

Abstract
Recent site‐resolved hydrogen exchange measurements have uncovered significant discrepancies between simulations and experimental data during protein folding, including the excessive intramolecular hydrogen bonds in simulations. This finding indicates a possibility that intramolecular charge–charge interactions have not included sufficient dielectric screening effect of the electronic polarization. Scaling down peptide atomic charges according to the optical dielectric constant is tested in this study. As a result, the number of intramolecular hydrogen bonds is lower than using unscaled atomic charges while reaching the same levels of helical contents or β‐hairpin backbone hydrogen bonds, because van der Waals interactions contribute substantially to peptide folding in water. Reducing intramolecular charge–charge interactions and hydrogen bonding increases conformational search efficiency. In particular, it reduces the equilibrium helical content in simulations using AMBER force field and the energy barrier in folding simulations using CHARMM force field.
Keywords: intramolecular hydrogen bonds, electronic polarization effect, atomic charge scaling, conformational search efficiency, computational costs
Introduction
As a landmark advance in scientific computing, folding of small proteins has become accessible in molecular dynamics simulations at the atomic level in millisecond timescale.1, 2, 3, 4 Recent site‐resolved hydrogen exchange and other biophysical measurements5 uncovered several significant discrepancies between the simulations6 and experimental data in regions of the energy surface outside of the native basin, including the excessive intramolecular hydrogen bonds in simulations. These findings suggest possible adjustments of the charge–charge interactions between intramolecular hydrogen bonding groups.
In widely applied all‐atom simulations, each atom is represented by a point charge with covalent bond constraints and a 6–12 Lennard‐Jones potential, widely called van der Waals interactions.7 Hydrogen bonding is represented by the Coulomb interactions between atomic charges of the polar groups. The structural polarization is represented by the charge redistribution of the atomic motion. The electronic polarization is currently not calculated in widely used force field methods for the purpose of saving computing time. Over the years, various polarizable models have been applied,8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18 representing the next level of molecular simulations. Given the size of the protein‐solvent system and the time scale of folding, the current computing speed has not reached the level for protein folding simulations with polarization calculations.
The continuum solvent model or semi‐microscopic model19, 20, 21 calculations usually include the average electronic polarization effect in the dielectric constants. In simulations with explicit solvent, the widely used water models, such as the SPC, TIP3P, and TIP4P models,22, 23, 24, 25 are mean field models with fixed atomic charges parameterized to reproduce equilibrium properties of liquid water. Based on the liquid water dipole moment of ∼3.0 D,26, 27, 28, 29 it is believed that the dipole moments of the widely used water models in the range of 2.1 to 2.4 D include the average dielectric screening effect of the electronic polarization.30, 31, 32 In widely used force field parameters of biomolecules, the total charge of a charged amino acid is equal to that of an electron or a proton, indicating that its atomic charges have not included the dielectric screening effect of the electronic polarization. Without the electronic polarization calculation, these atomic charges need to be scaled down according to the optical dielectric constant of ∼2.30, 31, 32 The excessive intramolecular hydrogen bonds found in folding simulations5 suggest that the atomic charges of neutral amino acids have not included sufficient dielectric screening effects of the electronic polarization and need to be scaled down according to a proper dielectric constant. Most non‐polar organic liquids, such as the liquid alkanes or benzene, have their dielectric constants in the range of 1.4–2.5,33, 34 which are largely from the electronic polarization contribution and are close to the optical dielectric constant of water from electronic polarization contribution at electrical field frequency greater than 10 THz.35, 36 A dielectric constant value of 1.5–2 has been suggested for macromolecules and proteins.37, 38 As a first approximation, the dielectric constant of 2 is assumed in this qualitative study to scale down peptide atomic charges, and the optimal scaling factor could be found in more quantitative studies in the future. Water parameters are not changed. A previous study39 has shown that peptide folding occurs in simulations without change–charge interactions and hydrogen bonding, suggesting a possibility of scaling down charge–charge interactions.
