

ABSTRACT
Mycobacterium tuberculosis primarily infects lung macrophages. However, a recent study showed that M. tuberculosis also infects and persists in a dormant form inside bone marrow mesenchymal stem cells (BM-MSCs) even after successful antibiotic therapy. However, the mechanism(s) by which M. tuberculosis survives in BM-MSCs is still not known. Like macrophages, BM-MSCs do not contain a well-defined endocytic pathway, which is known to play a central role in the clearance of internalized mycobacteria. Here, we studied the fate of virulent and avirulent mycobacteria in Sca-1+ CD44+ BM-MSCs. We found that BM-MSCs were able to kill avirulent Mycobacterium smegmatis and Mycobacterium bovis BCG but not the pathogenic species M. tuberculosis. Further mechanistic studies revealed that pathogenic M. tuberculosis dampens the antibacterial response of BM-MSCs by downregulating the expression of the cationic antimicrobial peptide cathelicidin. In contrast, avirulent mycobacteria were effectively killed by inducing the Toll-like receptor 2/4 (TLR2/4) pathway-dependent expression of cathelicidin, while small interfering RNA (siRNA)-mediated cathelicidin silencing increased the survival of M. bovis BCG in BM-MSCs. We also showed that M. bovis BCG infection caused increased expression levels of MyD88, phospho-interleukin-1 receptor-associated kinase 4 (pIRAK-4), and the p38 mitogen-activated protein kinase (MAPK) signaling pathway. Further downstream investigations demonstrated that IRAK-4–p38 activation increased the nuclear translocation of NF-κB, which subsequently induced the expression of cathelicidin and the cytokine interleukin-1β (IL-1β), resulting in the decreased survival of M. bovis BCG. On the other hand, inhibition of TLR2/4, pIRAK-4, p38, and NF-κB nuclear translocation decreased cathelicidin and IL-1β expression levels and therefore increased the survival of avirulent mycobacteria. This is the first report that demonstrates that virulent mycobacteria manipulate the TLR2/4–MyD88–IRAK-4–p38–NF-κB–Camp–IL-1β pathway to survive inside bone marrow stem cells.
KEYWORDS: mesenchymal stem cells, mouse bone marrow, mycobacteria, Toll-like receptors, p38, cathelicidin, Mycobacterium bovis BCG, Mycobacterium tuberculosis, MAPK
INTRODUCTION
Tuberculosis (TB), caused by Mycobacterium tuberculosis, is one of the world's major health problems. Through thousands of years of reciprocal coevolution, M. tuberculosis has acquired the abilities to establish active and latent infections and persist even in the presence of a fully functional immune system. Several studies have shown that innate immune responses play a critical role in the progression of M. tuberculosis infection. Macrophages, a vital part of the first host defense, are involved in eliciting innate immunity by performing various important antimycobacterial effector functions (1). However, over a period of time M. tuberculosis has acquired various immune evasion strategies that interfere with both innate and adaptive immunity. These strategies include blocking of phagolysosome fusion (2), autophagy inhibition (3), modulation of host cytokine production (4), inhibition of reactive oxygen and nitrogen species (5), and manipulation of antigen presentation to prevent or alter the quality of T-cell responses (6).
In addition to macrophages, dendritic cells and other cell types such as epithelial cells, fibrocytes, adipocytes, and endothelial cells, distributed in pulmonary and extrapulmonary niches, have also been described as possible hosts for M. tuberculosis. Recently, M. tuberculosis has also been shown to persist in a mesenchymal subpopulation of bone marrow (BM) stem cells (BMSCs) even after antibiotic treatment (7). The authors have shown that mesenchymal BMSCs may provide a favorable intracellular niche for the persistence of nonreplicating M. tuberculosis and therefore may be important for the maintenance of the dormant phase of the M. tuberculosis life cycle. Another study showed that mesenchymal stem cells (MSCs) are recruited at the site of infection and suppress T-cell responses by producing nitric oxide (8). MSCs are susceptible to infection by several other intracellular pathogens as well (9). Recent studies have shown that MSCs possess antimicrobial properties against Gram-positive and Gram-negative bacterial infections and also improve the survival of mice with bacterial infections (10).
Among the key effector molecules responsible for bacterial killing are antimicrobial proteins and polypeptides such as defensins, lactoferrin, and lysozyme (11). It is well established that MSCs express various soluble molecules (12) and Toll-like receptors (TLRs) (13) that are involved in inflammatory responses (14). The cathelicidin family is one of the main antimicrobial peptide (AMP) families in mammals (15). In humans, cathelicidin, encoded by CAMP, is synthesized as a propeptide known as human cationic antimicrobial peptide 18 (hCAP-18), which is then cleaved extracellularly into cathelin and C-terminal LL-37, with the latter exhibiting broad antimicrobial activity (16). hCAP-18/LL-37 is produced mainly by phagocytic leukocytes and epithelial cells, but it is also expressed in bone marrow (17, 18). Likewise, there is only one mouse gene encoding cathelicidin, Camp, which is highly homologous to the human gene. The mouse cathelicidin proform is processed to the mature biologically active peptide CRAMP (cathelicidin-related antimicrobial peptide) (19). LL-37 and CRAMP are amphipathic α-helical peptides that bind to negatively charged groups on the bacterial outer membrane, causing a disruption of the bacterial cell wall (20). Previously, it was reported that BM-MSCs exert antibacterial activity via the production of cathelicidin (18).
Although M. tuberculosis has been shown to persist in BM-MSCs (7), the mechanism by which it survives inside BM-MSCs is still not known. Here, we show that virulent M. tuberculosis strain H37Rv is able to survive inside BM-MSCs by downregulating the expression of cathelicidin, while avirulent mycobacteria such as Mycobacterium bovis BCG and Mycobacterium smegmatis were readily killed by BM-MSCs through the induction of cathelicidin. We furthermore show that the induction of cathelicidin was regulated via the TLR2/4-mediated induction of MyD88, phospho-interleukin-1 (IL-1) receptor-associated kinase 4 (pIRAK-4), and the p38 mitogen-activated protein kinase (MAPK) pathway. Further downstream signaling studies revealed that pIRAK-4 activation increased the expression levels of phospho-nuclear factor κB (NF-κB) and IL-1β. In summary, this is the first report that demonstrates the strategies employed by M. tuberculosis to survive in BM-MSCs and also how BM-MSCs are able to kill invading avirulent mycobacteria.
RESULTS
Characterization of cell surface markers of isolated mouse bone marrow mesenchymal stem cells.
Bone marrow cells were harvested from BALB/c mice (n = 10) and plated onto a 100-mm culture dish. Nonadherent cells were carefully removed after 6 h and replaced with fresh medium. Thereafter, this step was repeated every 24 h for up to 72 h of initial culture. The adherent cells were then washed with phosphate-buffered saline (PBS), and fresh medium was added every 2 to 3 days. On the fourth day, the initial adherent spindle-shaped cells appeared as individual cells. After 2 weeks, the culture became more than 80% confluent. Isolated BM-MSCs were characterized by using several positive (Sca-1 and CD44) and negative (CD31, c-Kit, CD45, and CD11b) markers (21). Bone marrow cells contain endothelial, myeloid, and hematopoietic cell lineages that express c-Kit, CD31, CD45, and CD11b, but BM-MSCs are negative for these markers. To exclude contaminating cell populations, we sorted the isolated BM-MSCs using positive (Sca-1 and CD44) and negative (CD31, c-Kit, and CD11b) markers. Flow cytometry analysis showed that BM-MSCs were negative for the CD31, CD45, c-Kit, and CD11b markers and were positively stained with antibodies against Sca-1 and CD44 (Fig. 1A). As shown in Fig. 1B, cell sorting using different positive and negative markers resulted in a cell population that was about 95% pure.
FIG 1.

Isolation of mouse BM-MSCs and mycobacterial infection in BM-MSCs. (A) Characterization of BM-MSCs using Sca-1, CD44, CD31, CD45, CD11b, and c-Kit markers by flow cytometry. Cells were stained with antibodies against the respective markers, and shifting of cells was analyzed by a histogram plot. (B) After positive and negative population analyses, BM-MSCs were sorted according to the CD44+ Sca1+ CD31− c-Kit− population. (C) BM-MSCs (red) were infected with GFP-expressing M. bovis BCG (green), and infected cells were observed under a fluorescence microscope. (D) M. bovis BCG bacterial counts per BM-MSC cell. Bacterial counts were determined by counting 100 cells. (E) BM-MSCs were infected with M. tuberculosis H37Rv (Mtb) for 3 h. Cells were washed, extracellular bacteria were killed by gentamicin treatment (20 μg/ml), cells were lysed, and the infection rate was determined by a CFU assay. The data shown are from three independent experiments.
Pathogenic and nonpathogenic mycobacteria are able to infect BM-MSCs.
