

Abstract
Background
Andersen-Tawil syndrome (ATS) is rare channelopathy caused by KCNJ2 mutation and probably KCNJ5. It is characterized by arrhythmias, neurological symptoms, and dysmorphic features. The present study retrospectively examined the characteristics of 11 unrelated families with ATS.
Methods
This study consisted of 11 probands positive for KCNJ2 variants and 33 family members (mean age 30.0 ± 17.3 years, female n=31). Additional genetic screening of 3 LQTS genes (KCNQ1, KCNH2, SCN5A) was performed in 9 families. Predictors of arrhythmias [premature ventricular beats > 2000/24 h, biventricular and polymorphic ventricular tachycardia (VT)], syncope, and/or cardiac arrest (CA) were evaluated.
Results
In KCNJ2 mutation carriers vs non-carriers (n=25 vs n=19) significant differences were observed in U-wave manifestations in V2–V4, Tpeak – Tend duration, QTUc duration (p<0.0001), dysmorphic features, and neurological symptoms. Compared to asymptomatic carriers (n=9), in those with arrhythmias and/or syncope and/or CA (n=16) micrognathia (p=0.004), periodic paralysis (p=0.019), palpitation (p=0.005), U-wave n V2–V4 (p=0.049) were more frequent; QTU (p=0.045) and Tpeak – Tend (p=0.014) were also longer (n=9). In the subgroup of carriers with syncope and/or cardiac arrest (n=10, 90% women), K897T-KCNH2 polymorphism (p=0.02), periodic paralysis (p=0.004), muscle weakness (p=0.04), palpitations (p=0.04), arrhythmias (biventricular VT, p=0.003; polymorphic VT, p=0.009) were observed more frequently. Tpeak – Tend duration was longer (p=0.007) and the percentage of patients with premature ventricular contraction >2000/24 hours was higher (p=0.005).
Conclusion
A higher risk of arrhythmia, syncope, and/or CA is associated with the presence of micrognathia, periodic paralysis, and prolonged Tpeak – Tend time. Our findings suggest that K897T may contribute to the occurrence of syncope.
Keywords: Andersen-Tawil syndrome, Long QT syndrome, Arrhythmia, K897T polymorphism
Introduction
Andersen-Tawil syndrome (ATS), classified as long QT syndrome type 7 (LQT7) is a rare disorder, sporadic or inherited in an autosomal dominant pattern [1,2]. The mutations causing ATS are located in KCNJ2 (GenBank accession no. NM_000891.2), which encodes the α-subunit of the potassium channel Kir2.1 a component of the inward rectifier IK1 [3,4]. Recent research showed that KCNJ5, encoding the G-protein-activated inwardly rectifying potassium channel 4 (Kir3.4), could be a second gene causing Andersen-Tawil syndrome. The inhibitory effects of mutant Kir3.4 on Kir2.1 channel may cause the clinical presentation in both skeletal and heart muscles [5].
The mutations are detectable in most cases. ATS confirmed by mutation in KCNJ2 gene is called type 1 [6]. The remaining cases are designated as ATS-type 2 and could be a result of mutation in other genes (e.g. KCNJ5) or the cause of the disorder is still unknown [7].
ATS is characterized by the clinical triad: ventricular arrhythmias, periodic paralysis or muscle weakness, distinctive facial and skeletal dysmorphism with variability of phenotype [2,8]. The management of patients with ATS continues to be a subject of debate. No randomized clinical therapeutic trials have been conducted on ATS. There are only few studies suggesting influence of flecainide on cardiac arrhythmias reduction but benefit of flecainide for prevention of medically significant cardiac arrhythmias is still unknown. Moreover, it is known that this drug causes many side effects often making treatment impossible [9,10].
A carrier could present only a part of the triad or even be asymptomatic [11]. It is still a question if the penetrance and expression of the ATS mutations, in particular arrhythmias, are relative to any factor. This study assessed symptoms and signs of ATS and explored possible risk factors of life-threatening cardiac arrhythmias in ATS. Additionally, influence of polymorphisms in three genes (KCNQ1, KCNH2, SCN5A), in which mutations result in the most prevalent LQT syndromes (LQT1, LQT2, and LQT3), were analyzed. The coexistence of ATS with K897T polymorphism in the KCNH2 gene resulted in more severe symptoms, which was suggested by us in a previous report [12].
Methods
Study design
This is a retrospective study of 11 unrelated probands with KCNJ2 mutations and their relatives (Fig. 1). Observations came from 4 centers in Poland and 1 center in Turkey. In every family, a pedigree analysis was done and, if it was possible, the type of inheritance was assessed. Clinical evaluation, electrocardiographic (ECG) analysis, and mutation screening were performed in all relatives who agreed to participate in the study.
