

ABSTRACT
The obligate intracellular parasite Toxoplasma gondii can actively infect any nucleated cell type, including cells from the immune system. The rapid transfer of T. gondii from infected dendritic cells to effector natural killer (NK) cells may contribute to the parasite's sequestration and shielding from immune recognition shortly after infection. However, subversion of NK cell functions, such as cytotoxicity or production of proinflammatory cytokines, such as gamma interferon (IFN-γ), upon parasite infection might also be beneficial to the parasite. In the present study, we investigated the effects of T. gondii infection on NK cells. In vitro, infected NK cells were found to be poor at killing target cells and had reduced levels of IFN-γ production. This could be attributed in part to the inability of infected cells to form conjugates with their target cells. However, even upon NK1.1 cross-linking of NK cells, the infected NK cells also exhibited poor degranulation and IFN-γ production. Similarly, NK cells infected in vivo were also poor at killing target cells and producing IFN-γ. Increased levels of transforming growth factor β production, as well as increased levels of expression of SHP-1 in the cytosol of infected NK cells upon infection, were observed in infected NK cells. However, the phosphorylation of STAT4 was not altered in infected NK cells, suggesting that transcriptional regulation mediates the reduced IFN-γ production, which was confirmed by quantitative PCR. These data suggest that infection of NK cells by T. gondii impairs NK cell recognition of target cells and cytokine release, two mechanisms that independently could enhance T. gondii survival.
KEYWORDS: natural killer cells, cytotoxicity, apicomplexa, parasite infection, cytokines, apicomplexan parasites, host-parasite relationship
INTRODUCTION
The obligate intracellular protozoan Toxoplasma gondii infects approximately 25% of the global human population (1). Although infection is principally asymptomatic, it can cause severe neurological complications in immunocompromised individuals, disseminated congenital infections in the developing fetus, and ocular manifestations in otherwise healthy individuals (1). In the early phase of T. gondii infection, inflammatory monocytic cells are recruited to the site of infection. Interleukin-12 (IL-12) and interferon gamma (IFN-γ) production ensures the establishment of the specific cell-mediated immune response leading to protection against recurrent infections via T cells and natural killer (NK) cells and later by B cell-mediated antibody production (2). While different cell types, e.g., epithelial cells or cells of the central nervous system (CNS), may provide a refuge for an intracellular pathogen, leukocytes also mediate immune surveillance and are essential for pathogen clearance. Paradoxically, the inherent migratory functions of leukocytes also make them a suitable target for pathogens so that the pathogens may use them as a Trojan horse to mediate their dispersion in the organism (3, 4).
A major effector mechanism of immune cells is their ability to kill pathogens in infected cells, thereby limiting the spread of an infectious agent. The driving of Th1 responses by NK cells and CD8+ T cells enhances the intracellular killing of T. gondii (5). Furthermore, killing of infected cells through perforin-mediated pathways could also protect hosts from T. gondii infection (6). However, recent observations on T cells, NK cells, and dendritic cells (DCs) vis à vis their infection by T. gondii have highlighted potential mechanisms by which this obligate intracellular parasite might evade cellular immunity and also might manipulate cell-mediated cytotoxicity to its own advantage (7, 8). Death receptor ligation in T. gondii-infected cells leads to the rapid egress of infectious parasites, an active process mediated through intracellular calcium flux as a consequence of caspase activation. Interestingly, upon interaction of CD8+ T cells with T. gondii-infected cells, the CD8+ T cells became infected themselves (7). Similar findings that CD8+ T cells could be infected by T. gondii were also observed in vivo using 2-photon microscopy (9). Similarly, perforin-dependent NK cell-mediated cytotoxicity of DCs induced parasite egress, which led to infection of NK cells both in vivo and in vitro (8). More recently, it has been shown that infection of NK cells in vivo may induce hypermotility in NK cells (10).
Since NK cells have important roles in immune responses to T. gondii (11, 12), in the present study, we examined the effect of T. gondii infection on NK cell effector function. We also identify potential molecular pathways targeted by the parasite that could affect NK cell functions.
RESULTS
NK cells infected by T. gondii exhibit reduced cytotoxicity in vitro.
Since we reported previously that T. gondii is efficiently transmitted from infected DCs to effector NK cells and T cells during the cytotoxicity of infected cells (7, 8), we investigated the functional consequences of these infections on NK cells. Since, in our previous study, IL-2-stimulated NK cells could become infected upon interaction with infected dendritic cells (8), we first infected IL-2-stimulated NK cells and tested for their cytotoxicity against YAC1 tumor cells in vitro. When NK cells exposed to T. gondii were compared with control unchallenged NK cells for their ability to kill YAC1 cells in a 51Cr release assay, there was a significant decrease in the killing of YAC1 cells by the T. gondii-exposed NK cells (Fig. 1A).
FIG 1.
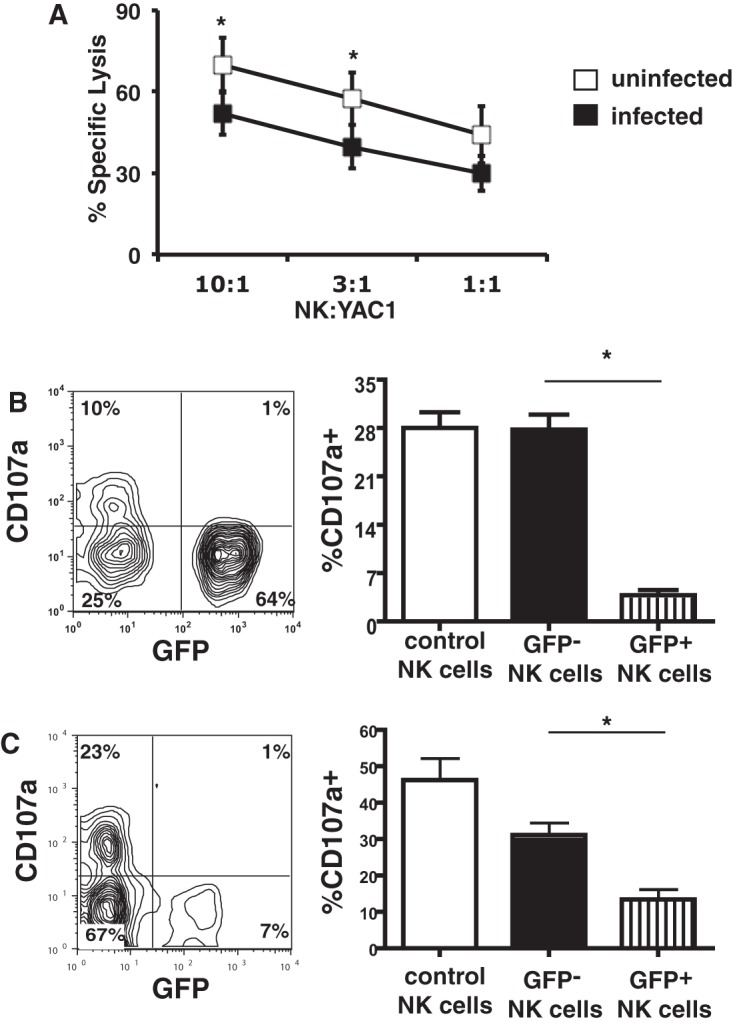
Infection of NK cells with T. gondii inhibits NK cell-mediated killing. (A) YAC1 cell killing by uninfected IL-2-stimulated NK cells or by NK cells infected with the RH-LDM T. gondii strain in the 51Cr release assay. The data represent means ± SEMs. *, P < 0.05, paired t test (n = 3 separate experiments). (B) Degranulation by IL-2-stimulated NK cells. (Left) Results of one representative experiment of degranulation by NK cells in the presence of YAC1 cells (10:1); (right) bar graph representing the percentage of CD107a+ cells by gating on the infected (GFP+) or uninfected (GFP−) NK cells separately. *, P < 0.01, ANOVA with the Bonferroni correction (n = 6 separate experiments). Control NK cells represent NK cells not exposed to T. gondii in culture. (C) Degranulation by NK cells following NK1.1 cross-linking. (Left) Results of one representative experiment; (right) bar graph representing the percentage of CD107a+ by gating on the infected (GFP+) or uninfected (GFP−) NK cells separately from all experiments. *, P < 0.01, ANOVA with the Bonferroni correction (n = 5 separate experiments). Control NK cells represent cells not exposed to T. gondii in culture.