To determine whether mycobacteria can infect BM-MSCs, the isolated BM-MSCs were sorted using Sca-1+ CD44+ and CD31− c-Kit− CD11b− antibodies and then infected with green fluorescent protein (GFP)-expressing M. bovis BCG and M. tuberculosis strains at a multiplicity of infection (MOI) of 1:5 for 2 to 3 h. The mycobacterium-exposed cells were washed and then treated with gentamicin to kill extracellular bacteria. Due to biosafety challenges associated with the biosafety level 3 (BSL3) category of M. tuberculosis for fluorescence microscopy, we measured the infection rate of M. tuberculosis by a CFU assay only. Fluorescence microscopic analysis and CFU enumeration assays showed that M. bovis BCG (Fig. 1C) and M. tuberculosis (Fig. 1E) were able to infect BM-MSCs. We counted 100 infected cells to determine the intracellular bacterial burden. Approximately 8 to 15 M. bovis BCG bacilli were present per cell (Fig. 1D), while M. tuberculosis infected BM-MSCs at an infection rate of 25 to 30% (Fig. 1E). Similar results were obtained for M. smegmatis infection (data not shown). In conclusion, the above-described results indicate that both virulent as well as avirulent mycobacteria were able to infect BM-MSCs.
BM-MSCs restrict intracellular growth of M. bovis BCG and M. smegmatis but not M. tuberculosis.
Previously, it was shown that M. tuberculosis can stay inside MSCs for longer periods of time after antibiotic treatment and can cause active TB at a later time point (7). In the next step, we studied the intracellular survival kinetics of M. tuberculosis, M. bovis BCG, and M. smegmatis in BM-MSCs. Interestingly, we observed a time-dependent decrease in the intracellular survival of avirulent M. bovis BCG and M. smegmatis strains such that approximately 2- and 6-fold decreases in the intracellular survival of M. bovis BCG (P < 0.001) (Fig. 2A) and M. smegmatis (P < 0.0001) (Fig. 2B), respectively, were observed after 24 h of infection. On the other hand, no such killing of the virulent M. tuberculosis strain was observed even after 48 h of infection (P < 0.05) (Fig. 2C). These results suggest that BM-MSCs are able to restrict the growth of avirulent strains, while virulent M. tuberculosis can survive inside the cells.
FIG 2.

Intracellular survival of pathogenic and nonpathogenic mycobacteria in BM-MSCs. (A) Intracellular survival of M. bovis BCG in BM-MSCs was determined by infecting cells for 3 h. Cells were lysed at different time points, and bacterial survival was determined by a CFU assay. (B) Intracellular survival of M. smegmatis in BM-MSCs was determined by infecting cells for 2 h. Cells were lysed at different time points, and bacterial survival was determined by a CFU assay. (C) Intracellular survival of M. tuberculosis H37Rv in BM-MSCs was determined by infecting cells for 3 h. Cells were lysed at different time points, and bacterial survival was determined by a CFU assay. The results shown are from three independent experiments (means ± SD). *, P < 0.05; ***, P < 0.0001.
M. tuberculosis survives inside the BM-MSCs by downregulating the expression of the cationic antimicrobial peptide CRAMP.
Next, we studied the mechanism of survival of M. tuberculosis in BM-MSCs. Previously, we showed that mouse macrophages are able to kill M. smegmatis by inducing the expression of Camp (22). We found that Camp−/− mouse macrophages were significantly impaired in their ability to kill mycobacteria. BM-MSCs are also known to express Camp (18). Therefore, we investigated whether BM-MSC Camp has any role in the determination of the intracellular survival of virulent and avirulent mycobacteria. First, we investigated the expression of Camp in both M. tuberculosis- and M. bovis BCG-infected BM-MSCs at both the transcriptional and translational levels. Interestingly, we observed a significant decrease in the expression level of cathelicidin at both the translational (Fig. 3A) and transcriptional (Fig. 3B) levels (P < 0.0001) in response to M. tuberculosis infection, while the expression of Camp was found to be upregulated in M. bovis BCG-infected cells after 24 h (P < 0.0001) (Fig. 3C). This corresponds well with the intracellular survival kinetics of M. bovis BCG (Fig. 2A) and M. tuberculosis (Fig. 2C). To prove the fact that M. tuberculosis indeed survives inside BM-MSCs by downregulating the expression of Camp, we exogenously added purified CRAMP (50 μM) and then determined the intracellular survival of M. tuberculosis in BM-MSCs. Previously, we showed that macrophages actively internalize human cathelicidin LL-37 and also determined that 50 μM LL-37 is the 50% inhibitory concentration (IC50) for M. tuberculosis (22). We observed significant killing of M. tuberculosis in CRAMP-treated BM-MSCs (P < 0.0001) (Fig. 3D). On the other hand, silencing of Camp by small interfering RNA (siRNA) (Fig. 3E) significantly increased the bacillary burden of M. bovis BCG compared to that in untreated and scrambled siRNA-treated cells (P < 0.0001) (Fig. 3F). Altogether, the above-described data clearly indicate that M. tuberculosis survives by downregulating the expression of cathelicidin and that avirulent mycobacteria are killed by the induction of cathelicidin expression in BM-MSCs.
FIG 3.

Role of cathelicidin in intracellular survival of mycobacteria in BM-MSCs. (A) BM-MSCs were infected with M. tuberculosis H37Rv at an MOI of 1:5 for 24 h. After 24 h, cell lysates were prepared, and the expression of CRAMP was determined by Western blotting using a cathelicidin-specific antibody. (B) BM-MSCs were infected with M. tuberculosis H37Rv. Total RNA was isolated, cDNA was prepared, and the level of the Camp transcript was determined by qRT-PCR at 6 and 24 h postinfection. Uninfected cells were used as a control. (C) BM-MSCs were infected with M. bovis BCG. Total RNA was isolated, cDNA was prepared, and Camp expression was studied by qRT-PCR at 6 and 24 h postinfection. Uninfected cells were used as a control. (D) BM-MSCs were treated with 50 μM purified cathelicidin-related antimicrobial peptide (CRAMP), and cells were infected with M. tuberculosis H37Rv. Cells were lysed at the indicated time points, and intracellular bacterial survival was determined by a CFU assay. Untreated cells were used as a control. (E) Silencing of Camp by siRNA treatment. Cells were transfected with scrambled siRNA and siRNA against Camp (SiCamp). (F) Intracellular survival of M. bovis BCG in scrambled siRNA- and SiCamp-treated BM-MSCs. The results shown are from three independent experiments (means ± SD). ***, P < 0.0001.
M. tuberculosis and M. bovis BCG infections modulate TLR and cytokine expression in BM-MSCs.
Previous studies have shown that bacterial challenge increases the expression levels of TLRs, which subsequently increase the production of AMPs to reduce the risk of microbial infections (23). It has been shown that cathelicidin expression is regulated mainly via TLR2 and TLR4 signaling cascades (24, 25). Therefore, we first determined the expression levels of TLR2 and TLR4 in M. tuberculosis- and M. bovis BCG-infected BM-MSCs. M. tuberculosis challenge significantly increased TLR2 expression levels at both 6 and 24 h postinfection (P < 0.0001) (Fig. 4A); however, in the case of M. bovis BCG infection, a significant increase in the transcript level of TLR2 was observed after 24 h of infection compared to that in uninfected cells (P < 0.001) (Fig. 4B). In the case of TLR4, a significant increase in the TLR4 transcript level was observed only after 6 h, followed by a moderate downregulation at later time points in M. tuberculosis-infected cells (P < 0.0001) (Fig. 4C). On the other hand, M. bovis BCG infection showed higher expression levels of TLR4 at both 6 and 24 h of infection (P < 0.0001) (Fig. 4D), indicating that virulent and avirulent mycobacteria modulate TLR expression in BM-MSCs.
FIG 4.

Determination of TLR expression in M. tuberculosis- and M. bovis BCG-infected BM-MSCs. BM-MSCs (2 × 105) were seeded onto a 24-well tissue culture plate and infected with M. tuberculosis and M. bovis BCG. After 6 and 24 h of infection, cells were harvested, and cDNA was prepared to check the expression of TLRs by qRT-PCR analysis. Shown are data from qRT-PCR analyses of the expression of TLR2 in M. tuberculosis-infected (A) and M. bovis BCG-infected (B) BM-MSCs and of TLR4 in M. tuberculosis-infected (C) and M. bovis BCG-infected (D) BM-MSCs. The results shown are from three independent experiments (means ± SD). **, P < 0.001; ***, P < 0.0001; ns, not significant.
