

Abstract
T4 phage lysozyme is an inverting glycoside hydrolase that degrades the murein of bacterial cell walls by cleaving the β‐1,4‐glycosidic bond. The substitution of the catalytic Thr26 residue to a histidine converts the wild type from an inverting to a retaining enzyme, which implies that the original general acid Glu11 can also act as an acid/base catalyst in the hydrolysis. Here, we have determined the neutron structure of the perdeuterated T26H mutant to clarify the protonation states of Glu11 and the substituted His26, which are key in the retaining reaction. The 2.09‐Å resolution structure shows that the imidazole group of His26 is in its singly protonated form in the active site, suggesting that the deprotonated Nɛ2 atom of His26 can attack the anomeric carbon of bound substrate as a nucleophile. The carboxyl group of Glu11 is partially protonated and interacts with the unusual neutral state of the guanidine moiety of Arg145, as well as two heavy water molecules. Considering that one of the water‐binding sites has the potential to be occupied by a hydronium ion, the bulk solvent could be the source for the protonation of Glu11. The respective protonation states of Glu11 and His26 are consistent with the bond lengths determined by an unrestrained refinement of the high‐resolution X‐ray structure of T26H at 1.04‐Å resolution. The detail structural information, including the coordinates of the deuterium atoms in the active site, provides insight into the distinctively different catalytic activities of the mutant and wild type enzymes.
Keywords: lysozyme, hydrolysis, perdeuteration, neutron crystallography
Short abstract
Abbreviations
- GH
glycoside hydrolase
- HEWL
hen egg‐white lysozyme
- NAG
N‐acetylglucosamine
- NAM
N‐acetylmuramic acid
- T4L
T4 phage lysozyme
- WT
wild type
Introduction
Lysozymes are glycoside hydrolases that are involved in the degradation of murein, a major bacterial cell‐wall component, which catalyze the hydrolytic cleavage of the β‐1,4‐glycosidic bond between N‐acetyl muramic acid (NAM) and N‐acetyl glucosamine (NAG).1 On the basis of the different anomeric structures of hydrolyzed products, the members are divided into two types: retaining and inverting enzymes, and are further classified in seven glycoside hydrolase (GH) families and subfamilies (GH19, GH22c, GH22i, GH23, GH24v, GH24l, and GH46) of the lysozyme superfamily, based on their amino‐acid sequences.2, 3 The most characterized member is hen egg‐white lysozyme (HEWL) of the subfamily GH22c (for the chicken type). HEWL is a retaining enzyme that catalyzes the hydrolysis by a double displacement mechanism, in which the scissile glycosidic bond is cleaved between the two catalytic residues, Glu35 which functions as a general acid/base and Asp52 as a nucleophile.4, 5 This retaining mechanism has been verified by neutron structure analyses on HEWL from the viewpoint of the protonation states of the catalytic residues.6, 7
In contrast to HEWL, T4 phage lysozyme (T4L) is an inverting enzyme, and is classified in the subfamily GH24v (for the viral type).3, 8 In spite of the almost entirely different amino‐acid sequences between T4L and HEWL, their three‐dimensional structures share a similar two‐domain folding pattern (Fig. 1).9 The highly conserved element within the active site is the glutamic acid residue Glu11 corresponding to Glu35 in HEWL. The position of the nucleophilic Asp52 residue in HEWL is occupied by Thr26 in T4L rather than the acidic residue Asp20, understandably because of its different hydrolysis mechanism. In the case of inverting enzymes, a general base residue abstracts a proton from a water located at the α‐side of the anomeric sugar carbon, accompanying with a general acid assistance for the sugar departure, in which the enzyme cleaves the scissile glycosidic bond in a single step.5
Figure 1.
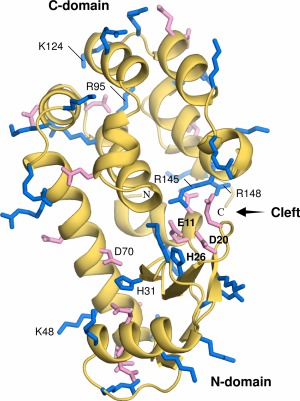
Ribbon diagram of the T26H mutant showing its two‐domain structure. The side‐chains of acidic (Asp and Glu) and basic (Arg, His and Lys) residues are shown with sticks and colored in pink and blue, respectively. The active site residues Glu11, Asp20 and His26 in the cleft are indicated in bold letters. Further amino acid residues described in the text are labeled.
Previously, we reported that the substitution of Thr26 in T4L with a nucleophilic histidine residue causes the hydrolytic product to change from the α‐ to β‐anomeric configuration. This clearly suggests that there is a change in the mechanism of hydrolysis.10, 11 Further structure‐based mutageneses and biochemical analyses showed that the T26H mutant is indeed a retaining enzyme that employs a double displacement mechanism, in which the imidazole ring of His26 acts as a nucleophile, leading to a covalent adduct intermediate. Subsequently, a water molecule deprotonated by Glu11 attacks the anomeric carbon of bound substrate from its equatorial direction (Supporting Information Fig. S1). It implies that the carboxyl group of Glu11 can acquire an anionic form to polarize the attacking water during the hydrolysis. In contrast, in the wild type (WT) enzyme, Glu11 functions as a general acid catalyst. Furthermore, the substitution of Thr26 by histidine in T4L not only induces the dramatic change in the hydrolysis mechanism but also facilitates transglycosylation more effectively than hydrolysis. In contrast, WT T4L has no transglycosidase activity whatever.
Here, to gain insight into the roles of Glu11 and His26 in the catalytic activities of the T26H mutant, we determined its neutron structure. The perdeuterated recombinant protein was prepared using a commercial deuterated culture media, and was used for crystallization.12, 13 The neutron structure of the perdeuterated T26H mutant has been determined at 2.09 Å‐resolution at room temperature using a crystal with a volume of less than 0.9 mm3. Indeed, the perdeuteration made it possible to reduce the crystal size and to shorten the data collection time, and still allow the visualization of protonation and hydration sites throughout the enzyme.12, 14 To complement the interpretation of the protonation states identified from the nuclear‐density maps, the high‐resolution structure of the same perdeuterated protein was also determined by X‐ray crystallography at 1.04‐Å resolution under cryogenic conditions.