Since the infection frequencies of the T. gondii-challenged NK cells were not 100% (range, 20 to 80%), it was important to determine whether the observed reduced cytotoxicity was a direct effect of the infection of the cells or whether some other extrinsic factor(s) affected the infected cells and bystander cells in the culture. To assess the degranulation of NK cells challenged with green fluorescent protein (GFP)-expressing T. gondii parasites by flow cytometry, we quantified the expression of the degranulation marker CD107a on the surface of the IL-2-stimulated NK cells when mixed with YAC1 cells. In the cultures that were exposed to T. gondii, the infected (GFP-positive [GFP+]) NK cells had significantly lower levels of CD107a expression on their cell surface than uninfected (GFP-negative [GFP−]) NK cells in the same culture (3.8% ± 1.8% versus 27.8% ± 5.3% CD107a+ cells; P < 0.01, analysis of variance [ANOVA] with the Bonferroni correction; n = 6 experiments) (Fig. 1B). The uninfected (GFP−) cells degranulated to the same extent as the control NK cells that were never exposed to T. gondii (27.8% ± 5.3% and 28% ± 5.7% CD107a+ cells, respectively; n = 6 experiments) (Fig. 1B).
Degranulation by NK cells can also be measured by cross-linking activating receptors, such as NK1.1, on their surface. To test if degranulation was also affected following NK1.1 cross-linking, splenocytes freshly isolated from C57BL/6 (B6).recombination activating gene-1−/− (B6.RAG1−/−) mice were challenged with T. gondii for 2 h, washed, and added to plates coated with anti-NK1.1 antibody. In this model, too, a reduced level of surface expression of CD107a was observed on the infected (GFP+) NK cells compared to the uninfected (GFP−) NK cells in the same culture (13.4% ± 6.3% versus 31.1% ± 7.8% CD107a+ cells; P < 0.01, ANOVA with the Bonferroni correction; n = 5) (Fig. 1C). There was no significant difference between the degranulation of the (GFP−) NK cells and the control NK cells that were never exposed to T. gondii (Fig. 1C).
T. gondii-infected NK cells form poor conjugates with target cells.
In order for NK cells to kill target cells, they must be able to form conjugates with the target cell. By mixing IL-2-stimulated NK cells exposed to T. gondii with YAC1 cells, we could compare how many infected (GFP+) and uninfected (GFP−) NK cells could form conjugates with YAC1 cells. While we observed that the NK cells challenged with T. gondii could form conjugates, the frequency of conjugate formation was significantly less than that observed with unchallenged NK cells (5.1% ± 2.1% versus 10.5% ± 2.7%; P < 0.05, Mann-Whitney test; n = 5 experiments) (Fig. 2A). By gating on the cells forming conjugates, infected (GFP+) NK cells made up 40.3% ± 18.2% of the cells in the conjugates, while uninfected bystander NK cells made up 58.1% ± 17.7% of the cells in the conjugates (n = 5) (Fig. 2B). Since the infection rate of the NK cells was 65.4% ± 10.3% (n = 5) (Fig. 2B; see also Fig. S1 in the supplemental material), infected NK cells forming conjugates were significantly underrepresented by approximately 30% (P < 0.01, ANOVA with the Bonferroni correction) (Fig. 2B).
FIG 2.

Impaired conjugate formation of T. gondii-infected NK cells with YAC1 cells. (A) (Left and middle) NK cell conjugate formation to YAC1 cells comparing control NK cells (left) with NK cells exposed to T. gondii (middle); (right) the graph compares the percentage of NK cells forming conjugates from both cultures. *, P < 0.05, Mann-Whitney test (n = 5 separate experiments). (B) (Left) Percentage of infected (GFP+) NK cells and uninfected (GFP−) NK cells found conjugated to YAC1 cells and percentage of infected (GFP+) NK cells and uninfected (GFP−) NK cells that did not form conjugates; (right) the graph represents the percentage of GFP+ NK cells that were conjugated and unconjugated relative to the percentage of infected NK cells added to the culture. *, P < 0.05, ANOVA (n = 5 separate experiments). (C) (Left and middle) Examination of the percentage of GFP− NK cells and GFP+ NK cells forming conjugates from the middle panel of panel A; among the 2.3% of cells forming conjugates, the majority were uninfected NK cells; (right) the graph compares the percentages of uninfected and infected cells conjugated to YAC1 cells from all experiments. *, P < 0.05, paired t test (n = 5 separate experiments).
To further demonstrate that there was a distinct underrepresentation in the frequency of infected (GFP+) NK cells forming conjugates, we also gated only on the infected or uninfected cells and examined how many cells formed conjugates. Using this gating, 7% ± 4% of the GFP+ NK cells formed conjugates, while 29% ± 8% of the GFP− NK cells in the culture formed conjugates (P < 0.01, Mann-Whitney test; n = 5) (Fig. 2C). This suggested that the lack of killing by NK cells was in part due to the reduced ability of infected NK cells to form conjugates with target cells.
Impaired IFN-γ production by T. gondii-infected NK cells.