Next, we compared the expression levels of several proinflammatory (tumor necrosis factor alpha [TNF-α] and IL-1β) and anti-inflammatory (IL-10 and transforming growth factor β [TGF-β]) cytokines in M. tuberculosis- and M. bovis BCG-infected BM-MSCs using quantitative real-time reverse transcription-PCR (qRT-PCR) and a Bioplex cytokine analysis kit. We observed significant downregulation of IL-1β at both the protein (P < 0.0001) (Fig. 5A) and RNA (P < 0.05) (Fig. 5B) levels at 6 h, whereas no significant difference was seen at 24 h for M. tuberculosis-infected and uninfected cells. TNF-α expression was found to be significantly downregulated at 6 h (P < 0.0001) (Fig. 5C), while at the transcriptional level, such downregulation was observed only after 24 h of M. tuberculosis infection (P < 0.05) (Fig. 5D). In the case of anti-inflammatory cytokines, we observed a moderate decrease in the level of TGF-β only at 6 h (P < 0.05) (Fig. 5E), whereas IL-10 was highly expressed at the protein level after 6 h (P < 0.0001) (Fig. 5F) and at the transcriptional level after 24 h (P < 0.0001) (Fig. 5G) in M. tuberculosis-exposed cells. The overall cytokine analysis indicated the downregulation of proinflammatory cytokines and the upregulation of anti-inflammatory cytokines after M. tuberculosis infection.
FIG 5.

Determination of cytokine expression levels in M. tuberculosis- and M. bovis BCG-infected BM-MSCs. BM-MSCs (2 × 105) were seeded onto a 24-well tissue culture plate and infected with M. tuberculosis and M. bovis BCG. After 6 and 24 h of infection, the supernatant was harvested for cytokine analysis by using a Bioplex kit assay, and cells were harvested for the preparation of cDNA to check cytokine transcript levels by qRT-PCR analysis. (A and B) IL-1β cytokine levels in M. tuberculosis-infected BM-MSCs. (C and D) TNF-α cytokine levels in M. tuberculosis-infected BM-MSCs. (E) TGF-β cytokine levels in M. tuberculosis-infected BM-MSCs. (F and G) IL-10 cytokine levels in M. tuberculosis-infected BM-MSCs. (H to K) mRNA transcript levels of IL-1β (H), TNF-α (I), TGF-β (J), and IL-10 (K) in M. bovis BCG-infected BM-MSCs. The results shown are from three independent experiments (means ± SD). *, P < 0.05; **, P < 0.001; ***, P < 0.0001.
In the case of M. bovis BCG infection, we observed a substantial increase in the transcript levels of IL-1β after 24 h of infection, whereas no difference in the expression level was observed at 6 h postinfection (P < 0.0001) (Fig. 5H). No significant differences in TNF-α expression levels were observed in infected and uninfected cells at the 6- and 24-h time points (Fig. 5I). Similarly, the expression levels of both TGF-β (P < 0.0001) (Fig. 5J) and IL-10 (P < 0.0001) (Fig. 5K) were found to be upregulated after 6 h of infection, whereas no differences were observed after 24 h of infection compared with the levels in uninfected cells. The above-described results indicated that M. tuberculosis and M. bovis BCG infections modulate the expression of pro- and anti-inflammatory cytokines at different stages of infection.
Inhibition of TLR2 and TLR4 downregulates Camp expression.
Based on the above-described results, our subsequent studies were focused on the investigation of the molecular mechanism(s) that is responsible for the induction of Camp expression that leads to the killing of intracellular bacilli. For this reason, all subsequent experiments were performed with M. bovis BCG as a model organism. As described above, we found the upregulation of both TLR2 and TLR4 in both M. tuberculosis- and M. bovis BCG-challenged BM-MSCs. Previously, several studies demonstrated the role of TLR2 and TLR4 in the regulation of Camp expression (26–28). In the next step, we attempted to identify the role of TLRs in the control of Camp expression in BM-MSCs. For this, BM-MSCs were first treated with TLR2 (anti-mouse CD282 at a 1:1,000 dilution) (catalog number 14-9024-80; eBioscience) (29) and TLR4 (TAK242 at 1 μM; Sigma) (30) inhibitors for 4 h, followed by M. bovis BCG infection. Following treatment with TLR2 and TLR4 blockers, the expression level of Camp decreased significantly compared with that in untreated and infected cells (P < 0.001) (Fig. 6A). However, treatment with a combination of TLR2 and TLR4 blockers did not further decrease Camp expression levels compared with treatment with TLR2 and TLR4 inhibitors alone. These results indicate that both TLR2 and TLR4 are involved in the modulation of Camp expression in response to M. bovis BCG infection in BM-MSCs.
FIG 6.

Camp expression in M. bovis BCG-infected BM-MSCs is regulated by the MyD88–IRAK-4–p38 MAPK pathway. (A) Determination of Camp expression in BM-MSCs infected with M. bovis BCG in the presence of TLR2 (anti-mouse CD282) and TLR4 (TAK242; 1 μM) blockers. Cells were pretreated with TLR2 and TLR4 inhibitors, and the Camp expression level was determined by qRT-PCR. Uninfected and untreated cells were used as a control. (B) Western blot analysis of MyD88, pIRAK-4, and IRAK-4 in uninfected and M. bovis BCG-infected BM-MSCs after 6 and 24 h of infection. (C) Determination of Camp expression levels in M. bovis BCG-infected and IRAK-4 inhibitor-treated BM-MSCs after 24 h. Untreated cells were used as a control. (D) Western blot analysis of p38 and pERK (p42/44) in uninfected and M. bovis BCG-infected BM-MSCs after 24 h of infection. (E) Expression of Camp in BM-MSCs infected with M. bovis BCG in the presence of the p38 inhibitor SB203580 (10 μM). (F) Intracellular survival of M. bovis BCG in the presence of a p38 inhibitor (SB203580; 10 μM), a TLR4 blocker (TAK242; 1 μM), a TLR2 blocker (anti-mouse CD282; 1:1,000), and an NF-κB nuclear translocation inhibitor (catalog number sc-3060; 20 μg/ml). (G) Expression of IL-1β in BM-MSCs infected with M. bovis BCG in the presence of the p38 inhibitor SB203580. (H) Expression of IL-1β in Camp-silenced BM-MSCs infected with M. bovis BCG. The results shown are from three independent experiments (means ± SD). **, P < 0.001; ***, P < 0.0001; ns, not significant.
Upregulation of Camp by TLRs is mediated via MyD88 and IL-1 receptor-associated kinase 4.
MyD88 is a universal adaptor protein that plays a critical role in TLR-mediated signal transduction (31). MyD88 knockout mice showed no cellular responses to the TLR2 (32, 33) and TLR4 (34) ligands, indicating that MyD88 is essential for the inflammatory responses mediated by TLR2 and TLR4. To corroborate that the modulation of Camp expression is mediated through TLR induction, we examined the expression of MyD88 in M. bovis BCG-infected BM-MSCs. Western blot analysis showed increased levels of MyD88 at both the 6- and 24-h time points in infected cells compared to uninfected cells (Fig. 6B).
The MyD88 adaptor protein links TLRs to IRAK-4. Binding of pathogen-associated molecular patterns (PAMPs) to TLRs triggers the recruitment of MyD88 to the cytoplasmic TIR domain of the TLR, which initiates the association and phosphorylation of IRAK-4 (35). Hence, we determined the expression levels of total IRAK-4 and phosphorylated IRAK-4 (pIRAK-4) in BM-MSCs following M. bovis BCG infection. Although the levels of total IRAK-4 remained unchanged in infected and uninfected BM-MSCs (Fig. 6B), the expression level of pIRAK-4 was found to be significantly increased in M. bovis BCG-infected cells at both 6 and 24 h compared to the levels in uninfected cells (Fig. 6B). To further confirm the involvement of IRAK-4, we determined the Camp expression levels in BM-MSCs treated with an IRAK-4 inhibitor (300 μM I5409; Sigma) (36). We observed a significant downregulation of Camp expression levels in IRAK-4 inhibitor-treated cells compared to those in untreated cells (P < 0.001) (Fig. 6C), indicating that Camp expression is regulated via IRAK-4.
Camp expression is regulated by the p38 MAPK pathway.