Results
Neutron structure determination of the T26H mutant
The neutron diffraction data were collected by the quasi‐Laue technique at room temperature.15 The neutron structure of T26H at 2.09‐Å resolution was determined by the joint X‐ray/neutron (X/N) refinement using X‐ray diffraction data from the same crystal which diffracted to 1.65‐Å resolution at room temperature (Supporting Information Table SI). The final coordinates are comprised of 3,018 protein atoms including 1,529 deuterium atoms. The mean isotropic B‐factor values of the T26H mutant are 39.4 Å2 for the whole constituent atoms, 34.2 Å2 for the main‐chain atoms, and 41.1 Å2 for the side‐chain atoms. One sodium ion and three chloride ions were identified based on their interatomic distances to surrounding atoms. A total of 86 heavy water molecules were assigned through the refinement of X‐ray data. Of these 56 waters were interpreted as full D2O molecules by the apparent nuclear‐density distributions for deuterium, and other two molecules were modelled as singly deuterated oxygen species due to the partial lack of nuclear densities. The remaining 28 waters did not show any residual nuclear densities and were assigned as single oxygen atoms.
The overall structure of the perdeuterated T26H mutant was nearly identical to that of the non‐deuterated T26H (PDB ID: 1QT8) previously determined by X‐ray crystallography.11 The root‐mean‐square deviation (rmsd) of the 144 Cα atoms was 0.20 Å and that of all the atoms was 0.64 Å, indicating that the protein deuteration did not cause any significant changes on its overall structure. Also, the side‐chain conformations were almost identical. The structure of the T26H mutant is similar to WT with an N‐ and C‐domain (Fig. 1).16 An active site cleft for hydrolysis is formed between the two domains, which is rich in hydrophilic residues and filled with solvent. The catalytic residue Glu11, acting as a general acid in the inverting reaction by WT, is located at the bottom of the cleft, and the Asp20 and His26 residues from the N‐domain are arranged opposite to Glu11 in the cleft.
Protonation states of acidic and basic amino acids
The T26H mutant contains 18 acidic residues (ten Asp and eight Glu) and 28 basic residues (13 Arg, 13 Lys, and two His). Since the crystal was grown in the presence of a high concentration of sodium‐potassium phosphate at pD 7.0, it was assumed in the starting model for the joint X/N refinement, that all of the acidic residues were deprotonated and all of the basic residues were protonated. However, a strong peak was observed around the carboxyl group of Asp20 in a F o − F c deuterium (D)‐omit nuclear‐density map, indicating that this residue is in its protonated form [Fig. 2(A)]. The Asp20 side‐chain with a mean B‐factor of 40.8 Å2 is located in the active site cleft, and its carboxyl Oδ1 atom forms hydrogen bonds with the main‐chain amides of Glu22 and Tyr24. The other carboxyl Oδ2 atom, assigned to be protonated, is exposed to the bulk solvent and interacts with a water molecule at a hydrogen bonding distance of 2.1 Å.
Figure 2.
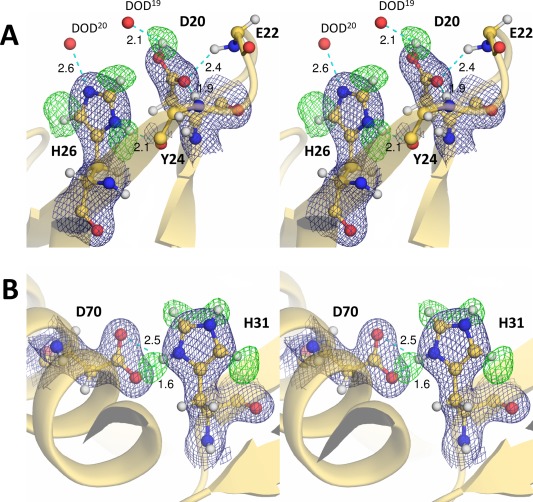
Nuclear‐density maps for the histidine and adjacent acidic residues. Each residue is represented as a ball‐and‐stick model. The 2F o – F c electron‐density maps (contoured at 1.5 σ) for the respective residues and their F o – F c deuterium (D)‐omit nuclear‐density maps (contoured at 3.2 σ) are shown as deep blue and green meshes, respectively. Hydrogen bonds are shown by cyan dashed lines, with distances in Å. (A) The carboxyl Dδ2 atom of Asp20 and the imidazole deuterium atoms of His26 were omitted from the Fourier synthesis calculation. (B) Only the imidazole deuterium atoms of His31 were omitted from the Fourier synthesis calculation. The doubly protonated imidazole ring of His31 forms a salt bridge with the deprotonated carboxylate group of Asp70.
The two histidine residues, His26 located in the active site cleft and His31 near the end of the cleft, differ in their protonation states. An apparent F o − F c peak corresponding to a deuterium atom bound to the Nδ1 nitrogen was observed besides the peaks for deuterium atoms bound to the Cɛ1 and Cδ2 carbons, indicating that the His26 side‐chain is singly protonated, corresponding to the neutral form of the imidazole ring [Fig. 2(A)]. The B‐factor for the imidazole ring of His26, including the deuterium atoms, is 34.6 Å2. The Nδ1 nitrogen is in a hydrogen bonding distance with the main‐chain carbonyl group of Tyr24. The residual electron density near the Nɛ2 atom, at a distance of 2.6 Å, was assigned as a mobile heavy water. The imidazole ring of His26 is located adjacent to the Asp20 side‐chain, but there is no specific interaction between them. The second histidine (His31), with a B‐factor of 33.0 Å2 for the imidazole ring, appeared to form a salt bridge with the deprotonated carboxyl group of Asp70 [Fig. 2(B)]. All four peaks corresponding to the deuterium atoms bound to Nδ1, Cɛ1, Nɛ2, and Cδ2 of His31 were observed in a F o − F c D‐omit nuclear‐density map, showing that the imidazole ring is in its doubly protonated form.
