piscataway, NJ) in PBS buffer at 25°C (at 0.2 mg/mL) shows that it has completely lost helical structure and exists primarily as disordered structure (Supporting Information Fig. S8). Thus, the PB10 peptide displayed on the surface of LS may also possess little structure because of the lack of stabilizing interactions. This assumption is further supported by the following two experimental observations. First, there is a general decreasing trend in the helix content in the noncrosslinked LS_PB10 constructs [Supporting Information Fig. S2(E)]. The second observation is that all four noncrosslinked LS_PB10 constructs showed almost the same thermal transitions as LS alone [Fig. 6(B); Supporting Information Fig. S5]. If the fused PB10 peptide had adopted helical structure on the surface of LS, it might have produced an alteration in the thermal melting curve. It is, therefore, possible that the unstructured PB10 peptide presented on the capsid of the LS elicited an antibody response which lacked the strong binding affinity to the cognate ricin molecule.
Finally, it is worth mentioning that a large immunogenic response to the LS itself in all mice immunized was detected presumably due to the strong immunogenicity of the scaffold. The possibly immunodominant role of the LS may somehow suppress the immunogenicity of the PB10 epitope which resulted in only moderate levels of anti‐ricin antibodies. A scaffold system with low or no immunogenicity is preferred for polyvalent antigen display.19 Therefore, efforts, such as mutation and guided computational design, could be undertaken to improve the immune response of the epitope displayed by minimizing or re‐directing the intrinsic immunogenicity of the LS scaffold.
Even though the end goal of designing neutralizing and protective ricin vaccine candidates based on LS was not shown within this work, we established the potential of LS from B. anthracis as a platform for antigen display. The next step is to incorporate a combination of more diverse linear epitopes, plus antigens that maintain the structural environment of the displayed epitopes.
Experimental Procedures
Plasmids and bacterial strains
The plasmid producing LS (pTBSG) was transformed into E. coli strain BL21 (λDE3) competent cells for protein expression. pTBSG plasmid and BL21(λDE3) were described previously.6, 30 The PB10 epitope was fused to the C terminus of LS with a linker. The chimeric gene fragment (LS_PB10_78) was cloned into a pET9a vector by Genscript® (Piscataway, NJ). The restriction sites used were BamHI and HindIII. The upstream T7 tag was later removed by PCR with the primers Forward 5′‐GGAGATATACATATGGTTTTTGAAGGTCATCTGGTGGGCACG‐3′ and Reverse 5′‐CATATGTA TATCTCCTTCTTAAAGTTAAACAAAATT‐3′. The resulting T7 tag free sequence was verified by DNA sequencing. The original LS_PB10 construct (LS_PB10_78) has a 14‐aa flexible linker‐“GGSGGSGGSGGSGG”. Three more LS_PB10 constructs with different linker sequences were also generated employing LS_PB10_78 as template by an In‐fusion HD Plus cloning kit (Takara Bio, CA). The primer sequences for the linker modification are summarized in Supporting Information Table S1. The LS_PB10 plasmids were transformed into E. coli strain TunerTM (DE3) pLysS (Novagen, WI) competent cells for protein expression.
Protein expression and purification
Recombinant proteins were expressed and purified using the protocol described below. Ten milliliters of starter culture was grown in LB media (P212121, MI) at 37°C overnight. The next morning, the 10 mL starter culture was transferred into a 1 L Terrific Broth media (P212121, MI) at 37°C. Protein expression was induced by addition of 1 mM IPTG after the OD600 of the culture reached approximately 0.8–1.0 and the media was incubated at 20°C overnight. After harvesting, the cell pellet was then resuspended in the hydrophobic interaction binding buffer (0.75 M ammonium sulfate, 20 mM phosphate, 5 mM EDTA, pH 7.0). The cells were lysed by sonication and the supernatant was collected after centrifugation at 15,000 g for 30 min. The supernatant was passed through a HiTrap Butyl HP column (5 mL; GE healthcare, Germany) which had been pre‐equilibrated with the hydrophobic interaction binding buffer. The column was then washed with the elution buffer (20 mM phosphate, 5 mM EDTA, pH 7.0) with a linear gradient elution method in a total volume of 200 mL. The elution fractions were analyzed by SDS‐PAGE and those containing the protein of interest were then dialyzed against anion exchange binding buffer (5 mM Tris, 5 mM EDTA, pH 8.0) overnight. The dialyzed sample was loaded onto three HiTrap Q HP columns (5 mL; GE healthcare, Germany) in tandem. The bound protein was eluted using the anion exchange elution buffer (1M NaCl, 5 mM Tris, 5 mM EDTA, pH 8.0).