Figure 1.
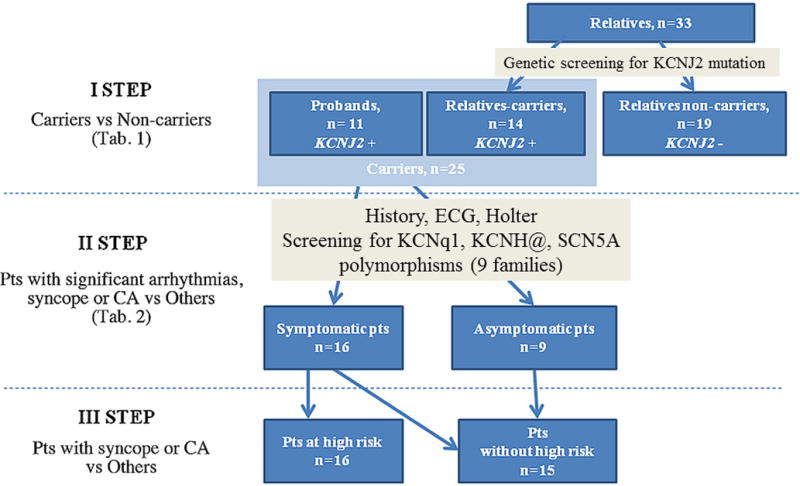
Study design.
CA, cardiac arrest; ECG, electrocardiogram
Clinical Evaluation
We analyzed retrospectively the history and medical records of each patient. The clinical evaluation consisted of: 1) detailed history (syncope, cardiac arrest, family history of sudden cardiac death), 2) assessment of dysmorphic features (clinodactyly, syndactyly, hypertelorism, low set ears, micrognathia), 3) cardiological exam (history, analysis of series of available ECGs); in every carrier of KCNJ2 mutation 24-hour Holter monitoring was performed additionally, 4) neurological symptoms (periodic paralysis and muscle weakness); in patients with syncope epilepsy was excluded, 5) the method of treatment.
ECG analysis
ECGs were analyzed and parameters characterizing the repolarization process were assessed manually by one cardiologist (P.K.) who was blinded to patients’ clinical status. The following ECG parameters were assessed: QT and QT+U interval duration, Tpeak – Tend duration, Tpeak – Upeak duration, U-wave time duration. Corrected QT (QTc) and corrected QT+U (QT+Uc) were calculated using the Bazett’s formula to adjust the intervals to heart rate. Additionally the presence of prominent U-wave in different territories: inferior leads (II, III, or aVF), anterior leads V2–V4 or lateral leads (V5) were analyzed. A U-wave was considered prominent if its amplitude was ≥ 0.15 mV. Electrocardiographic measurements of T-wave and U-wave (including QT+U interval) were made in leads with the highest amplitude, usually in V2 or V3. QT/QTc interval was calculated from lead II.
Mutation Screening
ATS mutation screening was performed in all relatives who agreed to participate in the study and had signed an informed consent. A 10 ml blood sample was collected on ethylenediaminetetraacetic acid. The genomic DNA was isolated with the phenol method. There was direct sequencing analysis of the whole translated region of KCNJ2 gene. DNA was amplified using site-specific primers and amplicons were subjected to bidirectional capillary-based sequencing using 3130XL Genetic Analyzer Applied Biosystem (Hitachi High Technologies Corporation, 882 Ichige, 312-8504, Japan). Data analysis was done by using DNA Sequencing Analysis Software version 5.3.1Applied Biosystem (Hitachi High Technologies Corporation, 882 Ichige, 312-8504, Japan).
Additionally, in 9 Polish families (18 subjects, 9 probands, female n=14) in ATS mutation carriers additional genetic screening of 3 major genes causing LQTS (KCNQ1, KCNH2, SCN5A) was done. The test was performed by polymerase chain reaction using exon-flanking intronic primers designed by our laboratory. Amplicons were analyzed by next generation sequencing by GS Junior System (Roche, Bradford, CT, USA) according to Amplicon Library Preparation Method Manual. Data were analyzed using GS Amplicon Variant Analyzer (AVA) software (Roche, Bradford, CT, USA).
Results
Study group
The study group consisted of 44 patients (female n=31), including 11 probands (female=11) with genetically confirmed KCNJ2 mutations, and 33 family members. In 14 subjects out of the family members the presence of KCNJ2 mutations was detected. Mean age was 30.0 ± 17.3 (median: 28.0, range: 3 – 64) years.