Interestingly, the guanidine moiety of Arg145 lacks nuclear density for the Dɛ atom when calculating the F o − F c D‐omit nuclear‐density map, indicating that Arg145 adopts its neutral form [Fig. 3(A)]. No obvious F o − F c D‐omit peak was found adjacent to the carboxyl group of Glu11 which interacts with the side‐chain of Arg145 and also forms hydrogen bonds with two heavy water molecules [Fig. 4(A)]. In contrast, the sigma‐A weighted 2F o − F c nuclear‐density map suggested that the Glu11 side‐chain was protonated [Fig. 4(B)]. The crystallographic occupancy of the carboxyl proton of Glu11 was estimated to be 0.6 so as to have approximately the same B‐factor as the carboxyl oxygen Oɛ2 to which the Dɛ2 atom is bound. One of the water‐binding sites adjacent to Glu11 shows relatively large density for one water molecule, which was interpreted as a molecule (DOD4) having two different orientations [Fig. 4(C)]. This water‐binding site is surrounded by the side‐chain carboxyl Oɛ2 and main‐chain carbonyl oxygen of Glu11 in addition to the phenolic hydroxyl group of Tyr18.
Figure 3.
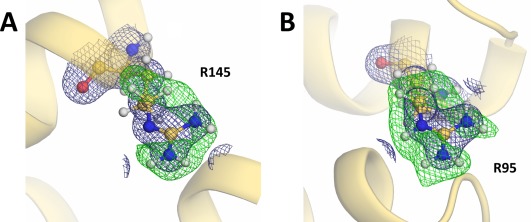
Nuclear‐density maps for the representative arginine residues. Each residue is represented as a ball‐and‐stick model. The 2F o – F c electron‐density maps (contoured at 1.5 σ) for the respective residues and their F o – F c deuterium (D)‐omit nuclear‐density maps (contoured at 3.2 σ) are shown as deep blue and green meshes, respectively. (A) The guanidine deuterium atoms of Arg145 in addition to the Dδ1 and Dδ2 atoms were omitted from the Fourier synthesis calculation. (B) The guanidine deuterium atoms of Arg95 in addition to the Dδ1 and Dδ2 atoms were omitted from the Fourier synthesis calculation. The guanidine moiety was assigned as a protonated guanidinium ion based on its D‐omit nuclear‐density map.
Figure 4.
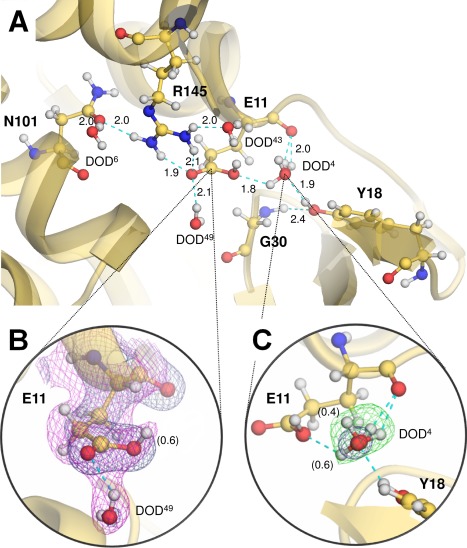
Protonation states of the active site residues. (A) Ball‐and‐stick representation of the active site residues. Hydrogen bonds are shown by cyan dashed lines, with distances in Å. (B) Nuclear‐density map for the catalytic Glu11 residue. The 2F o – F c electron‐density maps (deep blue) for Glu11 and a surrounding water molecule (DOD49) are contoured at 1.5 σ. The sigma‐A 2F o – F c nuclear‐density map (magenta) contoured at 1.0 σ suggests the protonated form of Glu11, in which its crystallographic occupancy was estimated to be 0.6. (C) Nuclear‐density map for DOD4 interacting with Glu11. The 2F o – F c electron‐density map (deep blue) for a heavy water (DOD4) interacting with Glu11 is contoured at 1.5 σ, and the F o – F c D‐omit nuclear‐density map (green) is contoured at 3.2 σ. This water‐binding site was assigned as a mixed state with two different orientations. Their relative occupancies are indicated in parentheses.
No significant peak in a F o − F c D‐omit nuclear‐density was observed close to the carboxyl groups of the remaining acidic residues, and consequently these residues were determined to be deprotonated. While the nuclear‐density maps at 2.09‐Å resolution made it possible to detect the protonated forms of the residues described above, it was difficult to verify the protonation states of lysines due to their flexibility. Only Lys48 and Lys124, having relatively low side‐chain B‐factors (39.1 Å2 and 37.9 Å2, respectively), were unambiguously assigned as positively charged protonated forms (data not shown). Visualizing the protonation states of arginines was also limited to the residues with relatively low B‐factors, namely Arg95 (30.1 Å2), Arg145 (33.2 Å2) and Arg148 (33.7 Å2). Comparing each of the F o − F c D‐omit nuclear‐density maps, the lack of a peak for deuterium at the Nɛ position was observed only for Arg145 (Fig. 3). The guanidine moiety of this arginine forms a hydrogen bond with the carboxyamide Oδ1 oxygen of Asn101 in addition to the carbonyl Oɛ1 oxygen of Glu11 [Fig. 4(A)].
High‐resolution X‐ray structure of the T26H mutant
A high‐resolution X‐ray structure of the perdeuterated T26H mutant was determined to complement the interpretation of the protonation states identified in the neutron structure analysis. The crystal structure was refined against diffraction data at 1.04‐Å resolution and converged to the R work/R free values of 15.9/19.3%. To deduce the respective C—O bond lengths for the carboxyl group of aspartic and glutamic acid, a conjugate‐gradient least‐squares (CGLS) refinement was performed that removed all geometrical restraints on the bond lengths and angles for the carboxyl moiety. The geometrical restraints for the imidazole group of histidine were also removed. The C—O bond lengths for the carboxyl group and the stereochemical parameters for the imidazole ring are listed in Table 1 and Table 2, respectively.
Table 1.