In addition to their cytotoxic function, NK cells are also a major source of IFN-γ secretion and other proinflammatory cytokines in innate immune responses (13). IFN-γ has been shown to be an important cytokine in the host defense against T. gondii (14), driving host defenses through the STAT1 pathway (15, 16). When freshly isolated NK cells were stimulated with IL-12 or IL-18 for 4 h after infection with the type I RH-LDM strain of T. gondii, the level of IFN-γ production by the infected (GFP+) NK cells was significantly reduced compared to that by uninfected (GFP−) NK cells in the cultures (12.1% ± 10.2% versus 44.7% ± 28% IFN-γ-positive [IFN-γ+] cells; P < 0.01, ANOVA with the Bonferroni correction; n = 7) (Fig. 3A). No significant differences in the levels of expression of IFN-γ between the challenged uninfected (GFP−) NK cells and control NK cells that were never exposed to T. gondii were observed (Fig. 3A). In addition, IL-12- and IL-18-stimulated NK cells infected with the type II PTG-GFPS65T strain also exhibited reduced levels of IFN-γ production compared with uninfected (GFP−) NK cells (15.6% ± 7.9% versus 33.5% ± 27.6% IFN-γ+ cells; P < 0.05, paired t test; n = 3) (Fig. 3B). Furthermore, upon NK1.1 cross-linking, the infected (GFP+) NK cells produced significantly less IFN-γ than uninfected (GFP−) NK cells (2.8% ± 1.9% versus 7.8% ± 1% IFN-γ+ cells; P < 0.05, Mann-Whitney test; n = 4) (Fig. 3C).
FIG 3.

T. gondii infection of NK cells inhibits IFN-γ production. (A) IFN-γ production by NK cells upon IL-12 and IL-18 stimulation following infection with the type I RH-LDM strain. (Left) Results of one representative experiment with infected (GFP+) or uninfected (GFP−) NK cells; (right) bar graph representing the percentage of IFN-γ+ cells gated on infected (GFP+) or uninfected (GFP−) NK cells separately from all experiments. *, P < 0.01, ANOVA with the Bonferroni correction (n = 7 separate experiments). Control NK cells represent NK cells not exposed to T. gondii in culture. (B) IFN-γ production by NK cells upon IL-12 and IL-18 stimulation following infection with the type II PTG-GFPS65T strain. (Left) Results of one representative experiment with infected (GFP+) or uninfected (GFP−) NK cells; (right) bar graph showing the percentage of IFN-γ+ cells gated on the infected (GFP+) or uninfected (GFP−) NK cells separately from all experiments. *, P < 0.05, paired t test (n = 3 separate experiments). (C) IFN-γ production by NK cells following NK1.1 cross-linking. (Left) Results of one representative experiment with infected (GFP+) or uninfected (GFP−) NK cells; (right) bar graph representing the percentage of IFN-γ+ cells gating on infected (GFP+) or uninfected (GFP−) NK cells separately from all experiments. *, P < 0.01, Mann-Whitney test (n = 4 separate experiments).
T. gondii infection of NK cells in vivo impairs their effector/cytotoxic functions.
B6.RAG1−/− mice were intraperitoneally (i.p.) inoculated with T. gondii-infected DCs to test if T. gondii infection affected NK cell functions in vivo. When NK cells collected from peritoneal exudates of infected mice were tested against YAC1 cells, we again found significantly reduced levels of degranulation in the infected (GFP+) NK cells compared to that for the uninfected (GFP−) cells (17.3% ± 9.7% versus 38.8% ± 7.1% CD107a+ cells; P < 0.01, Mann-Whitney test; n = 6 mice) (Fig. 4A). In addition, the level of IFN-γ production by the infected (GFP+) NK cells upon phorbol myristate acetate (PMA) and ionomycin stimulation was also significantly reduced compared to that for uninfected NK cells from the same mice (3.9% ± 3.2% versus 14.3% ± 1.7% IFN-γ+ cells; P < 0.01, Mann-Whitney test; n = 6 mice) (Fig. 4B). This suggested that T. gondii could affect NK cell function even in vivo.
FIG 4.

NK cells infected in vivo with T. gondii exhibit reduced levels of cytotoxicity and IFN-γ production. (A) Lack of degranulation by infected GFP+ NK cells collected from the peritoneal cavity of mice inoculated with infected DCs when mixed 10:1 with YAC1 cells. *, P < 0.05, Mann-Whitney test (n = 6 mice). (B) IFN-γ production by NK cells collected from the peritoneal cavity of mice inoculated with T. gondii-infected DCs and stimulated with PMA and ionomycin in vitro. *, P < 0.05, Mann-Whitney test (n = 6 mice).
TGF-β and SHP-1 are induced in T. gondii-infected NK cells but do not affect STAT4 phosphorylation.
Transforming growth factor β (TGF-β) is a cytokine with strong anti-inflammatory capabilities; inhibits cytokine production, cytotoxicity, and conjugate formation (17); and can be induced by T. gondii (18–20). When freshly isolated NK cells from B6.RAG1−/− mice were cultured overnight following T. gondii infection, there was a small but significant increase in the amount of TGF-β detected in the culture medium (Fig. 5A). When NK cells were stained for surface expression of latency-activating protein (LAP), which forms a heterodimer with TGF-β to form a latent TGF-β complex, to determine the expression of TGF-β on the NK cells, there was an increased level of expression of LAP on the infected (GFP+) NK cells compared with that on the uninfected (GFP−) NK cells from the same cultures (Fig. 5B).
FIG 5.

Expression of TGF-β and SHP-1 by infected NK cells. (A) Levels of TGF-β from cultures of freshly isolated NK cells from B6.RAG1−/− mice exposed to or not exposed to T. gondii measured by ELISA following overnight culture. *, P < 0.05, paired t test (n = 6). (B) (Left) Percent surface expression of LAP on freshly isolated NK cells following 2 h of infection with T. gondii and 4 h of culture in vitro (dashed line, uninfected cells; solid line, infected cells; shaded area, isotype control); (right) bar graph representing the increase in the level of LAP on infected (GFP+) NK cells relative to that on uninfected (GFP−) NK cells from three separate experiments. (C) (Left) Percent intracellular expression of SHP-1 on freshly isolated NK cells following 2 h of infection with T. gondii and 4 h of culture in vitro (dashed line, uninfected cells; solid line, infected cells; shaded area, isotype control); (right) bar graph representing the increase in the level of SHP-1 in infected (GFP+) NK cells relative to that in uninfected (GFP−) NK cells from six separate experiments.