Genetic study
We found 8 distinct KCNJ2 mutations in 11 probands and 14 family members (25 KCNJ2 mutation carriers). The following KCNJ2 mutations were detected: Arg218Gln (n=7), Arg67Trp (n=6), Arg82Trp (n=3), Cys154Tyr (n=3), Arg218Pro (n=2), Gly300Ala (n=2), Tyr68Asp (n=1), and Thr305Ala (n=1). The pedigree analysis showed that 6 mutations (Arg67Trp, Arg82Trp, Arg218Gln, Cys154Tyr, Gly300Ala, Arg218Pro) were inherited in autosomal dominant pattern and one appeared de novo (Thr305Ala). Inheritance in other families was impossible to evaluate because of death or lack of data from family members. Two common polymorphisms were detected in 7 pts (39%) patients: K897T (Lys897Thr) in KCNH2 (n=1) and H558R (His558Thr) in SCN5A (n=3), both (n=3).
Clinical Characteristics of KCNJ2 Mutation Carriers
The carriers group consisted of 25 patients (female n=19, mean age 30.8 ± 16.4, median 27, range: 7 – 63 years). Episodes of syncope were reported in 9 patients (36%). Frequency of syncope varied from one episode to multiple episodes during follow-up. The relationship between arrhythmias and episodes of syncope was not confirmed in most patients, even in patients with implantable cardioverter defibrillators (ICD); in all cases vasovagal syncope was excluded (high negative Calgary Syncope Symptom Score) as well as epilepsy. One female presented suspected cardiac arrest, who survived after short time of cardiopulmonary resuscitation (no ECG confirmation). She had 3 adequate ICD interventions during first year after implantation (polymorphic VT 230–270/min) on physical activity. One patient experienced adequate intervention during amiodarone therapy. Sudden cardiac deaths (SCD) were reported in 3 non-genotyped relatives: two of them in the family with Arg218Gln mutation and the coexisting K897T polymorphism and one in the family with Cys154Tyr mutation.
A 24-hours Holter monitoring revealed arrhythmia in 15 patints (60%), frequent premature ventricular contraction (PVC) >2000/24h in 14 patients (56%), polymorphic ventricular tachycardia (PVT) in 12 patients (48%), bidirectional ventricular tachycardia (BiVT) in 11 patients (44%) predominantly during activity.
Dysmorphic features were seen in 22 pts (88%) of KCNJ2 mutation carriers. Micrognathia occurred in 15 patients (60%), clinodactyly of fingers in 9 patients (36%), low set ears in 7 patients (28%), hypertelorism in 5 patients (20%), syndactyly of toes in 3 patients (12%), clinodactyly of toes in 2 patients (8%). Moreover, in one proband the new, previously undescribed, probable dysmorphic feature of ATS was found - lack of digital furrow on the 4th finger presented (Fig. 2A).
Figure 2.

(A) Lack of digital furrow; (B) Clinodacytly; (C) Micrognathia; (D) A short fragment of the Holter recording. Permanent, bidirectional ventricular tachycardia, frequency –135–140/min., which represents 30% to 50% of 24-hour monitoring (depending on the study) in the patient presented.
Neurological symptoms occurred in carriers – 10 patients (40%). Seven patients (28%) had a history of periodic paralysis, of whom 2 had additional muscle weakness. Three patients had a history of isolated muscle weakness. The frequency of periodic paralysis or muscle weakness ranged from a single lifetime episode, to monthly manifestations of various duration, and were induced by such factors as stress, exercise, menstruation, pre-menstruation period, or parturition. The clinical triad was observed in 9 patients (36%), isolated dysmorphy in 7 patients (28%), dysmorphy and cardiac symptoms in 5 patients (20%), no symptoms in 2 patients (8%), dysmorphy and neurological symptoms in 1 patient (4%), isolated cardiac symptoms in 1 patient (4%).
Characteristics of carriers and non-carriers of KCNJ2 mutations
The clinical and electrocardiographic characteristic of KCNJ2 gene carriers vs non-carriers are depicted in Table 1.
Table 1.