Geometrical Parameters for the Carboxyl Moiety of Acidic Residues and their Estimated Standard Deviations
| C—O bond lengths (Å) | B‐factors (Å2)a | |
|---|---|---|
| Asp10 | 1.231 (13) 1.274 (13) | 12.0 |
| Glu11 | 1.264 (18) 1.294 (17) | 14.9 |
| Asp20 | 1.191 (19) 1.279 (21) | 17.1 |
| Glu62 | 1.249 (17) 1.266 (15) | 14.8 |
| Glu64 | 1.247 (19) 1.248 (19) | 16.4 |
| Asp70 | 1.233 (14) 1.242 (13) | 12.3 |
| Asp92 | 1.259 (16) 1.283 (18) | 14.6 |
| Asp127 | 1.237 (19) 1.275 (18) | 16.0 |
| Anionic formb | 1.256 (15) | − |
| Protonated formb | 1.210 (16) 1.310 (17) | − |
Values are calculated for the atoms forming each carboxyl moiety.
Experimental data from Fisher et al. (2012).17
Table 2.
Geometrical Parameters of the Imidazole Ring of Histidines and their Estimated Standard Deviations
| Cɛ1—Nδ1 (Å) | Cɛ1—Nɛ2 (Å) | —Nδ1— (°) | —Nɛ2— (°) | B‐factors (Å2)a | |
|---|---|---|---|---|---|
| His26 | 1.341 (19) | 1.343 (20) | 107.4 (12) | 105.6 (13) | 13.6 |
| His31 | 1.290 (16) | 1.333 (18) | 109.9 (11) | 107.5 (11) | 12.7 |
| Nδ1 protonatedb | 1.338 (8) | 1.316 (9) | 108.1 (5) | 105.3 (6) | − |
| Doubly protonatedb | 1.325 (8) | 1.316 (9) | 109.3 (5) | 108.9 (6) | − |
Values are calculated for the atoms forming each imidazole ring.
Experimental data from Malinska et al. (2015).20
The C—O bond lengths are generally known to be 1.256 Å for the delocalized bonds in the carboxylate form, and to be 1.210 Å for the C=O bond and 1.310 Å for the C—OH bond, respectively, in the protonated carboxyl form, although these lengths can be influenced by the surrounding environment.17, 18 A significant difference between the C—O bond lengths was found for Asp20, but not for any other such residues in the protein (Table 1). The data clearly suggests that Asp20 has a protonated carboxyl group. The difference between the C—O bond lengths (1.264 Å and 1.294 Å) for Glu11 might be insufficient to discriminate between each protonation state, but the longer length is most comparable with the ideal value for a C—OH bond. It presumably implies that both the protonated and deprotonated forms of Glu11 occur in the crystal, resulting in somewhat unequal C—O bond lengths. Indeed, the observed bond lengths for Glu11 correspond well with those for a partially protonated Asp residue in human chitotriosidase.19
Hydrogen atoms bound to either or both of the two nitrogen atoms, Nδ1 and Nɛ2, of an imidazole ring can slightly affect the geometry of the five‐membered system.17, 20 If both nitrogen atoms are protonated, each bond length between Cɛ1 and its nitrogen neighbors is shortened compared to that of the singly protonated forms because the bonds gain partial double‐bond characteristics. Furthermore, their endocyclic angles at the Nδ1 and Nɛ2 atoms become similar and larger by more than 1° compared to those of the singly protonated imidazole, in which the ideal —Nδ1— angle is proposed as 109.3°, and the —Nɛ2— angle is 108.9° (Table 2). The bond lengths of Cɛ1—Nδ1 and Cɛ1—Nɛ2 in His31 are shorter than those of His26, and the angles at the Nδ1 and Nɛ2 atoms of His31 are larger than those of His26, suggesting that the protonation state of His31 is different from that of His26. Considering that the calculated angles at the Nδ1 and Nɛ2 atoms of His31 are in good agreement with the respective ideal values, the residue can be inferred to be in the doubly protonated form, as also observed in the neutron structure. Similarly, the calculated angles at the Nδ1 and Nɛ2 atoms of His26 are enough close to the ideal values of 108.1° and 105.3°, respectively, for the Nδ1 singly protonated form of imidazole, although the bond lengths of Cɛ1—Nδ1 and Cɛ1—Nɛ2 of His26 are not necessarily consistent with the ideal values.
Recently, molecular guanidine was crystallized, and the respective C—N bonds were evaluated based on the neutron and X‐ray structures which showed that the C=NH bond was ca. 0.06 Å shorter than the C—NH2 bond.21 Since the positive charge of the guanidinium ion can be delocalized over the three nitrogen atoms, the respective C—N bond lengths would be similar to each other. When applying an unrestrained refinement to the guanidine moiety of Arg145 in the T26H mutant, the C—N bonds were estimated as 1.282(19) Å for Cζ—Nɛ, 1.348(20) Å for Cζ—Nη1, and 1.344(15) Å for Cζ—Nη2. Thus, the obtained geometrical parameters strongly support the existence of the neutral form of Arg145 as identified in the neutron structure, because when the deprotonation occurs at the Nɛ atom of guanidine, one double bond is formed between the Cζ and Nɛ atoms in the guanidine group.
Discussion
The neutron structure at 2.09‐Å resolution allowed the determination of the protonation states of the catalytic residues located in the active site cleft. These observations were fully compatible with the bond lengths determined in an unrestrained refinement of the high‐resolution X‐ray structure. Previously, we reported that the substitution of Thr26 in the active site to a glutamic acid converts WT into an inactive enzyme. In the crystal structure of the T26E mutant, it was found that its hydrolyzed product, composed of a NAM‐NAG disaccharide with a peptidic fragment, was trapped by forming a covalent bond between the C‐1 anomeric carbon of NAM and the carboxyl oxygen of Glu26.9 From the structural comparison with WT, it was concluded that the anionic carboxyl group of Glu26 nucleophilically attacked the C‐1 carbon from the α‐side, as is the case with the attacking water in the inverting hydrolysis by WT. Accordingly, the His26 residue in the T26H mutant was also presumed to adopt its nucleophilic deprotonated form for the first step of the retaining reaction (Supporting Information Fig. S1).11 In the present neutron structure analysis of the T26H mutant, the catalytic His26 residue was identified as a singly protonated form. When the active site cleft of T26H is superimposed on that of the T26E‐adduct complex, the Nɛ2 atom of His26 is 2.7 Å away from the C‐1 carbon of NAM (Fig. 5), supporting that His26 can act as a nucleophile for hydrolysis (Scheme 1, right). The protonated form of the Asp20 side‐chain was also observed, but its carboxyl group does not directly interact with the adjacent imidazole group of His26 presumably due to their respective neutral forms. This observation is in good agreement with the result of mutational analyses in which it was found that Asp20 plays a less important role in hydrolysis by T26H.11
Figure 5.