Materials and Methods
The AMBER force field40 and software package41 are applied to molecular dynamics (MD) simulations of peptide folding with explicit solvent and periodic boundary conditions. The CHARMM force field42 and the NAMD program43 are applied to verify the qualitative results. As a first approximation, the peptide atomic charges of both force fields are scaled by a factor of to include the dielectric screening effect of the electronic polarization in intramolecular charge–charge interactions. The parameters of the Lennard‐Jones potential and covalent bond constraints are used without changes. The TIP3P water parameters, including atomic charges, are used without any changes.
Multiple constant volume simulations are carried out on an α‐helix forming peptide and a β‐hairpin forming peptide, starting from different unfolded structures generated from high temperature simulations at 900K. For comparison, the same numbers of simulations are carried out from the same starting structures using the standard force field atomic charges. The effect of scaling peptide atomic charges on peptide solution density is tested in NPT ensemble simulations. The cutoff distance is 8 Å, the time step 0.002 picoseconds (ps), and the bond length connecting to hydrogen atoms constrained using the SHAKE algorithm.44
Results
α‐helix folding
The α‐helix is a widely available basic secondary structure with a well‐defined geometry. Its folding has been studied extensively using computational methods.45, 46, 47, 48, 49 Early peptide folding simulations45, 50 were carried out successfully on synthetic peptide sequences. Among these peptides, a well characterized alanine‐based α‐helical peptide51 Ac(AAQAA)3Y‐NH2 is studied here. The peptide is solvated with 1529 water molecules in constant volume simulations with periodic boundary conditions. A group of 16 simulations is carried out at 273K for 60 ns starting from 16 unfolded structures with scaled (by ) peptide atomic charges of the AMBER force field. For comparison, another group of 16 simulations are carried out from the same starting structures using standard AMBER atomic charges.
In all simulations, helical segments are observed, including partial helices and two‐segment helices. These structures are interconverting during simulations. With scaled peptide atomic charges, the whole peptide folded into a single helix in 11 of the 16 simulations. A helical structure observed at 39 ns in the simulation #3 is shown in Figure 1, where the majority of backbone hydrogen bonds is in the (i, i + 4) α‐helix pattern and a small number of hydrogen bonds in the (i, i + 3) 310 helix pattern. The hydrogen‐oxygen distances are slightly larger because the strength of charge–charge interactions is reduced. With the standard AMBER atomic charges, the whole peptide folded into a single helix in 12 of the 16 simulations, and different types of structures are more stable, interconverting less frequently. A helical structure observed at 50 ns in the simulation #2 is shown in Figure 1, with more (i, i + 4) backbone hydrogen bonds. The structures are displayed using the molecular graphics software of Schrodinger LLC. The hydrogen bonds in the figure are based on the default hydrogen bond criteria of the software, which include the maximum oxygen‐hydrogen distance of 2.5Å, the minimum donor angle of 120°, and the minimum acceptor angle of 90°. The geometrical hydrogen bond criteria are used in this study because the energy‐based definitions are force field parameter dependent.
Figure 1.
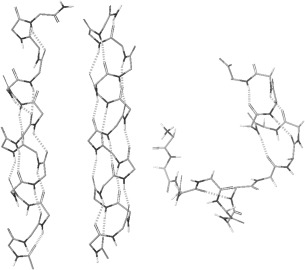
From left, a helical structure observed during simulations with scaled AMBER peptide atomic charges, a structure with the standard AMBER atomic charges, and a structure with scaled CHARMM peptide atomic charges. The C‐terminus is in the upper portion of each structure. Hydrogen bonds are shown as dotted lines. For a clear view of the backbone structure, non‐polar hydrogen atoms, side chains, and water molecules are not shown
As a measure of the progress of helix folding, the number of helical residues is calculated based on backbone dihedral angles. In literatures,46, 47, 48, 49, 52 several dihedral angle criteria of the helical conformation have been used, such as ϕ = −57° ± 30° and ψ = −47° ± 30°, ϕ = −65° ± 35°, and ψ = −42.5° ± 37.5°, ϕ = −65° ± 35°, and ψ = −37° ± 30°, and so forth. In this study, when the ϕ, ψ angles of two or more consecutive amino acid residues are within 30° from the standard α‐helical angles (ϕ = −57° and ψ = −47°), these residues are assumed to be in the helical conformation, which include the majority of α‐helix and 310 helix residues. The average percentage of helical residues over the 16 simulations at every 0.2 ns is shown in Figure 2 (left), as the helical contents. With scaled peptide atomic charges (solid line), it reaches an equilibrium value of 59.7% in 50 ns. With standard AMBER atomic charges (dash‐dotted line), the helical content is higher at 80.6%.