Each of the four purified LS_PB10 constructs was further stabilized by formaldehyde treatment. LS_PB10 samples were buffer exchanged into citrate‐phosphate buffer (pH 8.0) using spin filters (10K MW cutoff, EMD Millipore, Germany). Protein concentration was then adjusted to 1 mg/mL by dilution. Molar ratio excess amounts of formaldehyde (100 µL, 37% w/w, Fisher Scientific) were added to 0.5 mL protein samples (1 mg/mL) and thoroughly mixed. After 30 min, the reaction was quenched by adding 250 μL of 1 M Tris solution (pH 7.8). Protein samples were dialyzed against storage buffer (20 mM sodium phosphate, 0.1 M NaCl, 5mM EDTA, pH 7.0) and filtered through 0.1 μm syringe filters (Millex®, Germany). Protein samples (noncrosslinked and crosslinked) were analyzed by SDS‐PAGE under reducing condition (16.7 mM DTT). The gel was stained by Coomassie blue R‐250 dye (Teknova, CA) and imaged using an Alpha Imager HP system (ProteinSimple, San Jose, CA). LS_PB10 samples were purified from one batch of protein expression and were used for the following analysis.
Size exclusion chromatography and multi‐angle light scattering
LS and LS_PB10 samples were injected onto a Superose 6 10/300 GL column connected to an ÄKTA explorer (GE healthcare, IL, USA) equipped with UV absorbance and conductivity detectors. Protein samples were analyzed by SEC with PBS buffer (10 mM Na2HPO4, 1.8 mM KH2PO4, 2.7 mM KCl, 137 mM NaCl, pH 7.4) running at a flow rate of 0.5 mL/min. SEC purified protein samples were then analyzed for the measurement of absolute molecular weights by multi‐angle light scattering (MALS). The concentration of each protein was first measured by an Agilent 8453 UV‐visible spectrophotometer (Agilent Technologies, Santa Clara, CA) and the value was used for MW calculation. An automated Calypso sample delivery unit (Wyatt Technology, Santa Barbara, CA) was used to pump protein samples into the MALS detector (Wyatt Technology, Santa Barbara, CA) equipped with a 661 nm laser at a flow rate of 0.5 mL/min. The detector was equilibrated with PBS buffer until a stable baseline was obtained (< 0.0001 AU). Otherwise, Contrad 70 (Decon Labs, PA) was used to thoroughly clean the tubing and detector. One hundred microliter of the sample was sequentially injected into the detector 5 times, and 500 microliter two times followed by a prime using PBS buffer. A narrow segment from the highest plateau of the intensity‐time plot was selected and used for data analysis. Sample eluted from the MALS detector was fractionated and the concentration of the fraction corresponding to the selected segment for data analysis was measured to calculate the concentration factor. The absolute molecule weight was calculated by Zimm's equation.31 Three replicates were performed for each sample.
Dynamic light scattering
The hydrodynamic diameters of the LS and LS_PB10 samples at 25°C were measured by a Brookhaven ZetaPALS Analyzer (Brookhaven Instruments Corporation, Holtsville, NY). The instrument was equipped with a laser (653 nm) and the detector was placed at 90 degrees. Two hundred microliter of protein sample at a concentration of 0.2 mg/mL was added to a 1 × 0.2 cm quartz cuvette. For each run, three acquisitions were taken with an acquisition time of 30 sec. Three runs were measured for each sample. The viscosity value used for calculation was 1.019 cP. The data was analyzed using a polynomial distribution model and number‐averaged diameters were determined.