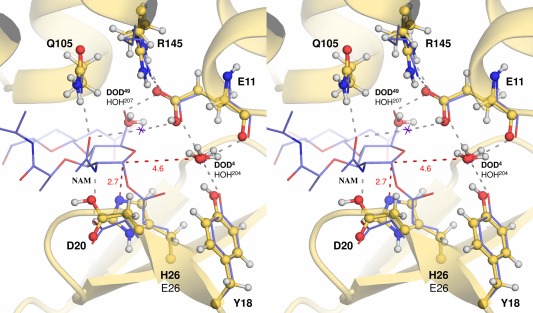
Superimposition of the active sites of the T26H mutant and the T26E‐adduct complex. The neutron structure of the T26H mutant (pale yellow ball‐and‐sticks labeled with bold letters) was superimposed to the T26E mutant structure complexed with a covalent adduct (PDB ID: 148L, violet lines labeled with black letters) with an rmsd of 0.56 Å for all the Cα atoms. Water molecules within hydrogen bonding distances of Glu11 are represented as red spheres for T26H and purple crosses for T26E, respectively. Hydrogen bonds observed in the T26E mutant complex are depicted with gray dashed lines. The respective distances from the anomeric C‐1 carbon of the adduct are indicated in red dashed lines, with the distances in Å.
Scheme 1.
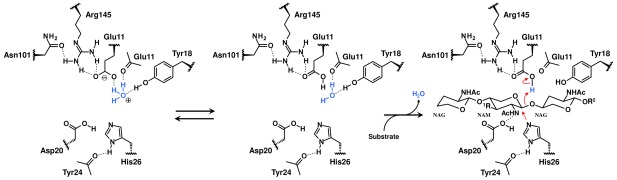
Proposed initial step of hydrolysis by the T26H mutant. To gain the ability to act as a proton donor in the initial step, the anionic carboxyl group of Glu11 extracts a proton from an adjacent hydronium ion and generates a water molecule. The protonation of Glu11 was partially induced even under neutral conditions in this study, presumably because the neutral guanidine moiety of Arg145 can serve to reduce the acidity of the carboxyl group of Glu11. After substrate binding, the singly protonated His26 nucleophilically attacks the anomeric carbon of bound substrate to proceed the hydrolysis via the double displacement mechanism.
In general, the retaining hydrolysis mechanism catalyzed by GHs is considered to be achieved through a double displacement mechanism involving two steps: a glycosylation and deglycosylation step (Supporting Information Fig. S1).5 In the first step, the enzyme is glycosylated by the concerted action of a general acid/base and nucleophile residue, Glu11 and His26 in the T26H mutant, that are arranged on opposite sides of the active site cleft. Concomitantly with the donation of a proton to the bound substrate from the general acid/base catalyst, the nucleophile attacks the anomeric carbon at the oxocarbenium ion‐like transition state, leading to the formation of a covalently linked glycosyl‐enzyme intermediate. Therefore, the general acid/base residue must adopt its protonated form for initiating the first step when the substrate approaches to the active site cleft, even though the pK a value for the carboxyl group is assumed to be around 4 in neutral solution.
In this neutron and high‐resolution X‐ray structure analysis of T26H, the Glu11 side‐chain was identified as having two forms, the neutral and anionic forms, suggesting that the pK a value of the carboxyl group was increased to a near neutral value because the crystal was grown at pD 7.0 [Fig. 4(A)]. Considering that the acidity of Glu11 can be more weakened by the interaction with neutral functional groups than with positively charged groups, it is chemically consistent that the guanidine moiety of Arg145 interacting with the Glu11 side‐chain predominantly adopts its neutral form. However, it was recently reported that arginine side‐chains are invariably protonated under physiological conditions of near neutral pH due to their high pK a values.22 The finding that Arg145 in T26H is apparently uncharged, would be highly unusual, although the Nη1 deprotonated Arg adjacent to the chromophore was observed in the neutron structure of photoactive yellow protein.23, 24 Presumably, the enzyme has a proton transfer pathway through the Asn101 side‐chain that allows for efficient extraction of protons from the Nɛ nitrogen of Arg145, but it has not been clarified yet. The remaining negative charge from the deprotonated carboxyl group of Glu11 could be compensated by some cation. One of the water‐binding sites adjacent to Glu11 was occupied by a DOD4 molecule, modelled in two different orientations. However, its trigonal‐pyramidal shaped nuclear density can also be assigned as the equilibrium state between a water and a hydronium ion,25 although it is difficult to definitely identify this in a 2.09‐Å resolution map [Fig. 4(C)]. Based on this observation, we suggest that the proton to be used for the protonation of Glu11 would be supplied by the hydronium ion (Scheme 1, left). Such proton transfer was also proposed for xylanase, a GH family 11 member.26 The resulting water molecule can be released to the bulk solvent when the substrate approaches to the active site cleft since it occupies a part of the subsite.
Some characteristic mechanisms for controlling the protonation states of the general acid or acid/base catalyst in GHs have been revealed by earlier neutron crystallographic studies. In endoglucanase of GH45, the general acid Asp114 is connected to the general base Asn92 located on the opposite side via side‐chain hydrogen bonds.27 When the Nδ2 nitrogen of the imidic acid form of Asn92 extracts a proton from the attacking water, the proton is immediately transferred to the scissile glycosidic bond of substrate through the hydrogen bond network. Meanwhile, in xylanase of GH11, the general acid/base Glu177 receives a proton from the bulk solvent by altering its side‐chain conformation which enables to increase the pK a value of Glu177.26 In the present neutron structure analysis of T26H, we found that the Arg145 residue interacting with Glu11 can play an important role in the protonation of the general acid/base in the retaining hydrolysis [Fig. 4(A)]. The neutral form of Arg145 facilitates the protonation of Glu11 by increasing the pK a value of the carboxyl group. Interestingly, the combination of the general acid residue and an arginine residue in the active site cleft is conserved in other inverting enzymes, chitinase of GH19 and chitosanase of GH46, of the lysozyme superfamily.3 Since the catalytic Glu11 residue of T26H originally acts as a general acid in the inverting hydrolysis by WT, it should not be surprising that the Glu‐Arg pair is not conserved in retaining enzymes of the lysozyme superfamily.