TGF-β can induce the expression of the phosphatase SHP-1 (21, 22). SHP-1 regulates lymphocyte activation by binding to ITIM motifs in the cytoplasmic tails of inhibitory molecules expressed on the surface of NK cells (23, 24). Overexpression of SHP-1 inhibits conjugate formation as well as interferes with cytokine signaling pathways (25). For this reason, NK cells were stained for SHP-1 after exposure to T. gondii. While SHP-1 could be detected at high levels in NK cells, the infected (GFP+) NK cells exhibited increased levels of expression of SHP-1 upon T. gondii infection compared to the uninfected NK cells in the same culture (Fig. 5C).
STAT4 signaling plays an important role in inducing IFN-γ (26) and, in addition, plays a role in defense against T. gondii (27). Since we observed increased SHP-1 levels in the infected cells, we stained infected cells for phosphorylated STAT4 (pSTAT4) to determine if increased amounts of SHP-1 could affect STAT4 phosphorylation and, as a consequence, reduce the level of IFN-γ production. Interestingly, there was already an increase in the level of pSTAT4 expression in infected (GFP+) NK cells compared to that in uninfected NK (GFP−) cells (mean fluorescence intensity [MFI] of pSTAT4, 21.2 ± 11.5 versus 30.4 ± 8.7; P < 0.05, paired t test; n = 4) (Fig. 6A) even without any IL-12 or IL-18 stimulation. In addition, there was also a nonsignificant increase in pSTAT4 levels in infected NK cells stimulated with IL-12 and IL-18 compared to control NK cells (MFI of pSTAT4, 53.8 ± 13.3 versus 43.2 ± 14.7) (Fig. 6B). Since pSTAT4 levels were not reduced between infected and uninfected NK cells, it did not appear that the reduced level of IFN-γ expression was due to the levels of STAT4 phosphorylation. In addition, we found that the level of phosphorylation of other STATs was increased in NK cells exposed to T. gondii (Fig. S2). Thus, NK cells, like other cells, may be affected by the rhoptry proteins (ROP) secreted by T. gondii, which stimulate STAT phosphorylation (28).
FIG 6.

STAT4 phosphorylation is not disrupted in infected NK cells, but the levels of IFN-γ transcripts are reduced. (A) (Left and middle) pSTAT4 expression in unstimulated control NK cells (left) and infected NK cells (middle). The results of one representative experiment of four that were conducted are shown. (Right) Bar graph of the MFI of pSTAT4 in infected and bystander NK cells from cultures exposed to T. gondii. *, P < 0.05, paired t test (n = 4). (B) (Left and middle) pSTAT4 expression in IL-12- and IL-18-stimulated control NK cells (left) and NK cells exposed to T. gondii (middle). The results of one representative experiment of four that were conducted are shown. (Right) Bar graph of the MFI of pSTAT4 in infected and bystander NK cells from cultures exposed to T. gondii (n = 4). (C) qPCR of IFN-γ RNA comparing the relative levels of expression of IFN-γ by uninfected NK cells and infected NK cells from the same culture stimulated with IL-12 and IL-18 and by NK cells that were not stimulated with IL-12 or IL-18 (control). (D and E) qPCR of miRNA146 (D) and miRNA155 (E) comparing the relative levels of expression by uninfected NK cells and infected NK cells following IL-12 and IL-18 stimulation in the same culture (n = 4 separate sortings). (F) qPCR of SOCS3 comparing the relative levels of expression by uninfected NK cells and infected NK cells from the same culture and by NK cells that were not stimulated with IL-12 and IL-18 (control).
Altered transcription of IFN-γ in T. gondii-infected NK cells.
When RNA was collected from infected and uninfected NK cells from B6.RAG1−/− mice stimulated with IL-12 and IL-18, we found by quantitative PCR (qPCR) that there was an approximately 6-fold decrease in the relative amount of the IFN-γ transcript in infected NK cells (Fig. 6C). Compared to the findings for unstimulated control NK cells, it was clear that IL-12 and IL-18 could still induce transcripts even in the infected cells, suggesting that IFN-γ could be induced in the infected cells (Fig. 6C). The reduced level of production of IFN-γ was not due to a reduction in the level of IL-18 receptor expression, as we found that both infected and uninfected NK cells expressed similar amounts of the IL-18 receptor (Fig. S3). In addition, we also found that the levels of tumor necrosis factor transcripts were also reduced in NK cells infected with T. gondii (Fig. S4). MicroRNA15/16 (miRNA15/16), miRNA29, miRNA146, and miRNA155 have been implicated in controlling IFN-γ expression in NK cells (29, 30); however, we found no difference in the levels of expression of these miRNAs between infected and uninfected cells (Fig. 6D and E and data not shown). These data suggest that the reduced level of expression of IFN-γ could be at the level of transcription but did not appear to be controlled by miRNA. Previous observations have shown that SOCS3 can be induced by T. gondii (31), and SOCS3 has been implicated in controlling cytotoxicity (32). However, we did not find any significant difference in the levels of expression of SOCS3 RNA between infected and uninfected NK cells (Fig. 6F). By Western blotting, we found that there were no changes in SOCS3 protein levels between infected and uninfected bystander NK cells (Fig. S2).
DISCUSSION
Previously, we found that NK cells rapidly become parasitized by T. gondii upon their interactions with infected dendritic cells (8). In the current study, we demonstrate that infection of NK cells with T. gondii can affect their ability to perform cytotoxicity and produce IFN-γ. In part, this downmodulatory effect on NK cells could be explained by the fact that infected cells were poor at conjugating with the target cells. However, we also found that cross-linking of NK1.1 on NK cells or stimulation with IL-12 and IL-18 also inhibited degranulation and/or cytokine production by NK cells, suggesting that there were also disturbances in intracellular signals in the infected cells. Together, these data suggest that the infection of NK cells by a pathogen might endow the pathogen with a further advantage in escaping immune responses because of the inability of NK cells to kill other infected cells.
TGF-β production is associated with T. gondii infection and affects immune responses not only to the parasite but also in a systemic manner (18–20, 33, 34). TGF-β induction has been associated with phosphatidylserine (PS) expression on the surface of the parasite and soluble factors (20, 35–37). In the present study, we observed only very minute increases in the amount of TGF-β in the cell cultures but could detect increased levels of LAP expression only on the surface of freshly isolated infected NK cells. However, only infected cells were affected, which suggests that if TGF-β were to play a role in inhibiting NK cell function, it likely does so only in an autocrine fashion.