Transmission electron microscopy
Protein samples were applied to a carbon grid (Ted Pella, Redding, CA) and left to air dry. Glutaraldehyde solution (1%, v/v, Sigma‐Aldrich) was then added to fix the sample. The grid was washed twice with ultrapure water. The sample was then negatively stained by adding 2% uranyl acetate (Sigma‐Aldrich) for 2 min. The morphology of the LS and LS_PB10_78 was studied using a FEI Tecnai F20 XT Transmission Electron Microscope equipped with a CCD camera (FEI TecnaiTM, Oregon). The particle size was calculated by averaging the diameters of 50 particles using ImageJ (https://imagej.nih.gov/ij/).
Sample preparation for analysis of protein conformations
Protein samples were dialyzed overnight at 4°C into PBS buffer. Samples for spectroscopic analysis (circular dichroism, intrinsic and extrinsic fluorescence) were prepared at a concentration of 0.2 mg/mL and 0.5 mg/mL for DSC analysis. Three replicates were performed for each technique.
Circular dichroism
The secondary structure of LS and LS_PB10 constructs was studied by recording circular dichroism (CD) spectra from 200 to 260 nm in 1‐nm increments with a Chirascan‐plus CD spectrometer equipped with a 4‐position, Peltier‐controlled cell holder (Applied Photophysics, UK). Measurements were made in a 1‐mm pathlength cuvette at 25°C. The content of secondary structure for each protein sample were calculated employing the online server BeStSel.25
Intrinsic fluorescence
The tertiary structure of LS and LS_PB10 constructs was studied by measuring tryptophan fluorescence emission spectrum at 25°C. The samples were excited at 295 nm and emission spectra were recorded from 310 nm to 400 nm by a steady‐state fluorometer (Photon Technology International, NJ). Data was collected at one nanometer interval with an integration time of 1 sec per data point. The excitation and emission slit widths were set to 4 nm. Spectra were buffer subtracted and normalized for comparison using in‐house software (Middaugh Suite).32
Extrinsic fluorescence
An Mx3005P qPCR system (Agilent Technologies, Santa Clara, CA) was used to perform extrinsic fluorescence melting studies of the LS and LS_PB10 constructs. Sypro orange dye (5000x, Invitrogen) was added as a sensitive probe to monitor protein structural change during thermal unfolding. Sypro orange was added into protein samples at a dilution factor of 1000. The excitation and emission wavelength were set at 492 nm and 610 nm, respectively. The temperature ramp was set from 25 to 99°C with an increment of 1°C per step and an equilibration time of 2 min at each temperature. T m values for LS and LS_PB10 constructs were calculated by a first derivative method employing Origin 8.0.
Differential scanning calorimetry
Differential scanning calorimetry was performed with a MicroCal VP‐Capillary DSC (Malvern, UK) to investigate the conformational stability of LS and LS_PB10. The temperature was ramped from 10 to 110°C at a scan rate of 1°C/min. The sample cell was equilibrated for 15 min at 10°C at the beginning of each run. The filtering period was set to be 16 sec. Four hundred microliter of sample was loaded into a 96‐well plate for each run. The plate was sealed and kept inside a sample container maintained at 5°C through all of the runs. Apparent transition temperatures were calculated using a non‐two‐state equilibrium model in Origin 7.0 (OriginLab; Northampton, MA).
Stability study in storage buffer
Purified LS_PB10 samples were stored in storage buffer (20mM phosphate, 5 mM EDTA, 0.1 M NaCl, pH 7.0). A long‐term stability study of LS_PB10 constructs in storage buffer was performed at 4 and 37°C for a period of 1 month. Samples were analyzed on days 15 and 30. The colloidal stability and chemical stability of LS_PB10 constructs were evaluated by DLS and reducing SDS‐PAGE, respectively.