Figure 2.
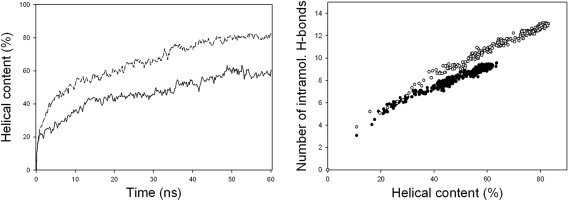
The left figure shows the average helical content over the 16 simulations with scaled AMBER peptide atomic charges (solid line) and with the standard AMBER atomic charges (dash‐dotted line). The right figure shows the average number of peptide intramolecular hydrogen bonds over the 16 simulations with scaled AMBER peptide atomic charges (filled circle) and with the standard AMBER atomic charges (open circle)
To address the issue of excessive intramolecular hydrogen bonds, the total number of peptide intramolecular hydrogen bonds is calculated with the same geometrical criteria as those used in Figure 1 and its average value over the 16 simulations at different helical contents is shown in Figure 2 (right). With the same helical content, the total number of the peptide intramolecular hydrogen bonds is lower with the scaled peptide atomic charges. The standard force field simulations have more peptide intramolecular hydrogen bonds at the same helical content during folding, as found in the experimental benchmarking study.5 In NPT ensemble simulations with scaled peptide atomic charges, the same qualitative features of helix folding are observed and the solution density is in the range of 1.00–1.01, showing that with a sufficient number of water molecules, the volume is mainly determined by water parameters.
Using scaled CHARMM peptide atomic charges and the NAMD program, helix folding of this peptide is tested in two simulations starting from unfolded structures. The peptide is solvated with 1570 water molecules in constant volume simulations with periodic boundary conditions. Helical turns and helical segments are observed within 30 ns in both simulations. A structure observed at 13.9 ns in the second simulation is shown in Figure 1. For comparison, simulations with standard CHARMM atomic charges are carried out from the same starting structures. With standard atomic charges, structures with one helical turn are observed, but the turn has not developed into longer helical segments during the simulations up to 100 ns. Peptide atomic charge scaling reduces energy barriers and the folding time in simulations using CHARMM force field, and reduces the equilibrium helical content in simulations using AMBER force field, making the difference smaller between the results from the two force fields.
β‐hairpin folding
Computational studies of another important secondary structure, the β‐sheet, are not as widely available as those of the α‐helix. With limited computing resources, a small peptide is the first choice for a folding simulation. Blanco et al.53 have successfully designed a small β‐hairpin peptide YQNPDGSQA, and Wu et al.54 have carried out a folding simulation of this peptide using standard AMBER parameters and an enhanced conformational search method.55
Using scaled (by ) AMBER peptide atomic charges, a group of 20 simulations of this peptide is carried out at 300K for 80 ns starting from 20 unfolded structures. For comparison, another group of 20 simulations are carried out from the same starting structures using the standard AMBER atomic charges. The peptide is solvated with 1273 water molecules in constant volume simulations with periodic boundary conditions. During simulations structures are recorded, and hydrogen bonds are calculated using the same geometrical criteria as in the helix folding section. With scaled peptide atomic charges, structures with 3 or 4 β‐hairpin backbone hydrogen bonds are observed in 11 of the 20 simulations. A structure observed at 61.62 ns in simulation #3 is shown in Figure 3. Its hydrogen bonding pattern is consistent with the model structure based on NOE data.53 With the standard AMBER atomic charges, structures with 1 or 2 β‐hairpin backbone hydrogen bonds are observed, such as the structure at 64.47 ns of the simulation #20 shown in Figure 3, but no structures with 3 or 4 β‐hairpin backbone hydrogen bonds consistent with the NOE based model structure are observed within 80 ns in any of the 20 simulations. A much longer simulation time is needed for β‐hairpin folding with standard AMBER atomic charges without enhanced conformational search techniques. β‐hairpin folding using the scaled peptide atomic charges shows higher conformational search efficiency.