Clinical presentation and ECG parameters in KCNJ2 mutation carriers vs non-carriers
| Carriers (n=25) | Non-Carriers (n=19) | p | |
|---|---|---|---|
| Dysmorphic feature and neurological symptoms (%) | |||
| Sex (female) | 76 | 68 | ns |
| Micrognathia | 60 | 26 | 0.026 |
| Hypertelorism | 20 | 5 | ns |
| Low set ears | 28 | 5 | ns |
| Clinodactyly of fingers | 36 | 5 | 0.02 |
| Syndaktyly of fingers | 0 | 0 | - |
| Clinodactyly of toes | 8 | 0 | - |
| Syndaktyly of toes | 12 | 0 | - |
| Periodic paralysis | 28 | 0 | - |
| Muscle weakness | 20 | 0 | - |
| Palpitation | 36 | 0 | - |
| ECG parameters (%) | |||
| U-wave | 96 | 57 | 0.002 |
| U wave inferior | 52 | 5 | 0.001 |
| U wave V2–V4 | 92 | 31 | <0.0001 |
| U wave V5 | 72 | 21 | 0.0008 |
| QT+U > 600 | 64 | 5 | 0.002 |
| QTc > 440 *(460**) | 28 | 5 | ns |
| ECG parameters (ms) | |||
| QT | 400 ± 38 | 367 ± 38 | 0.007 |
| QTc | 427 ± 26 | 404 ± 23 | 0.005 |
| QT+U | 628 ± 69 | 537 ± 86 | 0.002 |
| QT+Uc | 671 ± 35 | 598 ± 48 | <0.0001 |
| U-wave time | 198 ± 52 | 148 ± 43 | 0.009 |
| Tpeak – Tend | 139 ± 29 | 97 ± 19 | <0.0001 |
| Tpeak – Upeak | 238 ± 24 | 179 ± 48 | 0.003 |
for men,
for women
ECG, electrocardiographic.
Micrognathia (60% vs 26%, p=0.02) and fingers clinodactyly (36% vs 5%, p=0.02) were more common dysmorphic features observed in KCNJ2 mutation carriers vs non-carriers. The periodic paralysis and muscle weakness, syndactyly and clinodactyly of toes occurred only in subjects with KCNJ2 mutation carriers.
U-wave duration, Tpeak – Tend duration and Tpeak – Upeak duration were significantly longer in KCNJ2 mutation carriers vs non-carriers. Presence of prominent U-wave in any out of 12-lead ECG or separately in any territories (inferior: 52% vs 5%, anterior: 92% vs 31%, or lateral : 72% vs 21 %) was significantly more frequently registered in subjects with KCNJ2 mutation carriers vs non-carriers. QT interval (400±39 ms vs 367±38 ms, p=0.007) and corrected QT interval (427±26 ms vs 405±23 ms, p=0.006) were significantly longer in carriers vs non-carriers. Similarly, QT+U interval (628±69 ms vs 537±86 ms, p=0.002) and QT+Uc (672 ± 36 ms vs 598±49 ms, p<0.0001) were statistically significantly longer in KCNJ2 mutation carriers vs non-carriers. The percentage of subjects with QT+U interval > 600 ms was significantly higher (64% vs 5%, p=0.002) in KCNJ2 mutation carriers vs non-carriers. In turn, the percentage of subjects with prolonged QT interval and corrected QT interval, respectively QTc interval > 440 ms in men, and QTc > 460 ms in women, did not show statistical differences.
Risk factors of arrhythmic events, syncope, and CA in mutation carriers
The symptomatic KCNJ2 mutation carriers (n=16) with a history of syncope or cardiac arrest and/or amount of premature ventricular complexes > 2000 per day, the presence of polymorphic ventricular tachycardia (PVT) or bidirectional ventricular tachycardia (BiVT) were compared with the asymptomatic KCNJ2 mutation carriers (n=9) without above listed events (syncope or cardiac arrest or arrhythmias). The clinical and electrocardiographic characteristics of these two groups are depicted in Table 2.
Table 2.
Clinical presentation, gene polymorphisms, and electrocardiographic (ECG) parameters in KCNJ2 mutations carriers with a history of syncope or cardiac arrest (CA) and/or amount of premature ventricular complexes > 2000 per day, the presence of polymorphic ventricular tachycardia (PVT) or bidirectional ventricular tachycardia (BiVT) and in KCNJ2 carriers without above listed events.