TGF-β can lead to the induction of SHP-1 in cells, which might explain the upregulation of SHP-1 in the NK cells (21, 22). SHP-1 expression can play a role in viral (38) and other parasite (39) infections. SHP-1 is found intracellularly and binds to ITIM motifs on the tail of surface receptors. As a phosphatase, its primary role is to dephosphorylate phosphorylated signaling molecules, and so it plays an important role in modulating immune responses (24). In this regard, SHP-1 can inhibit cytotoxicity, cytokine production, and conjugate formation (24, 25, 40–45). T. gondii upregulates SHP-1 in NK cells, which may explain why these cells are inhibited from producing cytokines and inducing cytotoxicity. However, we could not see any effects on the phosphorylation of STAT4. Furthermore, we could see only mild increases in the levels of degranulation and IFN-γ production in NK cells treated with the SHP-1 and SHP-2 inhibitor sodium stibogluconate (data not shown), but this could be due to the short exposure time within the cultures. Thus, although the observation that the levels of SHP-1 increased is interesting, SHP-1 might not have any physiological effect on T. gondii-infected cells.
Although we observed decreased amounts of the IFN-γ transcript and decreased levels of IFN-γ production by infected cells, STAT4 phosphorylation did not seem to be impaired. In fact, even in unstimulated infected cells, there appeared to be a slight increase in the levels of pSTAT4. STAT phosphorylation has previously been observed in myeloid cells upon T. gondii infection and is associated with ROP molecules secreted by the parasite (28). Thus, it is interesting to speculate that STAT4 signaling could also be a target for ROP molecules in NK cells and thus affect its ability to signal or bind to its targets. However, ROP molecules may not be the whole explanation, since both the type I RH-LDM and the type II PTG-GFPS65T parasite lines appear to inhibit IFN-γ production similarly. Apart from ROP molecules, repression of STAT4-mediated transcription might be due to other molecules secreted by T. gondii. STAT1 signaling can be inhibited in infected cells and is caused by the Mi-2/NuRD complex induced by the Toxoplasma gondii inhibitor of STAT1-dependent transcription (TgIST) blocking STAT1 promoters (46). Thus, the 6-fold decrease in the IFN-γ RNA level observed in the infected cells could be due to STAT4 promoter blockage. This blockade may not be complete or may be leaky, which might explain why some cells respond and others do not. Finally, another point at which T. gondii infection of cells could inhibit IFN-γ production is through the induction of miRNA146 and miRNA155 (47), both of which can control IFN-γ production (29, 30). However, we found only small differences in miRNA146 or miRNA155 levels between infected and uninfected cells, suggesting that the infection probably did not affect this pathway of inhibiting IFN-γ production.
We could link a lack of killing by NK cells infected with T. gondii to reduced conjugation between target and effector cells. However, the mechanisms behind why the infected cells were poor at forming conjugates are unclear. In addition, it was also unclear why the infected cells that did form conjugates did not kill either. It is possible that, in forming the parasitophorous vacuole, alterations in trafficking of receptors or their signaling molecules could prevent conjugate formation or interfere with synapse formation, which is critical for granule release (48). Infected NK cells have been reported to be more motile (10). Thus, increased motility may also prevent the long and sustained conjugate formation necessary for killing. Therefore, multiple mechanisms may be involved in the inhibition of killing by the T. gondii-infected NK cells.
We have previously hypothesized that infection of killer cells by T. gondii would be beneficial to T. gondii so that it could evade immune responses since it did not appear to be targeted by other uninfected killer cells (8). However, the present study demonstrates that infection of NK cells by T. gondii can affect immune responses and be advantageous for the parasite in a multiplicity of ways. While our future research will focus on divining the pathways responsible, it is clear that altering the ability of NK cells to kill or release proinflammatory cytokines would (i) allow other infected cells to evade detection, which would allow infected cells, such as DCs, to make their way to immune-privileged sites; (ii) inhibit nitric oxide and other intracellular host defense processes that might be detrimental to parasite survival; and (iii) limit proinflammatory responses to ensure that the host does not succumb to mortality. In addition, we have observed that CD8+ T cells infected with T. gondii are also poor at killing and producing cytokines (unpublished observations), suggesting that the turning off of killer cells might confer an advantage to parasite survival. Altogether in this study, we present another layer by which T. gondii modulates the immune system to its advantage.
MATERIALS AND METHODS
Animals.
C57BL/6 (B6) and B6.recombination activating gene-1−/− (B6.RAG1)−/− (age, 6 to 10 weeks) were housed under standard conditions at the Department of Microbiology and Tumor and Cell Biology at the Karolinska Institutet and at the Karolinska University Hospital Huddinge, Stockholm, Sweden. All procedures were performed under institutional and national guidelines (ethical numbers from Stockholm County Council, N327/12, N135/15, N147/15).
Antibodies.
All anti-CD107a (clone 1D4B), anti-IFN-γ (clone XMG1.2), anti-latency-activating protein (anti-LAP; clone TW7-16B4), anti-NK1.1 (clone PK135), and anti-NKp46 (clone 29A1.4) antibodies were purchased from eBioscience (San Diego, CA) or BioLegend (San Diego CA). Anti-pSTAT4 (clone 38/p-STAT4) was purchased from BD Biosciences (San Diego CA). Anti-SHP-1 (catalog number ab32559) control rabbit antibody and goat anti-rabbit immunoglobulin antibody conjugated to DyLight 650 were purchased from Abcam (Cambridge, United Kingdom).
NK cell preparation.
NK cells were prepared by purifying DX5-positive (DX5+) cells from the spleens of B6 or B6.RAG1−/− mice by using a magnetically activated cell sorting (MACS) separation system (Miltenyi Biotech, Bergisch Gladbach, Germany), according to the manufacturer's guidelines. For the production of IL-2-stimulated NK cells, DX5+ cells were resuspended in complete alpha minimal essential medium (αMEM; 10 mM HEPES, 2 × 10−5 M 2-mercaptoethanol, 10% fetal calf serum [FCS], 100 U/ml penicillin, 100 U/ml streptomycin) and cultured in 1,000 U recombinant IL-2 (Immunotools, Friesoythe, Germany)/ml for 5 days.
Parasites and infection.
Green fluorescent protein (GFP)-expressing type I RH-LDM (49) and type II PTG-GFPS65T (50) T. gondii tachyzoites were maintained by serial 2-day passage in human foreskin fibroblast (HFF) monolayers. HFFs were propagated in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Paisley, United Kingdom) with 10% FCS (BioWhittaker, Verviers, Belgium), 20 μg/ml gentamicin, 2 mM glutamine, and 0.01 M HEPES (Invitrogen). For infection of NK cells or splenocytes in vitro, cells were harvested and incubated with freshly egressed GFP-expressing T. gondii tachyzoites at multiplicities of infection (MOI) of 3:1 for 2 h. After 2 h, the cells were washed twice by centrifugation at 150 × g for 10 min each time without brake to remove free parasites. The cells were then used in the experiments as outlined below. Infection frequencies were assessed by flow cytometry by counting of the number of GFP+ cells.