In the deglycosylation step, the second step of hydrolysis, the glycosyl‐enzyme intermediate suffers from hydrolytic cleavage at its linkage. The anionic carboxyl group that functioned as an acid catalyst in the first step acts as a base next by abstracting a proton from the incoming nucleophile, usually a water molecule (Supporting Information Fig. S1). When another disaccharide donor substrate approaches to the catalytic base instead of the water, the hydroxyl group of disaccharide is deprotonated and attacks the glycosyl‐enzyme linkage, leading to transglycosylation. The yields for transglycosylation are generally low because the glycosylated product itself can suffer from hydrolysis. From the structural comparison with the T26E mutant‐adduct complex, it is estimated that the C‐1 carbon of NAM is 4.6 Å away from the DOD4 oxygen, which could be a relatively long distance to attack the anomeric carbon (Fig. 5).11 The attacking water molecule may not be located at an optimum position for hydrolysis because the T26H mutant is an artificial retaining enzyme. It is difficult to deduce which water molecule can function as an attacking water from this neutron structure analysis due to its substrate‐free form. Further neutron structure analysis of the T26H‐substrate/product complex would be of interest to reveal how the protonation states of catalytic residues are affected by substrate/product binding and how the transglycosylation proceeds.
Materials and Methods
Protein expression and purification
The perdeuterated form of the T26H mutant was prepared using Escherichia coli BL21(DE3) cells (Novagen).12, 28 The cells grown at 310 K in a BioExpress deuterated medium (U‐D, 98%; Cambridge Isotope Laboratories) which was diluted with heavy water (D2O, 99.9%; Cambridge Isotope Laboratories) were induced with 1 mM isopropyl‐β‐D‐galactoside for 6 h. After harvesting the cells, they were disrupted by sonication, and the overexpressed product was purified following procedures described previously.12 Approximately 35 mg of purified protein was obtained from a 1‐L bacterial culture. The deuteration level of the purified sample was evaluated by measuring its molecular weight, using a 4800 Plus MALDI TOF/TOF Analyzer (Applied Biosystems), and a greater than 99% deuteration level was achieved under the conditions employed here. The purified protein was concentrated to approximately 40 mg mL−1 in 20 mM sodium phosphate buffer (pD 7.5) prepared with D2O.
Crystallization and data collection
To obtain larger single crystals, the reported condition12 was optimized by varying the concentrations of both the precipitant (sodium‐potassium phosphate) and the additive reagent (1,6‐hexanediol). After mixing equal volumes of the protein solution and the reservoir composed of 1.6 M sodium‐potassium phosphate (pD 7.0), 0.1 M NaCl, and 0.2 M 1,6‐hexanediol in D2O, 60 μL of the total mixture was equilibrated against 2 mL of the reservoir at 293 K. A relatively large crystal with a truncated cubic shape was grown up to 1.5 × 1.5 × 0.4 mm in size within a month.
The T26H mutant crystal with a volume of less than 0.9 mm3 was enclosed in a quartz capillary with a diameter of 2 mm for a neutron diffraction experiment at room temperature. The full neutron data set was collected using the quasi‐Laue technique on the IMAGINE instrument installed at the High Flux Isotope Reactor (HFIR), the Oak Ridge National Laboratory (ORNL), with a narrow‐bandpass of 3.3–4.5 Å (λ peak ≈ 3.9 Å, dλ/λ ≈ 30%) that delivers 4.6 × 107 neutrons cm−2 s−1 at the sample position.15 The quasi‐Laue images were measured from two crystal orientations to help fill the blind region and increase the completeness. A total of 10 images, with 19 h exposure each, were collected in less than eight days. The full neutron data set was indexed and integrated using LAUEGEN 29, 30 from the CCP4 suite,31 and wavelength‐normalized to account for the spectral distribution of the quasi‐Laue beam using LSCALE.32 Scaling and merging of the data set was performed using SCALA.31 The statistics for data collection are given in Supporting Information Table SI.
Neutron structure determination by joint X/N refinement
A full X‐ray data set was collected from the same crystal used in the neutron diffraction experiment. The X‐ray diffraction images were measured at room temperature at the beamline BL26B1 at SPring‐8 (Harima, Japan), processed and scaled using the HKL2000 software package.33 The crystal with the cell dimensions of a = b = 61.23 Å and c = 96.79 Å was sufficiently isomorphous to the neutron diffraction data, which was used for the refinement procedure. The X‐ray crystal structure was determined first with the PHENIX software package34 using the coordinates of the T26H mutant (PDB ID: 1QT8) as a starting model for molecular replacement. After several rounds of iterative manual rebuilding of the protein coordinates, ligand molecules and metal ions were interpreted based on 2F o – F c and F o – F c electron‐density maps. Model building and water picking were manually performed with COOT.35 Using this X‐ray structure as a starting model, a joint X/N refinement was performed with the phenix.refine program36 in PHENIX. The R free flags were generated to be consistent with both the X‐ray and neutron data sets. After an initial rigid‐body minimization, deuterium atoms were added to the protein model, except for solvent molecules. The maximum‐likelihood‐based refinement of individual coordinates was then performed for several cycles, followed by the respective refinements of isotropic displacement parameters and occupancies for alternative conformations. At this point, deuterium atoms for D2O were generated according to their positive peaks on a F o − F c nuclear‐density map, with manual adjustment of the coordinates. The bond lengths and angles of deuterium atoms were refined as individual atoms with isotropic displacement parameters. The protonated forms of dissociative functional groups were identified by inspection of the F o − F c nuclear‐density maps. The final coordinates were refined to the R work/R free values of 22.5/27.8% for the neutron structure and of 15.5/18.5% for the X‐ray structure. Finally, PROCHECK 37 was employed to evaluate the final molecular coordinates. The final refinement statistics are summarized in Supporting Information Table SI.