| Group with syncope, CA, PVC>2000/24h, PVT, BiVT (n=16) | Group with no syncope, CA, PVC>2000/24h, PVT, BiVT (n=9) | p | |
|---|---|---|---|
| Polymorphisms | |||
| K897T | 4 (14) | 0 (4) | ns |
| H558R | 5 (14) | 1 (4) | ns |
| Dysmorphic features and neurological symptoms (%) | |||
| Micrognathia | 81 | 22 | 0.004 |
| Hypertelorism | 25 | 11 | ns |
| Low set ears | 31 | 22 | ns |
| Clinodactyly of fingers | 31 | 44 | ns |
| Syndaktyly of fingers | 0 | 0 | ns |
| Clindaktyly of toes | 6 | 11 | ns |
| Syndaktyly of toes | 6 | 22 | ns |
| Periodic paralysis | 43 | 0 | 0.019 |
| Muscle weakness | 25 | 11 | ns |
| Palpitation | 56 | 0 | 0.005 |
| ECG parameters (%) | |||
| U-wave | 100 | 88 | ns |
| U- wave V2–V4 | 100 | 77 | 0.049 |
| U- wave V5 | 81 | 55 | ns |
| ECG parameters (ms) | |||
| QT | 407 ± 41 | 387 ± 31 | ns |
| QTc | 430 ± 27 | 422 ± 25 | ns |
| QT+U | 648 ± 58 | 588 ± 75 | 0.045 |
| QT+Uc | 679 ± 28 | 655 ± 45 | ns |
| U- wave time | 194 ± 58 | 207 ± 40 | ns |
| Tpeak - Tend | 150 ± 26 | 121 ± 25 | 0.014 |
| Tpeak - Upeak | 243 ± 24 | 230 ± 22 | ns |
Females were slightly more frequent in the first group (81% vs 67%, p=ns). Out of other analyzed variables only micrognathia (81% vs 22%, p=0.004), periodic paralysis (44% vs 0%, p=0.02), and palpitations (56% vs 0%, p=0.005) were observed significantly more frequently in this group.
The duration of the descending portion of T-wave, calculated as Tpeak – Tend, was significantly longer in the symptomatic patients, respectively 150±27 ms vs 121±25 ms, p=0.01. The presence of prominent U-wave was not different in both groups. In turn, the presence of U-wave in anterior leads (V2–V4) was significantly more frequently seen in the symptomatic vs the asymptomatic patients, respectively 100% vs 78%, p=0.049. QT and QTc intervals were not different in both analyzed subgroups. In turn, QT+U interval was significantly longer in the symptomatic patients (648±59 ms vs 588±76 ms, p=0.045).
K897T polymorphism was detected only in the group of symptomatic patients (4 out of 14 patients). Three subjects with K897T polymorphism had Arg218Gln mutation. All of them presented whole triad of ATS and losses of consciousness. In turn, H558R polymorphism was detected in 5 out of 14 symptomatic patients and in 1 out 4 asymptomatic patients (shown in Table 2).
Among KCNJ2 mutation carriers, we could detail a subgroup of patients at higher risk (n=10); defined as patients with syncope and/or cardiac arrest. This group was compared with KCNJ2 mutations carriers without syncope and/or cardiac arrest (n=15).
Women accounted for the majority of patients at higher risk, respectively 90% vs 67%, p=ns. In that group additional polymorphism in LQTS gene (K897T mutation) was significantly more frequently detected than in other patients, respectively 4/9 (44%) patients vs 0/9 (0%) patients, p = 0.02. In patients at higher risk periodic paralysis (60% vs 7%, p=0.004), muscle weakness (40% vs 7%, p=0.04) and palpitations (60% vs 20%, p=0.04) were observed significantly more frequently.
Moreover, in that group Tpeak – Tend duration time was significantly longer (respectively 158±28 ms vs 127±24 ms, p=0.007); the complex ventricular arrhythmias were more frequently registered (BiVT -80% vs 20%, p=0.003; PVT -80% vs 27%, p=0.009) and the percentage of patients with PVC > 2000/24 hours was significantly higher (respectively 90% vs 33%, p=0.005) in comparison to other patients.
Treatment
Most patients were treated without proper diagnosis before genetic study. The treatment was based on the preferences of physicians. Eleven out of twelve symptomatic patients received beta-blockers, 2 of them additionally received propafenone with no response. Flecainide was given to 2 patients and proved effective in one. Amiodarone was given to one patient. A pacemaker was implanted in 2 patients, because of bradycardia due to high doses of beta-blockers. Five patients were implanted with ICD, four as primary and one as secondary prevention after cardiac arrest. Two had appropriate ICD shocks appeared during follow-up (mean time: 9.6, median: 6, range: 1 – 27 years): one on beta blocker, and one during amiodarone therapy. Syncope and CA also appeared during beta-blocker therapy.