The MyD88-TLR interaction is known to recruit members of the IRAK family and initiates signaling through MAPK pathways (37). Moreover, previous studies showed that MAPK pathways are involved in the modulation of CRAMP expression (38). In order to identify the MAPK pathway that could be involved in the regulation of Camp expression, we checked the expression of MAPK proteins in M. bovis BCG-infected BM-MSCs. The MAPK family of protein kinases includes extracellular signal-regulated kinase (ERK) and the stress-activated p38 MAPK. We evaluated the levels of pERK (p42/p44) and p38 MAPK in response to M. bovis BCG infection. We observed the upregulation of p38 in infected cells compared to uninfected ones (Fig. 6D) and that the level of pERK was downregulated in response to M. bovis BCG infection compared to that in uninfected BM-MSCs (Fig. 6D). To prove that p38 upregulation is necessary for the induction of Camp expression, we examined Camp expression in the presence and absence of a p38 inhibitor (SB203580) in M. bovis BCG-challenged BM-MSCs. Following infection, the level of Camp expression was increased compared to that in uninfected cells, but this effect was aborted in the presence of the p38 inhibitor (P < 0.0001) (Fig. 6E). We also studied intracellular bacterial survival in TLR2, TLR4, p38, and NF-κB inhibitor-treated cells. Intriguingly, we found that only treatment with the p38 inhibitor increased the intracellular survival of the bacteria (P < 0.0001), whereas we did not observe any difference in bacterial burdens between other inhibitor-treated and untreated cells (Fig. 6F). We also found a significant downregulation of the transcript levels of IL-1β, which was previously shown to be highly expressed in M. bovis BCG-infected cells (Fig. 5H), in the presence of the p38 inhibitor compared to those in untreated cells (P < 0.0001) (Fig. 6G). A previous study showed that cathelicidin regulates the expression of IL-1β (39). Hence, we checked whether Camp silencing has any effect on the expression of IL-1β in infected BM-MSCs. We found that Camp silencing abrogated the expression of IL-1β in M. bovis BCG-infected cells compared to that in nonsilenced cells (P < 0.0001) (Fig. 6H). Altogether, these data suggest that the M. bovis BCG-induced upregulation of p38 MAPK is responsible for the observed increased expression levels of Camp and the IL-1β cytokine.
M. bovis BCG infection induces NF-κB activity in BM-MSCs.
In resting cells, NF-κB is sequestered in the cytoplasm by its inhibitor, IκBα, whereas stimulation with proinflammatory cytokines or certain bacterial infections causes the phosphorylation of IκBα, which is thereby targeted for proteasomal degradation (40). With the degradation of IκB, phosphorylated NF-κB–p65 is then freed to enter the nucleus, where it can activate the expression of target genes. Next, we examined the localization of activated NF-κB in M. bovis BCG-infected BM-MSCs using fluorescence microscopy. As shown in Fig. 7A, NF-κB was found to be readily translocated to the nucleus of M. bovis BCG-infected cells, whereas uninfected cells showed no nuclear localization of NF-κB. Western blot analysis also showed a significant increase in the level of nuclear NF-κB–p65 after 24 h of infection compared to that in uninfected BM-MSCs (Fig. 7B). To further prove that NF-κB translocation is important for Camp expression, we inhibited the nuclear translocation of NF-κB by treating BM-MSCs with an inhibitor (catalog number sc-3060; Santa Cruz Biotechnology Inc.). Interestingly, we found that treatment with an inhibitor downregulated the expression level of the Camp transcript (P < 0.0001) (Fig. 7C). These results suggest that M. bovis BCG infection induces the translocation of NF-κB to the cellular nucleus, where it controls Camp expression.
FIG 7.

Determination of NF-κB expression in M. bovis BCG-infected BM-MSCs. (A) Nuclear localization of NF-κB–p65 in uninfected and M. bovis BCG-infected BM-MSCs determined by fluorescence microscopy. Blue, nucleus; green, M. bovis GFP; red, NF-κB. (B) Western blot analysis of NF-κB–p65 in uninfected and M. bovis BCG-infected BM-MSCs after 6 and 24 h of infection. (C) mRNA expression of Camp in the presence of an NF-κB nuclear translocation inhibitor (catalog number sc-3060). The results shown are from three independent experiments (means ± SD). ***, P < 0.0001.
DISCUSSION
In recent years, accumulating evidence has shown that bone marrow acts as a reservoir for various immune cells, including MSCs, neutrophils, dendritic cells, B cells, T cells, and natural killer cells. In BM, MSCs play a central role in the regulation of various immunomodulatory activities that can either suppress or stimulate the functions of immune cells (41). For decades, it was presumed that macrophages are the primary host cells for M. tuberculosis, where it survives for extended period of time by manipulating the immune effector functions of macrophages (42). Recently, it was shown that M. tuberculosis also infects and persists in human BM-MSCs (7); however, the mechanisms responsible for the persistence of M. tuberculosis in BM are still not known. Depending upon the surrounding microenvironment, MSCs can either support or inhibit the growth of microorganisms (43, 44). For example, hepatitis B virus (45) and Staphylococcus aureus can infect and replicate in BM-MSCs (46), whereas the growth of Escherichia coli was found to be inhibited in MSCs (18). These results indicate that some pathogens are able to develop a strategy to survive inside MSCs. The current data show that both virulent as well as avirulent mycobacteria are able to infect mouse BM-MSCs. Interestingly, we found that nonpathogenic M. bovis BCG and M. smegmatis, but not M. tuberculosis, are killed by BM-MSCs and that the cationic antimicrobial peptide cathelicidin is necessary for TLR2/4–IRAK-4-dependent antimicrobial responses against mycobacteria.
We found that M. bovis BCG infection can induce the expression of Camp, which resulted in the killing of intracellular bacilli, while silencing of Camp increased bacterial survival. On the other hand, the virulent M. tuberculosis H37Rv strain was able to survive inside cells by downregulating the expression of Camp. Human MSCs produce an antimicrobial peptide, cathelicidin (hCAP-18/LL-37), which is known to inhibit bacterial growth (18). Previously, we showed that Camp−/− mouse macrophages were significantly impaired in their ability to kill nonpathogenic M. smegmatis (22). Previous reports also demonstrated that certain bacterial infections cause the downregulation of cathelicidin to aid in bacterial survival (47, 48). Altogether, these reports demonstrate the relationship between cathelicidin expression and the antibacterial activity of immune cells. Here, we establish that cathelicidin is one of the important mediators of intracellular mycobacterial killing in BM-MSCs.
The interactions between various mycobacterial ligands and cognate TLRs are the major determinants of the outcome of innate immune responses of macrophages. Previously, TLR2 and -4, the most prominent TLRs implicated in TB pathogenesis, were shown to interact with mycobacterial lipoproteins (49, 50) and proline-glutamic acid (PE) proteins (51). These interactions led to TLR activation, which subsequently induced the expression of cathelicidin in macrophages (52). The present study expands on these findings and demonstrates that M. tuberculosis and M. bovis BCG induce the expression of TLR2 and TLR4 during the infection process in BM-MSCs. Preincubation of BM-MSCs with TLR2 and TLR4 blockers markedly reduced Camp expression. These findings provide a direct link between M. tuberculosis- and M. bovis BCG-mediated TLR induction and cathelicidin expression. However, the specific mycobacterial components responsible for the induction of these two TLRs in BM-MSCs remain to be investigated. Previously, it was shown that the M. tuberculosis lipoprotein LpqH is involved in TLR2/1-mediated cathelicidin induction (26). These data suggest that several distinct mechanisms could contribute to TLR-induced cathelicidin expression.
Interactions between microbial ligands and TLRs lead to the induction of various inflammatory cytokines, such as TNF-α, IL-1β, IL-10, and TGF-β (53), which play a central role in the regulation of immune responses against bacteria. We found significantly higher expression levels of the proinflammatory cytokine IL-1β in M. bovis BCG-challenged BM-MSCs; however, the expression of IL-1β was found to be downregulated in M. tuberculosis-infected BM-MSCs. Moreover, M. tuberculosis infection significantly induced the expression of the anti-inflammatory cytokine IL-10, which is known to dampen the antibacterial effector function of immune cells (54). Previous reports showed that the induction of monocyte-derived macrophages with 1,25-dihydroxyvitamin, a cathelicidin inducer, increased the expression levels of IL-1β (55) and that M. tuberculosis lipoprotein induced the expression of cathelicidin, which subsequently led to the control of mycobacterial growth (28). Similarly, skin keratinocytes induced the expression of IL-1β via cathelicidin during infection by Gram-negative bacteria (56). These results indicate that the downregulation of IL-1β and the upregulation of IL-10 by M. tuberculosis may play a key role in the persistence of virulent mycobacteria in BM-MSCs. With M. bovis BCG infection, we did not observe any difference in the levels of TNF-α, IL-10, and TGF-β at later infection stages, indicating that the modulation of cathelicidin expression is independent of these cytokines, at least in BM-MSCs.