51Cr cytotoxicity assays.
YAC1 tumor cells were incubated for 1 h in the presence of 51Cr (Amersham, Oxford, United Kingdom) and then washed thoroughly in phosphate-buffered saline (PBS). After 4 h of NK cell-target cell coincubation, the cell culture supernatants were taken from these wells and analyzed by using a gamma radiation counter (Wallac Oy, Turku, Finland). Specific lysis was calculated according to the following formula: percent specific lysis = [(experimental release − spontaneous release)/(maximum release − spontaneous release)] × 100.
Detection of degranulation and IFN-γ production.
After 2 h of coculture with T. gondii as outlined above, NK cells were mixed with target cells at a ratio of 10:1 in the presence of anti-CD107a antibody (1:200). After 2 h, brefeldin A and monensin (BioLegend) were added to the culture and the culture was incubated for another 2 h. The cells were collected and stained for surface molecules.
To measure degranulation and IFN-γ production upon NK1.1 cross-linking, U-bottom plates were coated with 50 μg of anti-NK1.1 or control antibody for 1 h at 37°C before being left overnight at 4°C. The wells were then washed, and 1 × 106 B6.RAG1−/−-infected splenocytes were added to the wells. CD107a was added to the cultures for the duration of the assay (1:200). After 2 h, brefeldin A and monensin (BioLegend) were added to the culture and the culture was incubated for another 2 h. Cells were collected and stained for surface markers and then fixed in 1% paraformaldehyde. Intracellular IFN-γ was detected by permeabilization of the cells using permeabilization solution (BioLegend) and staining with anti-IFN-γ antibodies.
To measure IFN-γ expression following IL-12 and IL-18 stimulation, NK cells were stimulated with 100 ng/ml recombinant mouse IL-12 (Peprotech, Rocky Hill, NJ) and recombinant mouse IL-18 (Biosource, Brussels, Belgium), incubated, and stained for IFN-γ as described above. LAP expression was measured by staining for surface expression 4 h after infection of the NK cells. SHP-1 levels were measured by intracellular staining, as outlined above.
For the detection of pSTAT4, sorted NK cells were exposed to T. gondii RH-LDM for 2 h before stimulation with IL-12 and IL-18 as described above for 15 min before fixation. Staining for pSTAT4 was performed according to the manufacturer's guidelines (BD Biosciences).
In vivo infection of NK cells.
For in vivo infection of NK cells, bone marrow-derived dendritic cells (BMDCs) (51) were infected with T. gondii RH-LDM and inoculated i.p. After 48 h, the peritoneal cavity was washed and the cells were collected. DX5+ cells from the peritoneal wash were sorted with MACS beads and used in the experiments.
NK cell-target cell conjugation.
Target cells were incubated with CellTracker Orange (Thermo Fisher Scientific, Waltham, MA) for 15 min and then washed thoroughly. The target cells were added to effector cells at a 1:1 ratio in 200 μl. The cells were centrifuged for 30 s at 500 × g and then incubated at 37°C for 15 min. The cells were recovered, immediately put on ice, and stained for cell surface markers for 15 min. The cells were then washed in cold PBS and fixed in 1% paraformaldehyde.
TGF-β ELISA.
NK cells were infected with T. gondii RH-LDM as outlined above and then incubated overnight. Supernatants from these cultures were then analyzed using a TGF-β enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's guidelines (BioLegend).
qPCR analysis of infected NK cells.
NK cells infected with T. gondii RH-LDM were activated with IL-12 and IL-18 as outlined above and then sorted by flow cytometry on the basis of the expression of GFP. RNA was isolated from TRI Reagent (Sigma-Aldrich)-treated samples according to the manufacturer's instructions. Total RNA was reverse transcribed (RevertAid first-strand cDNA synthesis kit; Thermo Fisher Scientific), and the levels of IFN-γ and SOCS3 were quantified by real-time PCR (SYBR green Jumpstart Taq ready mix; Sigma-Aldrich). For microRNA (miRNA) quantification, RNA was reverse transcribed by use of a miScript reverse transcription kit (Qiagen), and the levels of miRNA146a and miRNA155 were quantified by real-time PCR (miScript SYBR green PCR kit with the miScript primer assay; Qiagen). The amount of transcripts in relation to the amount of hypoxanthine phosphoribosyltransferase (HPRT) or U6 (for miRNA) transcripts was calculated by the 2−(ΔΔCT) threshold cycle (CT) method and calibrated to the amount for uninfected cells. The specificity of the products obtained was confirmed by the use of dissociation curves.
Statistics.
All statistical analyses were performed using GraphPad Prism software (La Jolla, CA).
Supplementary Material
ACKNOWLEDGMENTS
We thank Martin Rottenberg for fruitful discussions and advice throughout this study.
This work was supported by the Swedish Cancer Society, the Swedish Foundation for Strategic Research, and a Stockholm County Council Theme Center Grant (to B.J.C.) and the Swedish Research Council (to A.B.).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00069-17.