Discussion
ATS is a rare channelopathy caused by KCNJ2 mutation and probably KCNJ5 [3,5]. It is characterized by arrhythmias, neurological symptoms, and dysmorphic features. KCNJ2 mutation could result in typical ATS phenotype with the entire ATS triad or atypical phenotype with one or no symptoms. Previous studies have reported that variability of the ATS phenotype can be influenced by the topological location of mutation in the Kir2.1 channel [11,13]. However, no data have been presented relative to risk factors predisposing to malignant arrhythmias. Few studies reported data concerning ATS manifestation in both sexes. In one report, a high penetrance of periodic paralysis in males was shown [14]. Another study showed that Arg67Trp mutation in one family caused different symptoms in male and female subjects; females exhibited arrhythmias whereas males exhibited periodic paralysis [4]. In contrast, another study reported that the phenotype of this mutation is not sex-specific [7]. In our cohort, in one family with the same mutation, female proband presented the clinical triad of ATS, yet a male relative displayed no feature of ATS. In another family, arrhythmia and dysmorphic features were presented only in the female proband, two females and one male relative had only dysmorphic features.
In the current study, every proband was female, suggesting that ATS may be more symptomatic in females.
In ATS, dysmorphic features affect in particular the face and fingers and manifest as clinodactyly, syndactyly, and micrognathia. In our group micrognathia was the most prevalent symptom. Moreover, it was statistically more prevalent in the group with arrhythmias, syncope, and/or CA. Interestingly, dysmorphic features appeared in family relatives without ATS mutations. This phenomenon is unexplained. It appears that dysmorphic features could not be the basis for diagnosis in relatives of ATS patients and other genetic or environmental factors contribute to these symptoms.
The neurological symptoms may be induced by various factors such as stress, exercise, menstruation, etc. and often are diagnostically problematic. Paradoxically, it could result in delaying a correct diagnosis [8]. Initially, patients from our group were treated for epilepsy or multiple sclerosis. The prevalence of paralysis is varied. It could manifest as the main medical problem for patients or be completely absent. Our study shows that neurological symptoms always coexist with arrhythmias. On the other hand, arrhythmia did not always occur with neurological symptoms.
Currently, cardiac manifestation of ATS is one of the most discussed issues. There are continuously reports concerning new insights into ECG interpretation [12,15]. Inclusion of this channelopathy into the group of LQT syndromes is still controversial. Previous studies report that some ATS patients present with normal QTc [5,10,11]. Therefore, in ATS, it is more appropriate to assess QT+U/QT+Uc interval additionally. Furthermore it was revealed that prolongation of final downslope of T-wave and Tpeak – Upeak interval were also characteristic for KCNJ2 mutation carriers [13].
In the presented research, the results concerning ECG parameters confirm previous reports. In KCNJ2 carriers only three individuals had prolonged QTc. In contrast, QT+Uc prolongation was presented by 88% carriers. Moreover, pathological U-wave in ECG, one of the most known sign of ATS, appeared in almost every carrier. Non-pathological U-wave was present in more than half of individuals without mutation. Additionally, in our study prolonged QT+Uc, Tpeak – Tend interval and presence of U-wave from V2 to V4 were the features predicting being a KCNJ2 mutation carrier.
Assessment of incidence of CA or SCD is relevant due to choice of treatment method and primary prevention. It is also difficult because of low prevalence of the syndrome, variable expression, and penetrance. Among patients with ATS, SCD risk is estimated as exceedingly low but they are still at a higher risk of developing life-threatening arrhythmias [16,17]. One of the characteristic features in the electrocardiogram of ATS patients is the “monotony” resulting from permanent ventricular arrhythmias, which in most cases are asymptomatic. Holter recordings could consist of over 50% PVC (Fig. 2D). Moreover, BiVT, as a special type of PVT, is detected [10]. In this study, 60% of KCNJ2 mutation carriers presented an arrhythmia (>2000 PVC or BiVT or PVT). We have not demonstrated a relation between arrhythmias and syncope in all of the patients. We observed adequate interventions only in two patients with inserted ICD, in both special circumstances for arrhythmia were present (amiodarone, physical effort after alcohol intake). Most of the reported episodes appeared during stress or physical activity and had no features of vasovagal syncope. Our data suggest that that the presence of micrognathia, periodic paralysis, palpitation, U-wave from V2 to V4, and Tpeak – Tend can predict the development of arrhythmias, syncope, and/or CA. Additionally, out of the features assessed, K897T polymorphism, periodic paralysis, muscle weakness, palpitation, prolonged Tpeak – Tend duration, complex ventricular arrhythmia were more prevalent in the group with syncope and/or CA.