In general, stimulation of cells with TLR ligands activates MyD88-dependent and -independent signaling pathways. The recruitment of MyD88 to TLRs triggers various signaling cascades via IRAKs, which subsequently initiate signaling through MAPK pathways. Within the IRAK family, IRAK-4 is the only family member indispensable for the induction of both inflammatory and antimicrobial responses. IRAK-4 undergoes autophosphorylation and activates downstream protein kinase cascades. We found upregulation of both MyD88 and phospho-IRAK-4 in M. bovis BCG-infected cells, while the inhibition of IRAK-4 abrogated M. bovis BCG-induced cathelicidin expression, indicating that the expression of cathelicidin is mediated through the MyD88–IRAK-4 signaling pathway, which subsequently controls bacterial growth. Previously reported findings also showed that mice deficient in MyD88 and IRAK-4 are highly susceptible to Staphylococcus aureus infection and succumb to infection, demonstrating the in vivo significance of MyD88 and IRAK-4 in pathogen-induced immune responses (32, 57). MAPK pathways are involved in the modulation of CRAMP expression (38). The level of p38 MAPK was increased in M. bovis BCG-infected BM-MSCs. Pretreatment with a p38 inhibitor subdued the expression of Camp and IL-1β, suggesting that the M. bovis BCG-induced upregulation of p38 MAPK is responsible for the increased expression levels of Camp and IL-1β. Moreover, treatment with a p38 inhibitor also increased the intracellular survival of M. bovis BCG, suggesting a critical role for p38 in the regulation of cathelicidin expression and the control of bacterial growth in BM-MSCs. It has been shown that the phosphorylation of p38 activates NF-κB and its subsequent translocation to the nuclear compartment of cells (58, 59). We found increased levels and nuclear localization of phospho-NF-κB in M. bovis BCG-infected BM-MSCs compared to those in uninfected cells. Pathak et al. showed that M. tuberculosis early secretory antigen target 6 (ESAT-6) inhibited TLR-mediated signaling and activation of NF-κB (60). That same study showed that the binding of ESAT-6 to TLR2 prevented the interaction between MyD88 and IRAK-4, thus abrogating NF-κB. Region of deletion 1 (RD1) is present in virulent M. tuberculosis strains but is deleted in M. bovis BCG. This region encompasses at least nine genes, including ESAT-6 and culture filtrate protein (CFP-10), that are involved in M. tuberculosis pathogenesis (61). Therefore, the observed activation of the MyD88–IRAK-4 signaling pathway and NF-κB could be attributed to the absence of ESAT-6 in M. bovis BCG. Moreover, activated NF-κB was also shown to activate Camp expression (62). In agreement with those results, we also observed decreased Camp expression levels after the inhibition of nuclear translocation of NF-κB in M. bovis BCG-infected cells. In summary, the present data are unique in that we demonstrate a distinct strategy that could be employed by pathogenic mycobacteria to persist inside bone marrow. Our proposed model for the mechanism by which the TLR2/4–MyD88–IRAK-4–p38–NF-κB–IL-1β pathway mediates a stimulatory effect on cathelicidin induction and mycobacterial killing is summarized in Fig. 8. However, further investigation is required to specifically determine the roles for TLR2 and TLR4 in cathelicidin regulation in bone marrow cells. These results also warrant further investigation of the induction of cathelicidin in BM as a promising alternative strategy for antimycobacterial therapy in BM.
FIG 8.
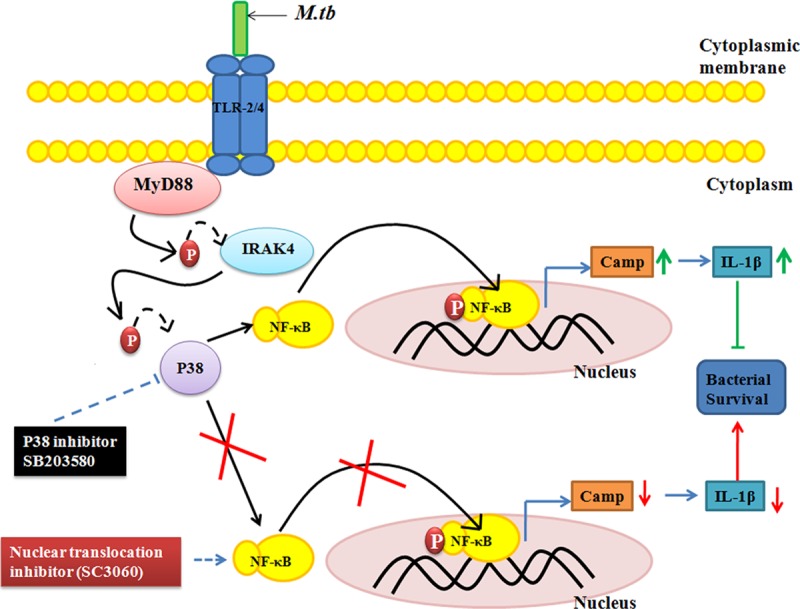
Schematic representation of the modulation of cathelicidin expression during mycobacterial infection in BM-MSCs. Mycobacterial infection activates TLR2/4 expression, which causes the activation of the downstream molecule MyD88 and the phosphorylation of IRAK-4. pIRAK-4 causes the phosphorylation of p38, which activates NF-κB. The translocation of NF-κB into the nucleus stimulates the expression of the cathelicidin antimicrobial peptide and the proinflammatory cytokine IL-1β, which subsequently leads to decreased bacterial survival inside BM-MSCs.
MATERIALS AND METHODS
Ethics statement.
All the animal experiments described in the present study were performed in strict accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals of the Committee for the Purpose of Control and Supervision on Experiments on Animals (CPCSEA) in India (63). All animal studies were conducted with adherence to experimental guidelines and procedures approved by the Institutional Animal Care and Use Committee (KSBT/IAEC/2014-15/MEET-1/A4) of the School of Biotechnology, KIIT University. All efforts were made to minimize suffering and ensure the highest ethical and human standards.
Bacterial strains.
Mycobacterium tuberculosis H37Rv and GFP-labeled M. bovis BCG were grown in Middlebrook 7H9 broth medium (Difco) supplemented with 10% OADC (oleic acid-albumin-dextrose-catalase) and 0.05% Tween 80 (Merck) at 37°C at 120 rpm. In order to stabilize GFP expression, the medium was supplemented with hygromycin (50 mg ml−1). Prior to infection, bacteria were washed with PBS to remove traces of hygromycin. M. smegmatis mc2155 cells were grown in Middlebrook 7H9 broth medium containing 0.05% Tween 80, 0.5% glucose, and 0.5% albumin at 37°C at 120 rpm.
Isolation of bone marrow mesenchymal stem cells from mice.
Six- to eight-week-old BALB/c mice were purchased from the National Centre for Laboratory Animal Sciences (NCLAS), Hyderabad, India. All mice were maintained in HEPA filter-bearing cages under 12-h light cycles in our animal facility and were given sterile chow and autoclaved water ad libitum. Mouse BM-MSCs were isolated according to a previously reported protocol (21). Briefly, animals were sacrificed, and an incision was made around the perimeter of the hind limb. Bones were kept in a sterile petri dish containing 1× PBS supplemented with 4% fetal bovine serum (FBS). BM-MSCs were collected from femoral and tibial bone marrow by inserting a 26-gauge syringe at the bone cavity and flushing it with Dulbecco's modified Eagle's medium (DMEM; Himedia, India) containing 10% heat-inactivated FBS, 2 mM l-glutamine, and 1× penicillin-streptomycin. The resulting cell suspension was filtered through a 70-mm filter mesh to remove any bone spicules or muscles, and the cells were gently resuspended in DMEM as mentioned above. Cells were cultured at 37°C in a 5% CO2 atmosphere on cell culture dishes. After 6 h of incubation, nonadherent cells were removed, medium was replaced with DMEM, and the cells were incubated for another 72 h. After 2 weeks, cells were trypsinized, collected in DMEM without penicillin-streptomycin, and seeded into 24-well plates at a density of 2 × 105 cells per well. The isolated BM-MSCs were characterized by flow cytometry using several positive (Sca-1 and CD44) and negative (CD31, c-Kit, CD45, and CD11b) markers (21).
Sorting of BM-MSCs by flow cytometry.
Isolated cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen) and a mixture of fluorochrome-labeled antibodies. Data were acquired on FACSAria II instrument using FACSDiva software (BD Biosciences), and compensation and data analyses were performed offline by using FlowJo software. The cell sorter was set up according to the manufacturer's guidelines with a 70-μm nozzle, and the cells were sorted at a very low flow rate of 1 to acquire a pure population. An unstained sample was run to set the voltages so that the cell population was properly positioned and exhibited low background fluorescence in all the channels. Forward scatter (FSC) and side scatter (SSC) were used to gate viable and single-cell events. Debris and dead cells were excluded from analysis by categorizing low-FSC events as debris and events with low FSC and high SSC as dead cells. A compact population was gated and further analyzed for the uptake of the impermeable DAPI stain to determine live versus dead cells. The DAPI-negative population was further analyzed for surface markers, i.e., CD44, CD45, Sca-1, CD31, CD11b, and c-Kit. Gates were created to collect the CD44+ Sca1+ CD31− CD11b− c-Kit− population. The sorted cells were transferred to 15-ml tubes and counted, viability was determined by trypan blue exclusion, and equal numbers of cells were seeded onto 6-well plates for infection assays.
BM-MSC infection assay.