Determination of high‐resolution X‐ray structure
The T26H mutant crystals, grown under the identical condition described above, were soaked in a cryoprotectant solution containing 30% (w/v) glucose in addition to the reservoir compositions. X‐ray diffraction data were collected under a stream of gaseous nitrogen at 100 K at the beamline BL5A at Photon Factory (Tsukuba, Japan). The data set was processed and scaled using the HKL2000 software package.33 The structure determination with the PHENIX software package34 was initiated by a rigid‐body refinement using the initial molecular‐replacement model. After several cycles of the refinements for individual coordinates and isotropic displacement parameters using SHELXL,38 in which the resolution range was limited from 20 Å to 1.2 Å, metal ions and ordered water molecules were manually incorporated to the model by COOT.35 Anisotropic displacement parameters for the protein model were then refined over the whole resolution range. The restraints of bond lengths and angles for the carboxyl and imidazole groups of protein were excluded in the next CGLS refinement. To converge their geometrical parameters to reasonable values, the unrestrained calculation was limited to the residues those were assigned as single conformations and with lower B‐factors below 17 Å2, as otherwise their coordinates were disordered during the unrestrained refinement. The estimated standard deviations for the bond lengths and angles were derived from one cycle of the full‐matrix least‐squares refinement. The statistics for data collection and structure refinement is given in Supporting Information Table SI. All graphic images of molecular structure were generated using PyMOL (The PyMOL molecular graphics system, version 1.7. Schrödinger, LLC).
Accession Numbers
The atomic coordinates of the perdeuterated T26H mutant have been deposited in the RCSB Protein Data Bank (PDB) under accession codes 5XPE for the neutron structure and 5XPF for the high‐resolution X‐ray structure.
Conflict of interests
All the authors declare that they have no conflicts of interest with respect to the publication of this article.
Supporting information
Supporting Information
Acknowledgments
The authors would like to acknowledge the beamline staffs at Photon Factory (proposal no. 2013G122) and SPring‐8 (proposal no. 2014B1995) synchrotron facilities for their support in data collection. MALDI‐TOF/MS analyses were performed at the Center for Instrumental Analysis at Ibaraki University. We thank Dr. K. Shimojo (JAEA, Japan) for his support in the measurements. Neutron diffraction experiments conducted at ORNL's HFIR (IPTS no. 11298.1) were supported by the NSF grant CHE‐0922719 and by the Scientific User Facilities Division, Office of Basic Energy Sciences, US Department of Energy. We are also grateful to the generous cooperation of Prof. Y. Higuchi (Univ. of Hyogo, Japan) in preparing this article.
Brief Statement: The substitution of the catalytic Thr26 residue in T4L to a histidine converts the wild type from an inverting to a retaining enzyme, which implies that the carboxyl group of Glu11 acts as an acid/base catalyst in the hydrolysis. To gain insight into the roles of Glu11 and the substituted His26 in the retaining reaction, we determined the neutron structure of the perdeuterated T26H mutant.
Contributor Information
Takeshi Hiromoto, Email: hiromoto@sci.u-hyogo.ac.jp.
Motoyasu Adachi, Email: adachi.motoyasu@qst.go.jp.
References
- 1. Chipman DM, Pollock JJ, Sharon N (1968) Lysozyme‐catalyzed hydrolysis and transglycosylation reactions of bacterial cell wall oligosaccharides. J Biol Chem 243:487–496. [PubMed] [Google Scholar]
- 2. Svensson B, Søgaard M (1993) Mutational analysis of glycosylase function. J Biotechnol 29:1–37. [Google Scholar]
- 3. Wohlkönig A, Huet J, Looze Y, Wintjens R (2010) Structural relationships in the lysozyme superfamily: significant evidence for glycoside hydrolase signature motifs. PLoS One 5:e15388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vocadlo DJ, Davies GJ, Laine R, Withers SG (2001) Catalysis by hen egg‐white lysozyme proceeds via a covalent intermediate. Nature 412:835–838. [DOI] [PubMed] [Google Scholar]
- 5. Cerqueira N, Brás N, Ramos MJ, Fernandes PA, Glycosidases: a mechanistic overview In: Chang CF, Ed. (2012) Carbohydrates: comprehensive studies on glycobiology and glycotechnology. Croatia: InTech, pp 117–134. [Google Scholar]
- 6. Niimura N, Minezaki Y, Nonaka T, Castagna JC, Cipriani F, Høghøj P, Lehmann MS, Wilkinson C (1997) Neutron Laue diffractometry with an imaging plate provides an effective data collection regime for neutron protein crystallography. Nat Struct Biol 4:909–914. [DOI] [PubMed] [Google Scholar]
- 7. Bon C, Lehmann MS, Wilkinson C (1999) Quasi‐Laue neutron‐diffraction study of the water arrangement in crystals of triclinic hen egg‐white lysozyme. Acta Crystallogr D55:978–987. [DOI] [PubMed] [Google Scholar]
- 8. Matthews BW, Remington SJ, Grütter MG, Anderson WF (1981) Relation between hen egg white lysozyme and bacteriophage T4 lysozyme: evolutionary implications. J Mol Biol 147:545–558. [DOI] [PubMed] [Google Scholar]
- 9. Kuroki R, Weaver LH, Matthews BW (1993) A covalent enzyme‐substrate intermediate with saccharide distortion in a mutant T4 lysozyme. Science 262:2030–2032. [DOI] [PubMed] [Google Scholar]
- 10. Kuroki R, Weaver LH, Matthews BW (1995) Structure‐based design of a lysozyme with altered catalytic activity. Nat Struct Biol 2:1007–1011. [DOI] [PubMed] [Google Scholar]