REFERENCES
- 1.Joynson DH, Wreghitt TJ. 2001. Toxoplasmosis: a comprehensive clinical guide. Cambridge University Press, Cambridge, United Kingdom. [Google Scholar]
- 2.Lang C, Gross U, Luder CG. 2007. Subversion of innate and adaptive immune responses by Toxoplasma gondii. Parasitol Res 100:191–203. doi: 10.1007/s00436-006-0306-9. [DOI] [PubMed] [Google Scholar]
- 3.Lambert H, Hitziger N, Dellacasa I, Svensson M, Barragan A. 2006. Induction of dendritic cell migration upon Toxoplasma gondii infection potentiates parasite dissemination. Cell Microbiol 8:1611–1623. doi: 10.1111/j.1462-5822.2006.00735.x. [DOI] [PubMed] [Google Scholar]
- 4.Fuks JM, Arrighi RB, Weidner JM, Kumar Mendu S, Jin Z, Wallin RP, Rethi B, Birnir B, Barragan A. 2012. GABAergic signaling is linked to a hypermigratory phenotype in dendritic cells infected by Toxoplasma gondii. PLoS Pathog 8:e1003051. doi: 10.1371/journal.ppat.1003051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hunter CA, Sibley LD. 2012. Modulation of innate immunity by Toxoplasma gondii virulence effectors. Nat Rev Microbiol 10:766–778. doi: 10.1038/nrmicro2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Denkers EY, Yap G, Scharton-Kersten T, Charest H, Butcher BA, Caspar P, Heiny S, Sher A. 1997. Perforin-mediated cytolysis plays a limited role in host resistance to Toxoplasma gondii. J Immunol 159:1903–1908. [PubMed] [Google Scholar]
- 7.Persson EK, Agnarson AM, Lambert H, Hitziger N, Yagita H, Chambers BJ, Barragan A, Grandien A. 2007. Death receptor ligation or exposure to perforin trigger rapid egress of the intracellular parasite Toxoplasma gondii. J Immunol 179:8357–8365. doi: 10.4049/jimmunol.179.12.8357. [DOI] [PubMed] [Google Scholar]
- 8.Persson CM, Lambert H, Vutova PP, Dellacasa-Lindberg I, Nederby J, Yagita H, Ljunggren HG, Grandien A, Barragan A, Chambers BJ. 2009. Transmission of Toxoplasma gondii from infected dendritic cells to natural killer cells. Infect Immun 77:970–976. doi: 10.1128/IAI.00833-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chtanova T, Han SJ, Schaeffer M, van Dooren GG, Herzmark P, Striepen B, Robey EA. 2009. Dynamics of T cell, antigen-presenting cell, and pathogen interactions during recall responses in the lymph node. Immunity 31:342–355. doi: 10.1016/j.immuni.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ueno N, Lodoen MB, Hickey GL, Robey EA, Coombes JL. 2015. Toxoplasma gondii-infected natural killer cells display a hypermotility phenotype in vivo. Immunol Cell Biol 93:508–513. doi: 10.1038/icb.2014.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gigley JP. 2016. The diverse role of NK cells in immunity to Toxoplasma gondii infection. PLoS Pathog 12:e1005396. doi: 10.1371/journal.ppat.1005396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yarovinsky F. 2014. Innate immunity to Toxoplasma gondii infection. Nat Rev Immunol 14:109–121. doi: 10.1038/nri3598. [DOI] [PubMed] [Google Scholar]
- 13.Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. 2008. Functions of natural killer cells. Nat Immunol 9:503–510. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki Y, Orellana MA, Schreiber RD, Remington JS. 1988. Interferon-gamma: the major mediator of resistance against Toxoplasma gondii. Science 240:516–518. doi: 10.1126/science.3128869. [DOI] [PubMed] [Google Scholar]
- 15.Gavrilescu LC, Butcher BA, Del Rio L, Taylor GA, Denkers EY. 2004. STAT1 is essential for antimicrobial effector function but dispensable for gamma interferon production during Toxoplasma gondii infection. Infect Immun 72:1257–1264. doi: 10.1128/IAI.72.3.1257-1264.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lieberman LA, Banica M, Reiner SL, Hunter CA. 2004. STAT1 plays a critical role in the regulation of antimicrobial effector mechanisms, but not in the development of Th1-type responses during toxoplasmosis. J Immunol 172:457–463. doi: 10.4049/jimmunol.172.1.457. [DOI] [PubMed] [Google Scholar]
- 17.Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. 2006. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol 24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- 18.Bermudez LE, Covaro G, Remington J. 1993. Infection of murine macrophages with Toxoplasma gondii is associated with release of transforming growth factor beta and downregulation of expression of tumor necrosis factor receptors. Infect Immun 61:4126–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hunter CA, Bermudez L, Beernink H, Waegell W, Remington JS. 1995. Transforming growth factor-beta inhibits interleukin-12-induced production of interferon-gamma by natural killer cells: a role for transforming growth factor-beta in the regulation of T cell-independent resistance to Toxoplasma gondii. Eur J Immunol 25:994–1000. doi: 10.1002/eji.1830250420. [DOI] [PubMed] [Google Scholar]
- 20.Seabra SH, de Souza W, Damatta RA. 2004. Toxoplasma gondii exposes phosphatidylserine inducing a TGF-beta1 autocrine effect orchestrating macrophage evasion. Biochem Biophys Res Commun 324:744–752. doi: 10.1016/j.bbrc.2004.09.114. [DOI] [PubMed] [Google Scholar]
- 21.Pallotta MT, Orabona C, Volpi C, Vacca C, Belladonna ML, Bianchi R, Servillo G, Brunacci C, Calvitti M, Bicciato S, Mazza EM, Boon L, Grassi F, Fioretti MC, Fallarino F, Puccetti P, Grohmann U. 2011. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat Immunol 12:870–878. doi: 10.1038/ni.2077. [DOI] [PubMed] [Google Scholar]
- 22.Choudhry MA, Sir O, Sayeed MM. 2001. TGF-beta abrogates TCR-mediated signaling by upregulating tyrosine phosphatases in T cells. Shock 15:193–199. doi: 10.1097/00024382-200115030-00006. [DOI] [PubMed] [Google Scholar]
- 23.Mahmood S, Kanwar N, Tran J, Zhang ML, Kung SK. 2012. SHP-1 phosphatase is a critical regulator in preventing natural killer cell self-killing. PLoS One 7:e44244. doi: 10.1371/journal.pone.0044244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lorenz U. 2009. SHP-1 and SHP-2 in T cells: two phosphatases functioning at many levels. Immunol Rev 228:342–359. doi: 10.1111/j.1600-065X.2008.00760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burshtyn DN, Shin J, Stebbins C, Long EO. 2000. Adhesion to target cells is disrupted by the killer cell inhibitory receptor. Curr Biol 10:777–780. doi: 10.1016/S0960-9822(00)00568-6. [DOI] [PubMed] [Google Scholar]
- 26.Lawless VA, Zhang S, Ozes ON, Bruns HA, Oldham I, Hoey T, Grusby MJ, Kaplan MH. 2000. Stat4 regulates multiple components of IFN-gamma-inducing signaling pathways. J Immunol 165:6803–6808. doi: 10.4049/jimmunol.165.12.6803. [DOI] [PubMed] [Google Scholar]
- 27.Cai G, Radzanowski T, Villegas EN, Kastelein R, Hunter CA. 2000. Identification of STAT4-dependent and independent mechanisms of resistance to Toxoplasma gondii. J Immunol 165:2619–2627. doi: 10.4049/jimmunol.165.5.2619. [DOI] [PubMed] [Google Scholar]