M. tuberculosis H37Rv, GFP-labeled M. bovis BCG, and M. smegmatis cultures in the mid-exponential growth phase were pelleted, washed in PBS (pH 7.4), and resuspended in antibiotic-free DMEM to a final optical density at 600 nm (OD600) of 0.1. Bacterial clumps were removed by ultrasonic treatment of bacterial suspensions for 15 min, followed by low-speed centrifugation for 2 min. BM-MSCs were seeded onto 24-well tissue culture plates at a density of 2 × 105 cells per well and incubated for 24 h. The cells were then infected with bacteria at an MOI of 1:10. In each experiment, after 3 h of infection, cells were washed, and DMEM plus gentamicin (20 μg ml−1) were added to kill extracellular bacteria. For M. smegmatis, a similar protocol was followed, except that cells were allowed to take up bacteria for 2 h.
For CFU assays, infected cells from 24-well plates were harvested at different time points, washed with PBS, and lysed with ice-cold 0.1% Triton X-100. Serial dilutions of lysed cells were prepared in PBS and plated onto 7H10 medium supplemented with OADC. Five microliters was plated in triplicate, M. smegmatis colonies were counted after 3 days, and M. bovis BCG and M. tuberculosis colonies were counted after 3 to 4 weeks.
Fluorescence microscopy.
To study the infection rate, BM-MSCs (5 × 104) were infected with GFP-labeled M. bovis BCG (MOI of 1:10) on glass coverslips in 24-well tissue culture plates for 2 to 3 h. The cells were then intensively washed with PBS to remove extracellular bacteria, fixed with a 4% paraformaldehyde solution in PBS for 20 min, and quenched by incubating the cells with glycine. BM-MSCs were stained with rhodamine-phalloidin (1:300) for 20 min. Phagocytosis of mycobacteria was assessed by counting cells containing green mycobacteria by fluorescence microscopy (Olympus BX61). Under each condition, triplicate experiments were performed, and at least 100 cells were counted per slide. In parallel, the cells were lysed, and the intracellular bacterial load was determined by a CFU assay.
Nuclear localization of NF-κB by fluorescence microscopy.
For fluorescence microscopy, 5 × 104 BM-MSCs were seeded onto 24-well tissue culture plates. The cells were then infected with GFP-labeled M. bovis BCG at an MOI of 1:10 as described above. After 24 h of infection, cells were fixed with methanol-acetone (1:1) at −20°C for 20 min and quenched by the addition of 1 mg/ml sodium borohydrate for 5 min. Subsequently, cells were permeabilized with 0.5% saponin in PBS for 15 min, washed, blocked with 5% bovine serum albumin (BSA) for 60 min, and then incubated overnight with primary antibody (NF-κB) (catalog number 8242; Cell Signaling) at 4°C. The cells were then washed and incubated with secondary antibody (goat anti-rabbit IgG) conjugated to Alexa Fluor 594. Cells were mounted with DAPI-containing ProLong Gold Antifade mounting solution (catalog number P-36931; Invitrogen) and analyzed by fluorescence microscopy (Olympus BX61).
Western blot analysis.
All antibodies except the antibody to CRAMP were purchased from Cell Signaling Technology. The expression levels of CRAMP (catalog number sc-66843; Santa Cruz), MyD88 (catalog number 4283), IRAK-4 (catalog number 4363), phospho-IRAK-4 (pIRAK-4) (catalog number 11927), p38 (catalog number 9212), phospho-p38 (catalog number 4511), and NF-κB (catalog number 8242) were checked by infecting BM-MSCs with M. bovis BCG for different times. After infection, cells were harvested, centrifuged at 5,000 rpm for 5 min, and washed three times with PBS. Protein samples were prepared by cell lysis using 1× SDS sample buffer (62.5 mM Tris-HCl [pH 6.8], 2% [wt/vol] SDS, 10% glycerol, 50 mM dithiothreitol [DTT], 0.15 [wt/vol] bromphenol blue) supplemented with a protease inhibitor cocktail and phenylmethylsulfonyl fluoride (PMSF). To study NF-κB expression, a nuclear protein extraction kit (catalog number ab113474; Abcam) was used for nuclear protein extraction. Proteins were electrophoresed in 12% SDS-PAGE gels and transferred to a polyvinylidene difluoride (PVDF) membrane (GE Healthcare, USA) overnight at 28 V. The blots were blocked by using 5% BSA or 5% skim milk in a solution containing 1× Tris-buffered saline and 1% Tween 20 (TBST) for 10 h at 4°C. The membranes were then incubated with primary antibodies (dilution, 1:1,000) overnight at 4°C. The next day, the membranes were washed three times by using 1× TBST for 5 min. The membranes were then incubated with secondary antibodies (dilution, 1:1,000) for 2 h. Membranes were washed by using 1× TBST, and X-ray film was developed by using a standard chemiluminescent solvent. Densitometry analysis was performed to quantify the expression of proteins by using ImageJ software. The protein expression values are assigned with respect to the loading control.
Silencing of Camp by siRNA.
Camp siRNA (catalog number sc-45283) and scrambled siRNAs (catalog number sc-37007) were purchased from Santa Cruz Biotechnology Inc. BM-MSCs were transfected with siRNAs according to the manufacturer's instructions (Santa Cruz Biotechnology Inc.).
Real-time PCR.
Total RNA from infected and/or treated BM-MSCs was isolated by using TRIzol reagent according to the manufacturer's protocol (Invitrogen). cDNA synthesis was performed by using a Verso cDNA synthesis kit (catalog number AB1453A) according to the manufacturer's instructions. The synthesized cDNA was used as a template for qRT-PCR amplification using gene-specific primers (Table 1). Primer efficiency was determined by performing PCR using different dilutions of cDNA of test samples and the control. All reactions were performed with a total reaction mixture volume of 10 μl by using SYBR green PCR master mix (KapaBiosystems, USA) and carried out in an ABI Realplex thermocycler (ABI, Eppendorf, Germany) with an initial denaturation step at 95°C for 10 min, a final denaturation step at 95°C for 30 s, an annealing step at 55.5°C for 30 s, and an extension step at 72°C for 20 s to generate 200-bp amplicons. All qRT-PCRs were performed on three biological replicates, and the data for each sample were expressed relative to the expression level of the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene.
TABLE 1.
Oligonucleotides used in this study

aFP, forward primer; RP, reverse primer.
Cytokine profiling.
BM-MSCs (2 × 105) were seeded onto 24-well tissue culture plate and infected with M. tuberculosis. After 3 h of infection, 20 mg/ml gentamicin-containing DMEM was added to kill the extracellular bacteria. After 6 and 24 h, the supernatant was harvested, and cytokine levels were estimated by using a Bioplex kit assay (Bio-Rad).
Statistical analysis.
Data are presented as means ± standard deviations (SD). Two-way analysis of variance was used to determine statistical significance between groups.
ACKNOWLEDGMENTS
This work was supported by a grant from the Department of Biotechnology, Government of India, to A.S. S.K.N. acknowledges the University Grant Commission, Government of India, for a Rajiv Gandhi national fellowship (RGNF-2013-14-ST-ORI-44090). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
We thank Sonawane laboratory members for fruitful suggestions and discussions. We are thankful to Sunil Raghav and Suchitra Maiti, ILS, Bhubaneswar, India, for helping in cytokine analysis.
We have no conflict of interest.