Figure 3.
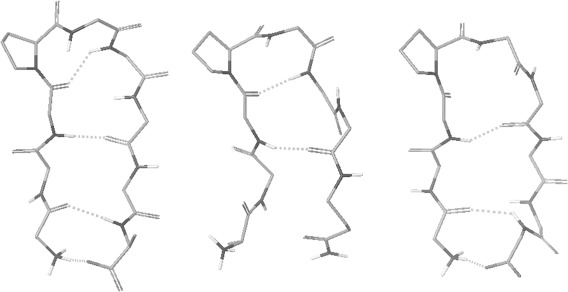
From left, a β‐hairpin structure observed during simulations with scaled AMBER peptide atomic charges, a structure with the standard AMBER atomic charges, and a structure with scaled CHARMM peptide atomic charges. The N‐terminus is on the left side of each molecule. Other descriptions about the atom display are the same as in Figure 1
Unlike for helical structures, the backbone dihedral angle‐based criteria are not widely available for β‐hairpin structures, except for the two residues at the β‐turn. The number of the β‐hairpin backbone hydrogen bonds consistent with the model structure based on NOE data53 is used as a measure of the β‐hairpin folding and its average values over the 20 simulations at every 0.2 ns are shown in Figure 4 (left). Their 80 ns average is 0.45 with the scaled peptide atomic charges (solid line) and 0.42 with standard AMBER atomic charges (dotted line). The average number of intramolecular hydrogen bonds over the 20 simulations vs. that of the backbone hydrogen bonds during the simulations is shown in Figure 4 (right). The filled circles show the average numbers of intramolecular hydrogen bonds using the scaled peptide atomic charges and the open circles show those using standard AMBER atomic charges, which is higher than the filled circles by 1.2 hydrogen bonds on average. With the same number of β‐hairpin backbone hydrogen bonds, reduced charge–charge interactions result in smaller total number of peptide intramolecular hydrogen bonds, because van der Waals interactions in the peptide‐solvent system contribute substantially to folding.39, 56, 57 Reduced hydrogen bonding in intermediate structures increases the conformational search efficiency.
Figure 4.

The left figure shows the average numbers of β‐hairpin backbone hydrogen bonds over the 20 simulations with scaled AMBER peptide atomic charges (solid line) and with the standard AMBER atomic charges (dotted line). The right figure shows the average peptide intramolecular hydrogen bonds over the 20 simulations with scaled AMBER peptide atomic charges (filled circle) and with the standard AMBER atomic charges (open circle)
Using scaled CHARMM peptide atomic charges and the NAMD program, β‐hairpin folding of this peptide is tested in two simulations at 300K. The peptide is solvated with 1002 water molecules in constant volume simulations with periodic boundary conditions. Staring from unfolded structures, structures with 3 β‐hairpin backbone hydrogen bonds are observed in both simulations. Figure 3 shows such a structure observed at 7.685 ns of the second simulation. From the same starting structures, two simulations are carried out with standard CHARMM atomic charges. Structures with one β‐hairpin backbone hydrogen bond appear multiple times, but structures with more than one β‐hairpin backbone hydrogen bond are not observed during the two simulations up to 120 ns. Using scaled CHARMM peptide atomic charges, energy barriers in β‐hairpin folding simulations are reduced, making the conformational search more efficient.