In‐vitro antibody binding
The binding kinetics of LS_PB10 constructs with a murine monoclonal IgG targeting the PB10 epitope was characterized with an Octet Red 96 system employing anti‐mouse IgG Fc capture biosensors (Pall ForteBio, Fremont, CA). The PB10 murine IgG was as described.15 The biosensors were hydrated for 20 min before use. Buffers, antibody samples, and LS_PB10 samples were loaded into a black bottom 96 well plate with a volume of 200 μL in each well. A stable baseline was obtained on the sensorgram for 300 sec. PB10 IgG (at 30 nM) prepared in kinetic buffer (PBS, 0.02% Tween 20, 0.2% BSA, pH 7.4) was then loaded onto the biosensors for a period of 300 sec. The biosensors with bound PB10 IgG were dipped into kinetic buffer for 300 sec to remove any nonbound PB10 IgG. The biosensors were then dipped into 4.17 nM LS_PB10 samples prepared in kinetic buffer for 300 sec to induce the association between PB10 IgG and LS_PB10 constructs. The biosensors were then dipped into kinetic buffer for 0.5 hr for dissociation. Ten regeneration cycles were performed to fully regenerate biosensors by incubating biosensors in 10 mM glycine (pH 1.7) for 10 sec followed by a 10‐sec incubation in kinetic buffer. The data were processed using Data Analysis 8.2 (ForteBIO, Fremont, CA).
Mouse immunization
The mouse immunization study was conducted at the Wadsworth Center. Animals were housed according to conventional specific‐pathogen‐free conditions and animal experiments were performed in strict accordance with the Center's Institutional Animal Care and Use Committee guidelines. Female Swiss Webster mice, 5–6 weeks of age, were purchased from Taconic Labs (Hudson, NY). LS and LS_PB10 constructs were diluted in a 20 mM sodium phosphate, 100 mM sodium chloride, 5 mM EDTA buffer (pH 7.0) and cohorts of Swiss Webster mice were I.P. immunized on days 0 and 24 with either 100 μg of a single LS_PB10 construct or LS as a nonprotective control; on day 60 mice were I.P. immunized with an increased dose of 250 μg LS_PB10 construct or LS to compensate for weight gain. Serum was collected by retro‐orbital bleed from each mouse to determine anti‐ricin antibody titers by ELISA. Mice were administered ricin by I.P. injection equal to 10‐times the LD50 based on average weight (4 μg/mouse) on Day 95. Mice were monitored and weighed daily for signs of intoxication and were euthanized if they became moribund.
ELISA
Ricin‐specific serum IgG titers were determined by ELISA, as previously described.15 Briefly, Nunc Maxisorb F96 microtiter plates (ThermoFisher Scientific) were coated and incubated with 1 μg/mL ricin toxin or 1 μg/mL lumazine synthase and then treated with dilutions of mouse serum. Horseradish peroxidase (HRP)‐labeled goat anti‐mouse IgG (SouthernBiotech) was used as the secondary reagent and the plates were developed using 3,3′,5,5′ tetramethylbenzidine (Kirkegaard & Perry Labs, Gaithersburg, MD). The plates were analyzed with a SpectroMax 250 spectrophotometer with Softmax Pro 5.4.5 software (Molecular Devices, Sunnyvale, CA).
Supporting information
Supporting Information.
Acknowledgments
The authors would like to thank Dr. Prem Thapa, from the KU Microscopy and Analytical Imaging Laboratory, for his help with TEM study. We would like to thank Nick Larson for many valuable discussions and his comments on this manuscript. This study was supported by NIAID grant NFP0076894. Additional financial support for Yangjie Wei was provided by a University Graduate Fellowship from the University of Kansas. We confirm that none of the authors have any conflicts of interest.