REFERENCES
- 1.Rosenberger CM, Gallo RL, Finlay BB. 2004. Interplay between antibacterial effectors: a macrophage antimicrobial peptide impairs intracellular Salmonella replication. Proc Natl Acad Sci U S A 101:2422–2427. doi: 10.1073/pnas.0304455101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hart PD, Young MR, Gordon AH, Sullivan KH. 1987. Inhibition of phagosome-lysosome fusion in macrophages by certain mycobacteria can be explained by inhibition of lysosomal movements observed after phagocytosis. J Exp Med 166:933–946. doi: 10.1084/jem.166.4.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. 2004. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 4.Denis M, Gregg EO, Ghandirian E. 1990. Cytokine modulation of Mycobacterium tuberculosis growth in human macrophages. Int J Immunopharmacol 12:721–727. doi: 10.1016/0192-0561(90)90034-K. [DOI] [PubMed] [Google Scholar]
- 5.Voskuil MI, Bartek IL, Visconti K, Schoolnik GK. 2011. The response of Mycobacterium tuberculosis to reactive oxygen and nitrogen species. Front Microbiol 2:105. doi: 10.3389/fmicb.2011.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang ST, Linderman JJ, Kirschner DE. 2005. Multiple mechanisms allow Mycobacterium tuberculosis to continuously inhibit MHC class II-mediated antigen presentation by macrophages. Proc Natl Acad Sci U S A 102:4530–4535. doi: 10.1073/pnas.0500362102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Das B, Kashino SS, Pulu I, Kalita D, Swami V, Yeger H, Felsher DW, Campos-Neto A. 2013. CD271(+) bone marrow mesenchymal stem cells may provide a niche for dormant Mycobacterium tuberculosis. Sci Transl Med 5:170ra13. doi: 10.1126/scitranslmed.3004912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raghuvanshi S, Sharma P, Singh S, Van Kaer L, Das G. 2010. Mycobacterium tuberculosis evades host immunity by recruiting mesenchymal stem cells. Proc Natl Acad Sci U S A 107:21653–21658. doi: 10.1073/pnas.1007967107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fiedler T, Salamon A, Adam S, Herzmann N, Taubenheim J, Peters K. 2013. Impact of bacteria and bacterial components on osteogenic and adipogenic differentiation of adipose-derived mesenchymal stem cells. Exp Cell Res 319:2883–2892. doi: 10.1016/j.yexcr.2013.08.020. [DOI] [PubMed] [Google Scholar]
- 10.Hall SR, Tsoyi K, Ith B, Padera RF Jr, Lederer JA, Wang Z, Liu X, Perrella MA. 2013. Mesenchymal stromal cells improve survival during sepsis in the absence of heme oxygenase-1: the importance of neutrophils. Stem Cells 31:397–407. doi: 10.1002/stem.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agerberth B, Grunewald J, Castanos-Velez E, Olsson B, Jornvall H, Wigzell H, Eklund A, Gudmundsson GH. 1999. Antibacterial components in bronchoalveolar lavage fluid from healthy individuals and sarcoidosis patients. Am J Respir Crit Care Med 160:283–290. doi: 10.1164/ajrccm.160.1.9807041. [DOI] [PubMed] [Google Scholar]
- 12.Bassi EJ, de Almeida DC, Moraes-Vieira PM, Camara NO. 2012. Exploring the role of soluble factors associated with immune regulatory properties of mesenchymal stem cells. Stem Cell Rev 8:329–342. doi: 10.1007/s12015-011-9311-1. [DOI] [PubMed] [Google Scholar]
- 13.Tomchuck SL, Zwezdaryk KJ, Coffelt SB, Waterman RS, Danka ES, Scandurro AB. 2008. Toll-like receptors on human mesenchymal stem cells drive their migration and immunomodulating responses. Stem Cells 26:99–107. doi: 10.1634/stemcells.2007-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delarosa O, Dalemans W, Lombardo E. 2012. Toll-like receptors as modulators of mesenchymal stem cells. Front Immunol 3:182. doi: 10.3389/fimmu.2012.00182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bals R, Wilson JM. 2003. Cathelicidins—a family of multifunctional antimicrobial peptides. Cell Mol Life Sci 60:711–720. doi: 10.1007/s00018-003-2186-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sorensen OE, Follin P, Johnsen AH, Calafat J, Tjabringa GS, Hiemstra PS, Borregaard N. 2001. Human cathelicidin, hCAP-18, is processed to the antimicrobial peptide LL-37 by extracellular cleavage with proteinase 3. Blood 97:3951–3959. doi: 10.1182/blood.V97.12.3951. [DOI] [PubMed] [Google Scholar]
- 17.Wu W, Kim CH, Liu R, Kucia M, Marlicz W, Greco N, Ratajczak J, Laughlin MJ, Ratajczak MZ. 2012. The bone marrow-expressed antimicrobial cationic peptide LL-37 enhances the responsiveness of hematopoietic stem progenitor cells to an SDF-1 gradient and accelerates their engraftment after transplantation. Leukemia 26:736–745. doi: 10.1038/leu.2011.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krasnodembskaya A, Song Y, Fang X, Gupta N, Serikov V, Lee JW, Matthay MA. 2010. Antibacterial effect of human mesenchymal stem cells is mediated in part from secretion of the antimicrobial peptide LL-37. Stem Cells 28:2229–2238. doi: 10.1002/stem.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gallo RL, Kim KJ, Bernfield M, Kozak CA, Zanetti M, Merluzzi L, Gennaro R. 1997. Identification of CRAMP, a cathelin-related antimicrobial peptide expressed in the embryonic and adult mouse. J Biol Chem 272:13088–13093. doi: 10.1074/jbc.272.20.13088. [DOI] [PubMed] [Google Scholar]
- 20.Bowdish DM, Davidson DJ, Scott MG, Hancock RE. 2005. Immunomodulatory activities of small host defense peptides. Antimicrob Agents Chemother 49:1727–1732. doi: 10.1128/AAC.49.5.1727-1732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soleimani M, Nadri S. 2009. A protocol for isolation and culture of mesenchymal stem cells from mouse bone marrow. Nat Protoc 4:102–106. doi: 10.1038/nprot.2008.221. [DOI] [PubMed] [Google Scholar]
- 22.Sonawane A, Santos JC, Mishra BB, Jena P, Progida C, Sorensen OE, Gallo R, Appelberg R, Griffiths G. 2011. Cathelicidin is involved in the intracellular killing of mycobacteria in macrophages. Cell Microbiol 13:1601–1617. doi: 10.1111/j.1462-5822.2011.01644.x. [DOI] [PubMed] [Google Scholar]
- 23.Redfern RL, Reins RY, McDermott AM. 2011. Toll-like receptor activation modulates antimicrobial peptide expression by ocular surface cells. Exp Eye Res 92:209–220. doi: 10.1016/j.exer.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoshioka M, Fukuishi N, Kubo Y, Yamanobe H, Ohsaki K, Kawasoe Y, Murata M, Ishizumi A, Nishii Y, Matsui N, Akagi M. 2008. Human cathelicidin CAP18/LL-37 changes mast cell function toward innate immunity. Biol Pharm Bull 31:212–216. doi: 10.1248/bpb.31.212. [DOI] [PubMed] [Google Scholar]
- 25.Yang CS, Shin DM, Kim KH, Lee ZW, Lee CH, Park SG, Bae YS, Jo EK. 2009. NADPH oxidase 2 interaction with TLR2 is required for efficient innate immune responses to mycobacteria via cathelicidin expression. J Immunol 182:3696–3705. doi: 10.4049/jimmunol.0802217. [DOI] [PubMed] [Google Scholar]
- 26.Shin DM, Yuk JM, Lee HM, Lee SH, Son JW, Harding CV, Kim JM, Modlin RL, Jo EK. 2010. Mycobacterial lipoprotein activates autophagy via TLR2/1/CD14 and a functional vitamin D receptor signalling. Cell Microbiol 12:1648–1665. doi: 10.1111/j.1462-5822.2010.01497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wan M, van der Does AM, Tang X, Lindbom L, Agerberth B, Haeggstrom JZ. 2014. Antimicrobial peptide LL-37 promotes bacterial phagocytosis by human macrophages. J Leukoc Biol 95:971–981. doi: 10.1189/jlb.0513304. [DOI] [PubMed] [Google Scholar]
- 28.Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, Ochoa MT, Schauber J, Wu K, Meinken C, Kamen DL, Wagner M, Bals R, Steinmeyer A, Zugel U, Gallo RL, Eisenberg D, Hewison M, Hollis BW, Adams JS, Bloom BR, Modlin RL. 2006. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 311:1770–1773. doi: 10.1126/science.1123933. [DOI] [PubMed] [Google Scholar]
- 29.Flo TH, Halaas O, Lien E, Ryan L, Teti G, Golenbock DT, Sundan A, Espevik T. 2000. Human Toll-like receptor 2 mediates monocyte activation by Listeria monocytogenes, but not by group B streptococci or lipopolysaccharide. J Immunol 164:2064–2069. doi: 10.4049/jimmunol.164.4.2064. [DOI] [PubMed] [Google Scholar]
- 30.Matsunaga N, Tsuchimori N, Matsumoto T, Ii M. 2011. TAK-242 (resatorvid), a small-molecule inhibitor of Toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol Pharmacol 79:34–41. doi: 10.1124/mol.110.068064. [DOI] [PubMed] [Google Scholar]
- 31.Takeda K, Akira S. 2004. TLR signaling pathways. Semin Immunol 16:3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 32.Takeuchi O, Hoshino K, Akira S. 2000. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol 165:5392–5396. doi: 10.4049/jimmunol.165.10.5392. [DOI] [PubMed] [Google Scholar]