- 11. Kuroki R, Weaver LH, Matthews BW (1999) Structural basis of the conversion of T4 lysozyme into a transglycosidase by reengineering the active site. Proc Natl Acad Sci 96:8949–8954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hiromoto T, Adachi M, Shibazaki C, Schrader TE, Ostermann A, Kuroki R (2015) Preparation and crystallization of perdeuterated T4 phage lysozyme for neutron diffraction study. JPS Conf Proc 8:033003. [Google Scholar]
- 13. O'Dell WB, Bodenheimer AM, Meilleur F (2016) Neutron protein crystallography: a complementary tool for locating hydrogens in proteins. Arch Biochem Biophys 602:48–60. [DOI] [PubMed] [Google Scholar]
- 14. Shu F, Ramakrishnan V, Schoenborn BP (2000) Enhanced visibility of hydrogen atoms by neutron crystallography on fully deuterated myoglobin. Proc Natl Acad Sci 97:3872–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Meilleur F, Munshi P, Robertson L, Stoica AD, Crow L, Kovalevsky A, Koritsanszky T, Chakoumakos BC, Blessing R, Myles DA (2013) The IMAGINE instrument: first neutron protein structure and new capabilities for neutron macromolecular crystallography. Acta Crystallogr D69:2157–2160. [DOI] [PubMed] [Google Scholar]
- 16. Baase WA, Liu L, Tronrud DE, Matthews BW (2010) Lessons from the lysozyme of phage T4. Protein Sci 19:631–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fisher SJ, Blakeley MP, Cianci M, McSweeney S, Helliwell JR (2012) Protonation‐state determination in proteins using high‐resolution X‐ray crystallography: effects of resolution and completeness. Acta Crystallogr D68:800–809. [DOI] [PubMed] [Google Scholar]
- 18. Ahmed HU, Blakeley MP, Cianci M, Cruickshank DWJ, Hubbard JA, Helliwell JR (2007) The determination of protonation states in proteins. Acta Crystallogr D63:906–922. [DOI] [PubMed] [Google Scholar]
- 19. Fadel F, Zhao Y, Cachau R, Cousido‐Siah A, Ruiz FX, Harlos K, Howard E, Mitschler A, Podjarny A (2015) New insights into the enzymatic mechanism of human chitotriosidase (CHIT1) catalytic domain by atomic resolution X‐ray diffraction and hybrid QM/MM. Acta Crystallogr D71:1455–1470. [DOI] [PubMed] [Google Scholar]
- 20. Malinska M, Dauter M, Kowiel M, Jaskolski M, Dauter Z (2015) Protonation and geometry of histidine rings. Acta Crystallogr D71:1444–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sawinski PK, Meven M, Englert U, Dronskowski R (2013) Single‐crystal neutron diffraction study on guanidine, CN3H5. Cryst Growth Des 13:1730–1735. [Google Scholar]
- 22. Fitch CA, Platzer G, Okon M, Garcia‐Moreno E B, McIntosh LP (2015) Arginine: its pK a value revisited. Protein Sci 24:752–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Henry GD, Sykes BD (1995) Determination of the rotational‐dynamics and pH‐dependence of the hydrogen‐exchange rates of the arginine guanidine group using NMR spectroscopy. J Biomol NMR 6:59–66. [DOI] [PubMed] [Google Scholar]
- 24. Yamaguchi S, Kamikubo H, Kurihara K, Kuroki R, Niimura N, Shimizu N, Yamazaki Y, Kataoka M (2009) Low‐barrier hydrogen bond in photoactive yellow protein. Proc Natl Acad Sci 106:440–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cuypers MG, Mason SA, Blakeley MP, Mitchell EP, Haertlein M, Forsyth VT (2013) Near‐atomic resolution neutron crystallography on perdeuterated Pyrococcus furiosus rubredoxin: implication of hydronium ions and protonation state equilibria in redox changes. Angew Chem Int Ed 52:1022–1025. [DOI] [PubMed] [Google Scholar]
- 26. Wan Q, Parks JM, Hanson BL, Fisher SZ, Ostermann A, Schrader TE, Graham DE, Coates L, Langan P, Kovalevsky A (2015) Direct determination of protonation states and visualization of hydrogen bonding in a glycoside hydrolase with neutron crystallography. Proc Natl Acad Sci 112:12384–12389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nakamura A, Ishida T, Kusaka K, Yamada T, Fushinobu S, Tanaka I, Kaneko S, Ohta K, Tanaka H, Inaka K, Higuchi Y, Niimura N, Samejima M, Igarashi K (2015) Newton's cradle proton relay with amide–imidic acid tautomerization in inverting cellulase visualized by neutron crystallography. Sci Adv 1:e1500263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Poteete AR, Sun DP, Nicholson H, Matthews BW (1991) Second‐site revertants of an inactive T4 lysozyme mutant restore activity by restructuring the active site cleft. Biochemistry 30:1425–1432. [DOI] [PubMed] [Google Scholar]
- 29. Helliwell M, Gomez de Anderez D, Habash J, Helliwell JR, Vernon J (1989) A comparison of Laue and monochromatic X‐ray analyses: the determination of the hydrogen‐atom positions of an organic small‐molecule crystal. Acta Crystallogr B45:591–596. [Google Scholar]
- 30. Campbell JW, Hao Q, Harding MM, Nguti ND, Wilkinson C (1998) LAUEGEN version 6.0 and INTLDM . J Appl Crystallogr 31:496–502. [Google Scholar]
- 31. Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr D67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Arzt S, Campbell JW, Harding MM, Hao Q, Helliwell JR (1999) LSCALE: the new normalization, scaling and absorption correction program in the Daresbury Laue software suite. J Appl Crystallogr 32:554–562. [Google Scholar]
- 33. Minor W, Otwinowski Z (1997) Processing of X‐ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. [DOI] [PubMed] [Google Scholar]
- 34. Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse‐Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr D66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Crystallogr D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 36. Afonine PV, Mustyakimov M, Grosse‐Kunstleve RW, Moriarty NW, Langan P, Adams PD (2010) Joint X‐ray and neutron refinement with phenix.refine . Acta Crystallogr D66:1153–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26:283–291. [Google Scholar]
- 38. Sheldrick GM (2008) A short history of SHELX . Acta Crystallogr A64:112–122. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information