References
- 1. Karch CP, Burkhard P (2016) Vaccine technologies: From whole organisms to rationally designed protein assemblies. Biochem Pharmacol 120:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Liu W, Sohn HW, Tolar P, Pierce SK (2010) It's all about change: The antigen‐driven initiation of b‐cell receptor signaling. Cold Spring Harb Perspect Biol 2:a002295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baschong W, Hasler L, Häner M, Kistler J, Aebi U (2003) Repetitive versus monomeric antigen presentation: Direct visualization of antibody affinity and specificity. J Struct Biol 143:258–262. [DOI] [PubMed] [Google Scholar]
- 4. Jain NK, Sahni N, Kumru OS, Joshi SB, Volkin DB, Middaugh CR (2015) Formulation and stabilization of recombinant protein based virus‐like particle vaccines. Advan Drug Deliv Rev 93:42–55. [DOI] [PubMed] [Google Scholar]
- 5. Zhao L, Seth A, Wibowo N, Zhao C‐X, Mitter N, Yu C, Middelberg AP (2014) Nanoparticle vaccines. Vaccine 32:327–337. [DOI] [PubMed] [Google Scholar]
- 6. Wahome N, Cooper A, Thapa P, Choudhari S, Gao FP, Volkin DB, Middaugh CR, Production of well‐characterized virus‐like particles in an escherichia coli‐based expression platform for preclinical vaccine assessments. In: Thomas S, Ed. (2016) Vaccine Design: Methods and Protocols, Volume 2: Vaccines for Veterinary Diseases, Humana Press, New York, pp 437–457. [DOI] [PubMed] [Google Scholar]
- 7. Ladenstein R, Fischer M, Bacher A (2013) The lumazine synthase/riboflavin synthase complex: Shapes and functions of a highly variable enzyme system. FEBS J 280:2537–2563. [DOI] [PubMed] [Google Scholar]
- 8. Min J, Kim S, Lee J, Kang S (2014) Lumazine synthase protein cage nanoparticles as modular delivery platforms for targeted drug delivery. RSC Adv 4:48596–48600. [Google Scholar]
- 9. Jardine J, Julien J‐P, Menis S, Ota T, Kalyuzhniy O, McGuire A, Sok D, Huang P‐S, MacPherson S, Jones M (2013) Rational hiv immunogen design to target specific germline b cell receptors. Science 340:711–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Morgunova E, Illarionov B, Saller S, Popov A, Sambaiah T, Bacher A, Cushman M, Fischer M, Ladenstein R (2010) Structural study and thermodynamic characterization of inhibitor binding to lumazine synthase from bacillus anthracis. Acta Cryst D66:1001–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang X, Meining W, Fischer M, Bacher A, Ladenstein R (2001) X‐ray structure analysis and crystallographic refinement of lumazine synthase from the hyperthermophile aquifex aeolicus at 1.6 å resolution: Determinants of thermostability revealed from structural comparisons. J Mol Biol 306:1099–1114. [DOI] [PubMed] [Google Scholar]
- 12. Audi J, Belson M, Patel M, Schier J, Osterloh J (2005) Ricin poisoning: A comprehensive review. J Am Med Assoc 294:2342–2351. [DOI] [PubMed] [Google Scholar]
- 13. Wolfe DN, Florence W, Bryant P (2013) Current biodefense vaccine programs and challenges. Human Vaccines Immunotherap 9:1591–1597. [DOI] [PubMed] [Google Scholar]
- 14. Roxas‐Duncan VI, Smith LA, Progress in the development of vaccines against ricin intoxication. In: Cherwonogrodzky JW, Ed. (2014) Ricin Toxin. Bentham Science Publishers, United Arab Emirates, pp 110–129.