- 33.Takeuchi O, Kawai T, Muhlradt PF, Morr M, Radolf JD, Zychlinsky A, Takeda K, Akira S. 2001. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int Immunol 13:933–940. doi: 10.1093/intimm/13.7.933. [DOI] [PubMed] [Google Scholar]
- 34.Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. 1999. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 11:115–122. doi: 10.1016/S1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- 35.Picard C, Casanova JL, Puel A. 2011. Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IkappaBalpha deficiency. Clin Microbiol Rev 24:490–497. doi: 10.1128/CMR.00001-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Powers JP, Li S, Jaen JC, Liu J, Walker NP, Wang Z, Wesche H. 2006. Discovery and initial SAR of inhibitors of interleukin-1 receptor-associated kinase-4. Bioorg Med Chem Lett 16:2842–2845. doi: 10.1016/j.bmcl.2006.03.020. [DOI] [PubMed] [Google Scholar]
- 37.Kim TW, Staschke K, Bulek K, Yao J, Peters K, Oh KH, Vandenburg Y, Xiao H, Qian W, Hamilton T, Min B, Sen G, Gilmour R, Li X. 2007. A critical role for IRAK4 kinase activity in Toll-like receptor-mediated innate immunity. J Exp Med 204:1025–1036. doi: 10.1084/jem.20061825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Babolewska E, Pietrzak A, Brzezinska-Blaszczyk E. 2014. Cathelicidin rCRAMP stimulates rat mast cells to generate cysteinyl leukotrienes, synthesize TNF and migrate: involvement of PLC/A2, PI3K and MAPK signaling pathways. Int Immunol 26:637–646. doi: 10.1093/intimm/dxu069. [DOI] [PubMed] [Google Scholar]
- 39.Elssner A, Duncan M, Gavrilin M, Wewers MD. 2004. A novel P2X7 receptor activator, the human cathelicidin-derived peptide LL37, induces IL-1 beta processing and release. J Immunol 172:4987–4994. doi: 10.4049/jimmunol.172.8.4987. [DOI] [PubMed] [Google Scholar]
- 40.Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. 1995. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science 267:1485–1488. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- 41.Abdi R, Fiorina P, Adra CN, Atkinson M, Sayegh MH. 2008. Immunomodulation by mesenchymal stem cells: a potential therapeutic strategy for type 1 diabetes. Diabetes 57:1759–1767. doi: 10.2337/db08-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Giacomini E, Iona E, Ferroni L, Miettinen M, Fattorini L, Orefici G, Julkunen I, Coccia EM. 2001. Infection of human macrophages and dendritic cells with Mycobacterium tuberculosis induces a differential cytokine gene expression that modulates T cell response. J Immunol 166:7033–7041. doi: 10.4049/jimmunol.166.12.7033. [DOI] [PubMed] [Google Scholar]
- 43.Gupta N, Krasnodembskaya A, Kapetanaki M, Mouded M, Tan X, Serikov V, Matthay MA. 2012. Mesenchymal stem cells enhance survival and bacterial clearance in murine Escherichia coli pneumonia. Thorax 67:533–539. doi: 10.1136/thoraxjnl-2011-201176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hackstein H, Lippitsch A, Krug P, Schevtschenko I, Kranz S, Hecker M, Dietert K, Gruber AD, Bein G, Brendel C, Baal N. 2015. Prospectively defined murine mesenchymal stem cells inhibit Klebsiella pneumoniae-induced acute lung injury and improve pneumonia survival. Respir Res 16:123. doi: 10.1186/s12931-015-0288-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ma R, Xing Q, Shao L, Wang D, Hao Q, Li X, Sai L, Ma L. 2011. Hepatitis B virus infection and replication in human bone marrow mesenchymal stem cells. Virol J 8:486. doi: 10.1186/1743-422X-8-486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guerra AD, Cantu DA, Vecchi JT, Rose WE, Hematti P, Kao WJ. 2015. Mesenchymal stromal/stem cell and minocycline-loaded hydrogels inhibit the growth of Staphylococcus aureus that evades immunomodulation of blood-derived leukocytes. AAPS J 17:620–630. doi: 10.1208/s12248-015-9728-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johansson J, Gudmundsson GH, Rottenberg ME, Berndt KD, Agerberth B. 1998. Conformation-dependent antibacterial activity of the naturally occurring human peptide LL-37. J Biol Chem 273:3718–3724. doi: 10.1074/jbc.273.6.3718. [DOI] [PubMed] [Google Scholar]
- 48.Bergman P, Johansson L, Asp V, Plant L, Gudmundsson GH, Jonsson AB, Agerberth B. 2005. Neisseria gonorrhoeae downregulates expression of the human antimicrobial peptide LL-37. Cell Microbiol 7:1009–1017. doi: 10.1111/j.1462-5822.2005.00530.x. [DOI] [PubMed] [Google Scholar]
- 49.Pecora ND, Gehring AJ, Canaday DH, Boom WH, Harding CV. 2006. Mycobacterium tuberculosis LprA is a lipoprotein agonist of TLR2 that regulates innate immunity and APC function. J Immunol 177:422–429. doi: 10.4049/jimmunol.177.1.422. [DOI] [PubMed] [Google Scholar]
- 50.Blumenthal A, Kobayashi T, Pierini LM, Banaei N, Ernst JD, Miyake K, Ehrt S. 2009. RP105 facilitates macrophage activation by Mycobacterium tuberculosis lipoproteins. Cell Host Microbe 5:35–46. doi: 10.1016/j.chom.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tiwari BM, Kannan N, Vemu L, Raghunand TR. 2012. The Mycobacterium tuberculosis PE proteins Rv0285 and Rv1386 modulate innate immunity and mediate bacillary survival in macrophages. PLoS One 7:e51686. doi: 10.1371/journal.pone.0051686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Campbell GR, Spector SA. 2012. Toll-like receptor 8 ligands activate a vitamin D mediated autophagic response that inhibits human immunodeficiency virus type 1. PLoS Pathog 8:e1003017. doi: 10.1371/journal.ppat.1003017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zheng W, Zheng X, Liu S, Ouyang H, Levitt RC, Candiotti KA, Hao S. 2012. TNFalpha and IL-1beta are mediated by both TLR4 and Nod1 pathways in the cultured HAPI cells stimulated by LPS. Biochem Biophys Res Commun 420:762–767. doi: 10.1016/j.bbrc.2012.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Redford PS, Murray PJ, O'Garra A. 2011. The role of IL-10 in immune regulation during M. tuberculosis infection. Mucosal Immunol 4:261–270. doi: 10.1038/mi.2011.7. [DOI] [PubMed] [Google Scholar]
- 55.Eklund D, Persson HL, Larsson M, Welin A, Idh J, Paues J, Fransson SG, Stendahl O, Schon T, Lerm M. 2013. Vitamin D enhances IL-1beta secretion and restricts growth of Mycobacterium tuberculosis in macrophages from TB patients. Int J Mycobacteriol 2:18–25. doi: 10.1016/j.ijmyco.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 56.Reinholz M, Ruzicka T, Schauber J. 2012. Cathelicidin LL-37: an antimicrobial peptide with a role in inflammatory skin disease. Ann Dermatol 24:126–135. doi: 10.5021/ad.2012.24.2.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Suzuki N, Suzuki S, Duncan GS, Millar DG, Wada T, Mirtsos C, Takada H, Wakeham A, Itie A, Li S, Penninger JM, Wesche H, Ohashi PS, Mak TW, Yeh WC. 2002. Severe impairment of interleukin-1 and Toll-like receptor signalling in mice lacking IRAK-4. Nature 416:750–756. doi: 10.1038/nature736. [DOI] [PubMed] [Google Scholar]
- 58.Olson CM, Hedrick MN, Izadi H, Bates TC, Olivera ER, Anguita J. 2007. p38 mitogen-activated protein kinase controls NF-κB transcriptional activation and tumor necrosis factor alpha production through RelA phosphorylation mediated by mitogen- and stress-activated protein kinase 1 in response to Borrelia burgdorferi antigens. Infect Immun 75:270–277. doi: 10.1128/IAI.01412-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Karunakaran S, Ravindranath V. 2009. Activation of p38 MAPK in the substantia nigra leads to nuclear translocation of NF-κB in MPTP-treated mice: implication in Parkinson's disease. J Neurochem 109:1791–1799. doi: 10.1111/j.1471-4159.2009.06112.x. [DOI] [PubMed] [Google Scholar]
- 60.Pathak SK, Basu S, Basu KK, Banerjee A, Pathak S, Bhattacharyya A, Kaisho T, Kundu M, Basu J. 2007. Direct extracellular interaction between the early secreted antigen ESAT-6 of Mycobacterium tuberculosis and TLR2 inhibits TLR signaling in macrophages. Nat Immunol 8:610–618. doi: 10.1038/ni1468. [DOI] [PubMed] [Google Scholar]
- 61.Guinn KM, Hickey MJ, Mathur SK, Zakel KL, Grotzke JE, Lewinsohn DM, Smith S, Sherman DR. 2004. Individual RD1-region genes are required for export of ESAT-6/CFP-10 and for virulence of Mycobacterium tuberculosis. Mol Microbiol 51:359–370. doi: 10.1046/j.1365-2958.2003.03844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li G, Domenico J, Jia Y, Lucas JJ, Gelfand EW. 2009. NF-κB-dependent induction of cathelicidin-related antimicrobial peptide in murine mast cells by lipopolysaccharide. Int Arch Allergy Immunol 150:122–132. doi: 10.1159/000218115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pereira S, Veeraraghavan P, Ghosh S, Gandhi M. 2004. Animal experimentation and ethics in India: the CPCSEA makes a difference. Altern Lab Anim 32:411–415. [DOI] [PubMed] [Google Scholar]