Discussion
The recent experimental benchmarking study5 found excessive intramolecular hydrogen bonds in protein folding simulations, indicating a possible overestimate of intramolecular charge–charge interactions. Based on the approximate dielectric screening effect of the electronic polarization, peptide atomic charges are scaled down in this study. As a result, the number of intramolecular hydrogen bonds is lower than using unscaled atomic charges while reaching the same levels of helical contents or β‐hairpin backbone hydrogen bonds, because van der Waals interactions in the peptide‐solvent system contribute substantially to peptide folding.39, 56, 57 Reducing intramolecular charge–charge interactions lowers the stability of hydrogen bonded intermediate structures and energy barriers in the folding landscape, making the conformational search more efficient. The faster folding favors higher cooperativity in folding kinetics. These results are in better agreement with experimental observations in folding energetics and kinetics.5 The peptide atomic charge scaling factor may be further optimized in more quantitative studies in the future, including possible adjustments of Lennard‐Jones parameters of peptide atoms. More accurate dielectric screening at each atomic position in every protein‐solvent structure during folding simulations can be calculated using polarizable models or quantum mechanical models when sufficient computing resources become available.
Acknowledgments
The author is grateful to Dr. R. L. Baldwin for very helpful discussions and valuable suggestions.
Significance Statements Based on findings of the experimental benchmarking work, peptide atomic charges are scaled down to include the dielectric screening effect of the electronic polarization. This treatment reduces the excessive intramolecular hydrogen bonds, and increases conformational search efficiency in simulations. In particular, it reduces equilibrium helical contents in simulations with AMBER force field and folding energy barriers with CHARMM force field.
References
- 1. Voelz VA, Bowman GR, Beauchamp K, Pande VS (2010) Molecular simulation of ab initio protein folding for a millisecond folder NTL9(1–19). J Am Chem Soc 132:1526–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shaw DE, Maragakis P, Lindorff‐Larsen K, Piana S, Dror RO, Eastwood MP, Bank JA, Jumper JM, Salmon JK, Shan Y, Wriggers W (2010) Atomic‐level characterization of the structural dynamics of proteins. Science 330:341–346. [DOI] [PubMed] [Google Scholar]
- 3. Bowman GR, Voelz VA, Pande VS (2011) Atomistic folding simulations of the five‐helix bundle protein λ(6–85). J Am Chem Soc 133:664–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Piana S, Lindorff‐Larsen K, Shaw DE (2011) How robust are proteins folding simulations with respect to force field parameterization?. Biophys J 100:L47–L49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Skinner JJ, Yu W, Gichana EK, Boxa MC, Hinshaw JR, Freed KF, Sosnick TR (2014) Benchmarking all‐atom simulations using hydrogen exchange. Proc Natl Acad Sci USA 111:15975–15980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lindorff‐Larsen K, Piana S, Dror RO, Shaw DE (2011) How fast‐folding proteins fold. Science 334:517–520. [DOI] [PubMed] [Google Scholar]
- 7. Schulz GE, Schirmer RH (1990) Principles of protein structure. New York: Springer‐Verlag, pp 30–33. [Google Scholar]
- 8. Barnes P, Finney JL, Nicholas JD, Quinn JE (1979) Cooperative effects in simulated water. Nature 282:459–464. [Google Scholar]
- 9. Sung S, Jordan PC (1986) Structures and energetics of monovalent ion‐water microclusters. J Chem Phys 85:4045–4051. [Google Scholar]
- 10. Cieplak P, Caldwell JW, Kollman PA (2001) Molecular mechanics models for organic and biological systems going beyond the atom centered two body additive approximation. J Comput Chem 22:1048–1057. [Google Scholar]