Mutations in KCNQ1 are responsible for LQT1, in KCNH2 for LQT2, in SCN5A for LQT3 [18,19]. All of them can be associated with life-threatening arrhythmias including torsades de pointes. The role of SNPs in human organisms, in particular LQT syndromes, is unclear. It was revealed that K897T polymorphism in KCNH2 caused prolonged QT in women [20]. In our study, in one family, arrhythmia was revealed in all patients with a coexisting K897T polymorphism. In this family two unexplained SCDs occurred. Our data suggest that K897T is a risk factor for the occurrence of syncope, but an unambiguous assessment is not possible because of small group size.
In conclusion, although our study group is relatively small, the data presented point to a higher risk for patients presenting with the entire ATS triad, in particular micrognathia, periodic paralysis, and the specific changes in ECG. ATS manifestation appears to be more severe in females and the K897T polymorphism appears to be associated with a higher risk for syncope. These hypotheses will clearly require additional studies.
Acknowledgments
The authors are greatly indebted to the patients and their families.
Funding: The Turkish part of the research was supported by NHLBI of the NIH (HL47678, CA).
Footnotes
Disclosures: The Authors declare that there is no conflict of interest’.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Andersen ED, Krassilnikoff PA, Overvad H. Intermittent muscular weakness, extrasystoles, and multiple developmental anomalies. Acta Paediatr. 1971;60:559–64. doi: 10.1111/j.1651-2227.1971.tb06990.x. [DOI] [PubMed] [Google Scholar]
- 2.Tawil R, Ptacek LJ, Pavlakis SG, DeVivo DC, Penn AS, Ozdemir C, Griggs RC. Andersen’s syndrome: Potassium-sensitive periodic paralysis, ventricular ectopy, and dysmorphic features. Ann Neurol. 1994;35:326–30. doi: 10.1002/ana.410350313. [DOI] [PubMed] [Google Scholar]
- 3.Plaster NM, Tawil R, Tristani-Firouzi M, Canún S, Bendahhou S, Tsunoda A, Donaldson MR, Iannaccone ST, Brunt E, Barohn R, Clark J, Deymeer F, George AL, Jr, Fish FA, Hahn A, et al. Mutations in Kir2. 1 cause the developmental and episodic electrical phenotypes of Andersen’s Syndrome. Cell. 2001;105:511–9. doi: 10.1016/s0092-8674(01)00342-7. [DOI] [PubMed] [Google Scholar]
- 4.Andelfinger G, Tapper AR, Welch RC, Vanoye CG, George AL, Benson DW. KCNJ2 mutation results in Andersen syndrome with sex-specific cardiac and skeletal muscle phenotypes. Am J Hum Genet. 2002;71:663–8. doi: 10.1086/342360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kokunai Y, Nakata T, Furuta M, Sakata S, Kimura H, Aiba T, Yoshinaga M, Osaki Y, Nakamori M, Itoh H, Sato T, Kubota T, Kadota K, Shindo K, Mochizuki H, et al. A Kir3.4 mutation causes Andersen-Tawil syndrome by an inhibitory effect on Kir2. 1. Neurology. 2014;82:1058–64. doi: 10.1212/WNL.0000000000000239. [DOI] [PubMed] [Google Scholar]
- 6.Statland JM, Tawil R, Venance SL. Andersen-Tawil syndrome. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews® [Internet] Seattle (WA): University of Washington, Seattle; 2004. Nov 22, pp. 1993–2017. updated 2015 Sep 3. [PubMed] [Google Scholar]
- 7.Donaldson MR, Jensen JL, Tristani-Firouzi M, Tawil R, Tawil R, Bendahhou S, Suarez WA, Cobo AM, Poza JJ, Behr E, Wagstaff J, Szepetowski P, Pereira S, Mozaffar T, Escolar DM, et al. PIP2 binding residues of Kir2. 1 are common targets of mutations causing Andersen syndrome. Neurology. 2003;60:1811–6. doi: 10.1212/01.wnl.0000072261.14060.47. [DOI] [PubMed] [Google Scholar]
- 8.Donaldson MR, Yoon G, Fu YH, Ptacek LJ. Andersen-Tawil syndrome: a model of clinical variability, pleiotropy, and genetic heterogeneity. Ann Med. 2004;36(suppl 1):92–7. doi: 10.1080/17431380410032490. [DOI] [PubMed] [Google Scholar]