- 28.Saeij JP, Coller S, Boyle JP, Jerome ME, White MW, Boothroyd JC. 2007. Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature 445:324–327. doi: 10.1038/nature05395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leong JW, Sullivan RP, Fehniger TA. 2014. MicroRNA management of NK-cell developmental and functional programs. Eur J Immunol 44:2862–2868. doi: 10.1002/eji.201444798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lindsay MA. 2008. MicroRNAs and the immune response. Trends Immunol 29:343–351. doi: 10.1016/j.it.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 31.Whitmarsh RJ, Gray CM, Gregg B, Christian DA, May MJ, Murray PJ, Hunter CA. 2011. A critical role for SOCS3 in innate resistance to Toxoplasma gondii. Cell Host Microbe 10:224–236. doi: 10.1016/j.chom.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu X, Wang Q, Deng B, Wang H, Dong Z, Qu X, Kong B. 2012. Monocyte chemoattractant protein-1 secreted by decidual stromal cells inhibits NK cells cytotoxicity by up-regulating expression of SOCS3. PLoS One 7:e41869. doi: 10.1371/journal.pone.0041869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schluter D, Bertsch D, Frei K, Hubers SB, Wiestler OD, Hof H, Fontana A, Deckert-Schluter M. 1998. Interferon-gamma antagonizes transforming growth factor-beta2-mediated immunosuppression in murine Toxoplasma encephalitis. J Neuroimmunol 81:38–48. doi: 10.1016/S0165-5728(97)00156-2. [DOI] [PubMed] [Google Scholar]
- 34.Buzoni-Gatel D, Debbabi H, Mennechet FJ, Martin V, Lepage AC, Schwartzman JD, Kasper LH. 2001. Murine ileitis after intracellular parasite infection is controlled by TGF-beta-producing intraepithelial lymphocytes. Gastroenterology 120:914–924. doi: 10.1053/gast.2001.22432a. [DOI] [PubMed] [Google Scholar]
- 35.Clemente AM, Severini C, Castronovo G, Tanturli M, Perissi E, Cozzolino F, Torcia MG. 2014. Effects of soluble extracts from Leishmania infantum promastigotes, Toxoplasma gondii tachyzoites on TGF-beta mediated pathways in activated CD4(+) T lymphocytes. Microbes Infect 16:778–787. doi: 10.1016/j.micinf.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 36.D'Angelillo A, De Luna E, Romano S, Bisogni R, Buffolano W, Gargano N, Del Porto P, Del Vecchio L, Petersen E, Romano MF. 2011. Toxoplasma gondii dense granule antigen 1 stimulates apoptosis of monocytes through autocrine TGF-beta signaling. Apoptosis 16:551–562. doi: 10.1007/s10495-011-0586-0. [DOI] [PubMed] [Google Scholar]
- 37.Santos TA, Portes JDA, Damasceno-Sa JC, Caldas LA, Souza W, Damatta RA, Seabra SH. 2011. Phosphatidylserine exposure by Toxoplasma gondii is fundamental to balance the immune response granting survival of the parasite and of the host. PLoS One 6:e27867. doi: 10.1371/journal.pone.0027867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Christophi GP, Hudson CA, Gruber R, Christophi CP, Massa PT. 2008. Promoter-specific induction of the phosphatase SHP-1 by viral infection and cytokines in CNS glia. J Neurochem 105:2511–2523. doi: 10.1111/j.1471-4159.2008.05337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Forget G, Matte C, Siminovitch KA, Rivest S, Pouliot P, Olivier M. 2005. Regulation of the Leishmania-induced innate inflammatory response by the protein tyrosine phosphatase SHP-1. Eur J Immunol 35:1906–1917. doi: 10.1002/eji.200526037. [DOI] [PubMed] [Google Scholar]
- 40.Yusa S, Campbell KS. 2003. Src homology region 2-containing protein tyrosine phosphatase-2 (SHP-2) can play a direct role in the inhibitory function of killer cell Ig-like receptors in human NK cells. J Immunol 170:4539–4547. doi: 10.4049/jimmunol.170.9.4539. [DOI] [PubMed] [Google Scholar]
- 41.Rhee I, Veillette A. 2012. Protein tyrosine phosphatases in lymphocyte activation and autoimmunity. Nat Immunol 13:439–447. doi: 10.1038/ni.2246. [DOI] [PubMed] [Google Scholar]
- 42.Iype T, Sankarshanan M, Mauldin IS, Mullins DW, Lorenz U. 2010. The protein tyrosine phosphatase SHP-1 modulates the suppressive activity of regulatory T cells. J Immunol 185:6115–6127. doi: 10.4049/jimmunol.1000622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vyas YM, Maniar H, Dupont B. 2002. Cutting edge: differential segregation of the SRC homology 2-containing protein tyrosine phosphatase-1 within the early NK cell immune synapse distinguishes noncytolytic from cytolytic interactions. J Immunol 168:3150–3154. doi: 10.4049/jimmunol.168.7.3150. [DOI] [PubMed] [Google Scholar]
- 44.Stebbins CC, Watzl C, Billadeau DD, Leibson PJ, Burshtyn DN, Long EO. 2003. Vav1 dephosphorylation by the tyrosine phosphatase SHP-1 as a mechanism for inhibition of cellular cytotoxicity. Mol Cell Biol 23:6291–6299. doi: 10.1128/MCB.23.17.6291-6299.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu WM, Wang S, Keegan AD, Williams MS, Qu CK. 2005. Abnormal Th1 cell differentiation and IFN-gamma production in T lymphocytes from motheaten viable mice mutant for Src homology 2 domain-containing protein tyrosine phosphatase-1. J Immunol 174:1013–1019. doi: 10.4049/jimmunol.174.2.1013. [DOI] [PubMed] [Google Scholar]
- 46.Olias P, Etheridge RD, Zhang Y, Holtzman MJ, Sibley LD. 2016. Toxoplasma effector recruits the Mi-2/NuRD complex to repress STAT1 transcription and block IFN-gamma-dependent gene expression. Cell Host Microbe 20:72–82. doi: 10.1016/j.chom.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cannella D, Brenier-Pinchart MP, Braun L, van Rooyen JM, Bougdour A, Bastien O, Behnke MS, Curt RL, Curt A, Saeij JP, Sibley LD, Pelloux H, Hakimi MA. 2014. miR-146a and miR-155 delineate a microRNA fingerprint associated with Toxoplasma persistence in the host brain. Cell Rep 6:928–937. doi: 10.1016/j.celrep.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bryceson YT, Long EO. 2008. Line of attack: NK cell specificity and integration of signals. Curr Opin Immunol 20:344–352. doi: 10.1016/j.coi.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barragan A, Sibley LD. 2002. Transepithelial migration of Toxoplasma gondii is linked to parasite motility and virulence. J Exp Med 195:1625–1633. doi: 10.1084/jem.20020258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim K, Eaton MS, Schubert W, Wu S, Tang J. 2001. Optimized expression of green fluorescent protein in Toxoplasma gondii using thermostable green fluorescent protein mutants. Mol Biochem Parasitol 113:309–313. doi: 10.1016/S0166-6851(01)00212-2. [DOI] [PubMed] [Google Scholar]
- 51.Norbury CC, Chambers BJ, Prescott AR, Ljunggren HG, Watts C. 1997. Constitutive macropinocytosis allows TAP-dependent major histocompatibility complex class I presentation of exogenous soluble antigen by bone marrow-derived dendritic cells. Eur J Immunol 27:280–288. doi: 10.1002/eji.1830270141. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.