- 11. Kaminski GA, Stern HA, Berne BJ, Friesner RA, Cao XY, Murphy RB, Zhou R, Halgren TA (2002) Development of a polarizable force field for proteins via Ab Initio quantum chemistry: first generation model and gas phase tests. J Comput Chem 23:1515–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yu H, Hansson T, van Gunsteren WF (2003) Development of a single self‐consistent polarizable model for liquid water. J Chem Phys 118:221. [Google Scholar]
- 13. Lamoureux G, Roux B (2003) Modeling induced polarization with classical Drude oscillators: theory and molecular dynamics simulation algorithm. J Chem Phys 119:3025. [Google Scholar]
- 14. Patel S, Brook CL (2004) CHARMM fluctuating charge force field for proteins: I parameterization and application to bulk organic liquid simulations. J Comput Chem 25:1–15. [DOI] [PubMed] [Google Scholar]
- 15. Patel S, Markerell AD, Brook CL (2004) CHARMM fluctuating charge force field for proteins: II protein/solvent properties from molecular dynamics simulations using a nonadditive electrostatic model. J Comput Chem 25:1504–1514. [DOI] [PubMed] [Google Scholar]
- 16. Ren P, Ponder JW (2004) Temperature and pressure dependence of the AMOEBA water model. J Phys Chem B 108:13427–13437. [Google Scholar]
- 17. Ren P, Wu C, Ponder JW (2011) Polarizable atomic multipole‐based molecular mechanics for organic molecules. J Chem Theory Comput 7:3143–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shi Y, Xia Z, Zhang J, Best R, Wu C, Ponder JW, Ren P (2013) The polarizable atomic multipole‐based AMOEBA force field for proteins. J Chem Theory Comput 9:4046–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gilson MK, Honig HB (1986) The dielectric constant of a folded protein. Biopolymers 25:2097–2119. [DOI] [PubMed] [Google Scholar]
- 20. Gilson MK, Sharp KA, Honig HB (1988) Calculating electrostatic interactions in bio‐molecules: method and error assessment. J Comput Chem 9:327–335. [Google Scholar]
- 21. Dorman VL, Jordan PC (2004) Ionic permeation free energy in gramicidin: a semimicroscopic perspective. Biophys J 86:3529–3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jorgensen WL (1981) Transferable intermolecular potential functions for water, alcohols, and ethers. Application to liquid water. J Am Chem Soc 103:335–340. [Google Scholar]
- 23. Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans J, Interaction models for water in relation to protein hydration In: Pullman B, Ed. (1981) Intermolecular forces. Reidel, Dordrecht, p. 331–338. [Google Scholar]
- 24. Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935. [Google Scholar]
- 25. Fennell CJ, LI L, Dill KA (2012) Simple liquid models with corrected dielectric constants. J Phys Chem B 116:6936–6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dang LX, Chang TM (1997) Molecular dynamics study of water clusters, liquid, and liquid‐vapor interface of water with many‐body potential. J Chem Phys 106:8149–8159. [Google Scholar]
- 27. Silvestrelli PL, Parrinello M (1999) Structural, electronic, and bonding properties of liquid water from first principle. J Chem Phys 111:3572–3581. [Google Scholar]
- 28. Badyal YS, Saboungi ML, Price DL, Shastri SD, Haeffner DR, Soper AK (2000) Electronic distribution in water. J Chem Phys 112:9206–9209. [Google Scholar]
- 29. Gubskaya AV, Kusalik PG (2002) The total molecular dipole moment for liquid water. J Chem Phys 117:5290–5303. [Google Scholar]
- 30. Leontyev IV, Vener VM, Rostov IV, Basilevsky MV, Newton DE (2003) Continuum level treatment of electronic polarization in the framework of molecular simulations of solvation effects. J Chem Phys 119:8024–8037. [Google Scholar]
- 31. Leontyev IV, Stuchebrukhov A (2011) Accounting for electronic polarization in the non‐polarizable force field. Phys Chem Chem Phys 13:2613–2626. [DOI] [PubMed] [Google Scholar]
- 32. Schroder C (2012) Comparing reduced partial charge models with polarizable simulations of ionic liquids. Phys Chem Chem Phys 14:3089–3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Atkins PW (1978) Physical chemistry. San Francisco: W. H. Freeman and Company, p. 317. [Google Scholar]