- 15. O'Hara JM, Neal LM, McCarthy EA, Kasten‐Jolly JA, Brey RN, Mantis NJ (2010) Folding domains within the ricin toxin a subunit as targets of protective antibodies. Vaccine 28:7035–7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kumar S, Ochoa W, Singh P, Hsu C, Schneemann A, Manchester M, Olson M, Reddy V (2009) Tomato bushy stunt virus (tbsv), a versatile platform for polyvalent display of antigenic epitopes and vaccine design. Virology 388:185–190. [DOI] [PubMed] [Google Scholar]
- 17. Klein JS, Jiang S, Galimidi RP, Keeffe JR, Bjorkman PJ (2014) Design and characterization of structured protein linkers with differing flexibilities. Protein Eng Des Sel 27:325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tissot AC, Renhofa R, Schmitz N, Cielens I, Meijerink E, Ose V, Jennings GT, Saudan P, Pumpens P, Bachmann MF (2010) Versatile virus‐like particle carrier for epitope based vaccines. PLoS One 5:e9809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lu Y, Chan W, Ko BY, VanLang CC, Swartz JR (2015) Assessing sequence plasticity of a virus‐like nanoparticle by evolution toward a versatile scaffold for vaccines and drug delivery. Proc Natl Acad Sci 112:12360–12365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Poon B, Hsu J, Gudeman V, Chen I, Grovit‐Ferbas K (2005) Formaldehyde‐treated, heat‐inactivated virions with increased human immunodeficiency virus type 1 env can be used to induce high‐titer neutralizing antibody responses. J Virol 79:10210–10217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Poon B, Safrit J, McClure H, Kitchen C, Hsu J, Gudeman V, Petropoulos C, Wrin T, Chen I, Grovit‐Ferbas K (2005) Induction of humoral immune responses following vaccination with envelope‐containing, formaldehyde‐treated, thermally inactivated human immunodeficiency virus type 1. J Virol 79:4927–4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Klockenbusch C, Kast J (2010) Optimization of formaldehyde cross‐linking for protein interaction analysis of non‐tagged integrin β1. BioMed Res Int 2010:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Metz B, Kersten GF, Hoogerhout P, Brugghe HF, Timmermans HA, De Jong A, Meiring H, ten Hove J, Hennink WE, Crommelin DJ (2004) Identification of formaldehyde‐induced modifications in proteins reactions with model peptides. J Biol Chem 279:6235–6243. [DOI] [PubMed] [Google Scholar]
- 24. Jensen S, Thompson L, Harry E (2005) Cell division in bacillus subtilis: Ftsz and ftsa association is z‐ring independent, and ftsa is required for efficient midcell z‐ring assembly. J Bacteriol 187:6536–6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Micsonai A, Wien F, Kernya L, Lee Y‐H, Goto Y, Réfrégiers M, Kardos J (2015) Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc Natl Acad Sci 112:E3095–E3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kennedy‐Darling J, Smith LM (2014) Measuring the formaldehyde protein–DNA cross‐link reversal rate. Analyt Chem 86:5678–5681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rutenber E, Katzin BJ, Ernst S, Collins EJ, Mlsna D, Ready MP, Robertus JD (1991) Crystallographic refinement of ricin to 2.5 å. Proteins 10:240–250. [DOI] [PubMed] [Google Scholar]
- 28. Wen Y, Waltman A, Han H, Collier JH (2016) Switching the immunogenicity of peptide assemblies using surface properties. ACS Nano 10:9274–9286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lu H, Wang J, Bai Y, Lang JW, Liu S, Lin Y, Cheng J (2011) Ionic polypeptides with unusual helical stability. Nat Commun 2:206. [DOI] [PubMed] [Google Scholar]
- 30. Studier FW, Moffatt BA (1986) Use of bacteriophage t7 rna polymerase to direct selective high‐level expression of cloned genes. J Mol Biol 189:113–130. [DOI] [PubMed] [Google Scholar]
- 31. Zimm BH (1948) Apparatus and methods for measurement and interpretation of the angular variation of light scattering; preliminary results on polystyrene solutions. J Chem Phys 16:1099–1116. [Google Scholar]
- 32. Hu L, Trefethen JM, Zeng Y, Yee L, Ohtake S, Lechuga‐Ballesteros D, Warfield KL, Aman MJ, Shulenin S, Unfer R (2011) Biophysical characterization and conformational stability of ebola and marburg virus‐like particles. J Pharm Sci 100:5156–5173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information.