- 9.Sumitomo N, Shimizu W, Taniguchi K, Hiraoka M. Calcium channel blocker and adenosine triphosphate terminate bidirectional ventricular tachycardia in a patient with Andersen-Tawil syndrome. Heart Rhythm. 2008;5:498–9. doi: 10.1016/j.hrthm.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 10.Miyamoto K, Aiba T, Kimura H, Hayashi H, Ohno S, Yasuoka C, Tanioka Y, Tsuchiya T, Yoshida Y, Hayashi H, Tsuboi I, Nakajima I, Ishibashi K, Okamura H, Noda T, et al. Efficacy and safety of flecainide for ventricular arrhythmias in patients with Andersen-Tawil syndrome with KCNJ2 mutations. Heart Rhythm. 2015;12:596–603. doi: 10.1016/j.hrthm.2014.12.009. [DOI] [PubMed] [Google Scholar]
- 11.Kimura H, Zhou J, Kawamura M, Itoh H, Mizusawa Y, Ding WG, Wu J, Ohno S, Makiyama T, Miyamoto A, Naiki N, Wang Q, Xie Y, Suzuki T, Tateno S, et al. Phenotype variability in patients carrying KCNJ2 mutations. Circ Cardiovasc Genet. 2012;5:344–53. doi: 10.1161/CIRCGENETICS.111.962316. [DOI] [PubMed] [Google Scholar]
- 12.Jagodzińska M, Szperl M, Ponińska J, Kosiec A, Gajda R, Kukla P, Biernacka EK. Coexistence of Andersen-Tawil syndrome with polymorphisms in hERG1 Gene (K897T) and SCN5A Gene (H558R) in one family. Ann Noninvasive Electrocardiol. 2015;21:189–95. doi: 10.1111/anec.12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang L, Benson DW, Tristani-Firouzi M, Ptacek LJ, Tawil R, Schwartz PJ, George AL, Horie M, Andelfinger G, Snow GL, Fu YH, Ackerman MJ, Vincent GM. Electrocardiographic features in Andersen-Tawil syndrome patients with KCNJ2 mutations: characteristic T-U-wave patterns predict the KCNJ2 genotype. Circulation. 2005;111:2720–6. doi: 10.1161/CIRCULATIONAHA.104.472498. [DOI] [PubMed] [Google Scholar]
- 14.Kostera-Pruszczyk A, Potulska-Chromik A, Pruszczyk P, Bieganowska K, Miszczak-Knecht M, Bienias P, Szczałuba K, Lee HY, Quinn E, Ploski R, Kaminska A, Ptáček LJ. Andersen-Tawil syndrome: Report of 3 novel mutations and high risk of symptomatic cardiac involvement. Muscle Nerve. 2014;51:192–6. doi: 10.1002/mus.24293. [DOI] [PubMed] [Google Scholar]
- 15.Kukla P, Biernacka EK, Baranchuk A, Jastrzebski M, Jagodzinska M. Electrocardiogram in Andersen-Tawil Syndrome. New electrocardiographic criteria for diagnosis of type-1 Andersen-Tawil syndrome. Curr Cardiol Rev. 2014;10:222–8. doi: 10.2174/1573403X10666140514102528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Airey KJ, Etheridge SP, Tawil R, Tristani-Firouzi M. Resuscitated sudden cardiac death in Andersen-Tawil syndrome. Heart Rhythm. 2009;6:1814–7. doi: 10.1016/j.hrthm.2009.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delannoy E, Sacher F, Maury P, Mabo P, Mansourati J, Magnin I, Camous JP, Tournant G, Rendu E, Kyndt F, Haïssaguerre M, Bézieau S, Guyomarch B, Le Marec H, Fressart V, et al. Cardiac characteristics and long-term outcome in Andersen-Tawil syndrome patients related to KCNJ2 mutation. Europace. 2013;15:1805–11. doi: 10.1093/europace/eut160. [DOI] [PubMed] [Google Scholar]
- 18.Levine E, Rosero SZ, Budzikowski AS, Moss AJ, Zareba W, Daubert J. Congenital long QT syndrome: Considerations for primary care physicians. Cleve Clin J Med. 2008;75:591–600. doi: 10.3949/ccjm.75.8.591. [DOI] [PubMed] [Google Scholar]
- 19.Thomas D, Kiehn J, Katus HA, Karle CA. Defective protein trafficking in hERG-associated hereditary long QT syndrome (LQT2): molecular mechanisms and restoration of intracellular protein processing. Cardiovasc Res. 2003;60:235–41. doi: 10.1016/j.cardiores.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 20.Pietila E, Fodstad H, Niskasaari E, Laitinen PPJ, Swan H, Savolainen M, Kesäniemi YA, Kontula K, Huikuri HV. Association between HERG K897T polymorphism and QT interval in middle-aged Finnish women. J Am Coll Cardiol. 2002;40:511–4. doi: 10.1016/s0735-1097(02)01979-4. [DOI] [PubMed] [Google Scholar]