- 34.(2015) CRC Handbook of chemistry and physics. Cleveland: CRC Press, Vol. 6, 187–208. [Google Scholar]
- 35. Hasted JB, Liquid water: dielectric properties In: Franks F, Ed. (1972) Water, a comprehensive treatise. Plenum, NY, Vol. 1, pp. 255–309. [Google Scholar]
- 36. Zafar MS, Hasted JB, Chamberlain J (1973) Submillimeter wave dielectric dispersion in water. Nature 243:106–109. [Google Scholar]
- 37. Gilson M (1995) Theory of electrostatic interactions in macromolecules. Curr Opin Struct Biol 5:216–223. [DOI] [PubMed] [Google Scholar]
- 38. Warshel A, Sharma PK, Koto M, Parson WW (2006) Modeling electrostatic effects of proteins. Biochim Biophys Acta 1764:1647–1676. [DOI] [PubMed] [Google Scholar]
- 39. Sung S (2015) Peptide folding driven by van der Waals interactions. Protein Sci 24:1383–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Jr. Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA (1995) A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc 117:5179–5197. [Google Scholar]
- 41. Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE, III DeBolt S, Ferguson D, Seibel G, Kollman PA (1995) AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comp Phys Commun 91:1–41. [Google Scholar]
- 42. Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M (1983) CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comp Chem 4:187–217. [Google Scholar]
- 43. Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ryckaert JP, Ciccotti G, Berendsen HJC (1997) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n‐alkanes. J Comput Phys 23:327–341. [Google Scholar]
- 45. Sung S (1994) Helix folding simulations with various initial conformations. Biophys J 66:1796–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sung S (1995) Folding simulations of alanine‐based peptides with lysine residues. Biophys J 68:826–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Garcia AE, Sanbonmatsu KY (2002) Alpha‐helix stabilization by side chain shielding of backbone hydrogen bonds. Proc Natl Acad Sci USA 99:2782–2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Best RB, Hummer G (2009) Optimized molecular dynamics force fields applied to the helix‐coil transition of polypeptides. J Phys Chem B 113:9004–9015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Henry ER, Best RB, Eaton WA (2013) Comparing a simple theoretical model for protein folding with all‐atom molecular dynamics simulations. Proc Natl Acad Sci USA 110:17880–17885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang H, Sung S (2000) Molecular dynamics simulations of three‐strand β‐sheet folding. J Am Chem Soc 122:1999–2009. [Google Scholar]
- 51. Scholtz JM, York EJ, Steward JM, Baldwin RL (1991) A natural water‐soluble, α‐helical peptide: the effect of ionic strength on the helix‐coil equilibrium. J Am Chem Soc 113:5102–5104. [Google Scholar]
- 52. Daggett V, Levitt M (1992) Molecular dynamics simulations of helix denaturation. J Mol Biol 223:1121–1138. [DOI] [PubMed] [Google Scholar]
- 53. Blanco FJ, Jimenez MA, Herranz J, Rico M, Santoro J, Nieto JL (1993) NMR evidence of a short linear peptide that folds into a β‐hairpin in aqueous solution. J Am Chem Soc 115: 5887–5888. [ΧρoσσΡɛϕ][10.1021/φα00066α092] [Google Scholar]
- 54. Wu X, Wang S, Brooks BR (2002) Direct observation of folding and unfolding of a β‐hairpin in explicit water through computer simulation. J Am Chem Soc 124:5282–5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wu X, Wang S (1998) Self‐guided molecular dynamics simulation for efficient conformational search. J Phys Chem B 102:7238–7250. [Google Scholar]
- 56. Baldwin RL (2014) Dynamic hydration shell restores Kauzmann's 1959 explanation of how the hydrophobic factor drives protein folding. Proc Natl Acad Sci USA 111:13052–13056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jorgensen WL, Gao J, Ravimohan C (1985) Monte Carlo simulations of alkanes in water: hydration numbers and the hydrophobic effect. J Phys Chem 89:3470–3473. [Google Scholar]