

ABSTRACT
The interleukin-17 (IL-17) family of cytokines (IL-17A to IL-17F) is involved in many inflammatory diseases. Although IL-17A is recognized as being involved in the pathophysiology of Helicobacter pylori-associated diseases, the role of other IL-17 cytokine family members remains unclear. Microarray analysis of IL-17 family cytokines was performed in H. pylori-infected and uninfected gastric biopsy specimens. IL-17C mRNA was upregulated approximately 4.5-fold in H. pylori-infected gastric biopsy specimens. This was confirmed by quantitative reverse transcriptase PCR in infected and uninfected gastric mucosa obtained from Bhutan and from the Dominican Republic. Immunohistochemical analysis showed that IL-17C expression in H. pylori-infected gastric biopsy specimens was predominantly localized to epithelial and chromogranin A-positive endocrine cells. IL-17C mRNA levels were also significantly greater among cagA-positive than cagA-negative H. pylori infections (P = 0.012). In vitro studies confirmed an increase in IL-17C mRNA and protein levels in cells infected with cagA-positive infections compared to cells infected with either cagA-negative or cag pathogenicity island (PAI) mutant. Chemical inhibition of IκB kinase (IKK), mitogen-activated protein extracellular signal-regulated kinase (MEK), and Jun N-terminal kinase (JNK) inhibited induction of IL-17C proteins in infected cells, whereas p38 inhibition had no effect on IL-17C protein secretion. In conclusion, H. pylori infection was associated with a significant increase in IL-17C expression in human gastric mucosa. The role of IL-17C in the pathogenesis of H. pylori-induced diseases remains to be determined.
KEYWORDS: Helicobacter pylori, interleukin-17C, cytotoxin-associated gene A, mononuclear cell infiltration, Ingenuity Pathway Analysis
INTRODUCTION
Gastric mucosal infection with Helicobacter pylori is characterized by the induction of a number of proinflammatory cytokines, including interleukin-8 (IL-8) (1), IL-6 (2), and tumor necrosis factor α (TNF-α) (3), thought to be involved in the development of H. pylori-induced gastric inflammation (4, 5). The IL-17 family of cytokines, which includes IL-17A, B, C, D, E (also known as IL-25), and F, are involved in the pathogenesis of many inflammatory diseases (6–9). IL-17A is the best characterized and is primarily produced by T helper type 17 (Th17) cells (10, 11). Several lines of evidence suggest that IL-17A plays an important role in the pathophysiology of H. pylori-associated disease (12, 13). For example, Caruso et al. (14) showed that IL-17A-producing T cells were increased in H. pylori-infected gastric mucosa and that IL-17A was overexpressed. Shi et al. (15) suggested that H. pylori infection activated the Th17/IL-17 pathway in a mouse model which resulted in recruitment of inflammatory cells into the gastric mucosa. Recently, Serrano et al. (16) showed that downregulation of Th17 responses was associated with a reduction in gastritis in H. pylori-infected children. The functions of IL-17 cytokine family members other than IL-17A in H. pylori infection remain poorly understood. It is thought that IL-17C is produced mainly by epithelial cells and may play a role in promoting cytokine and chemokine production in intestinal epithelial cells and keratinocytes (7–9).
Both IL-17A and IL-17C have been associated with protection against microbial infections (17, 18). Stimulation of epithelial cells with either whole bacteria or with agonists to Toll-like receptor 2 (TLR2), TLR4, or TLR5 in in vitro experiments in cultured cells and in a mouse model resulted in rapid induction of expression of IL-17C (17–21). Despite accumulating evidence suggesting an important biological role for IL-17C, the biological role of IL-17C during H. pylori infection of gastric epithelium remains unclear. Here, we demonstrate increased expression of IL-17C in H. pylori-infected human gastric mucosa, and we explore the possible relationship between IL-17C expression and the pathogenesis of H. pylori-induced gastritis.
RESULTS
Study subjects and H. pylori cagA genotypes.
Samples from 136 subjects from Bhutan (median age, 40 years; age range, 16 to 92 years) and 157 subjects from the Dominican Republic (median age, 47 years; age range, 17 to 91 years) were included in this study (Table 1). These subjects were previously reported in studies of the effect of H. pylori infection on IL-8 and IL-10 mRNA levels (22) and TLR10 expression (23) in the gastric mucosa.
TABLE 1.
Characteristics of the study subjects and H. pylori cagA genotype
| Parameter | Value(s) for subjects from: |
|||||
|---|---|---|---|---|---|---|
| Bhutan |
Dominican Republic |
|||||
| H. pylori negative (n = 50) | H. pylori positive (n = 86) | P value | H. pylori negative (n = 65) | H. pylori positive (n = 92) | P value | |
| Age, yr (range) | 45 (18–78) | 34 (16–92) | 0.006 | 48 (17–91) | 46 (17–82) | 0.246 |
| Male [no. (%)] | 20 (40) | 40 (47) | 0.460 | 25 (38) | 30 (33) | 0.450 |
| cagA status [no. (%)] | ||||||
| Negative | 0 (0) | 14 (30) | ||||
| Western type | 2 (3) | 47 (70) | ||||
| East Asian type | 68 (97) | 0 (0) | ||||
| PMN, grade [no. (%)] | ||||||
| 0 | 41 (82) | 0 (0) | 58 (89) | 6 (6) | ||
| 1 | 8 (16) | 42 (49) | 6 (9) | 53 (58) | ||
| 2 | 1 (2) | 37 (43) | 1 (2) | 30 (33) | ||
| 3 | 0 (0) | 7 (8) | 0 (0) | 3 (3) | ||
| Mean, median | 0.200, 0 | 1.593, 2 | <0.001 | 0.123, 0 | 1.326, 1 | <0.001 |
| MNC, grade [no. (%)] | ||||||
| 0 | 16 (32) | 0 (0) | 39 (60) | 1 (1) | ||
| 1 | 33 (66) | 20 (23) | 20 (31) | 26 (28) | ||
| 2 | 1 (2) | 55 (64) | 5 (8) | 56 (61) | ||
| 3 | 0 (0) | 11 (13) | 1 (1) | 9 (10) | ||
| Mean, median | 0.700, 1 | 1.895, 2 | <0.001 | 0.508, 0 | 1.793, 2 | <0.001 |
| Atrophy, grade [no. (%)] | ||||||
| 0 | 6 (12) | 1 (1) | 24 (37) | 11 (12) | ||
| 1 | 35 (70) | 46 (53) | 39 (60) | 67 (73) | ||
| 2 | 6 (12) | 35 (41) | 2 (3) | 14 (15) | ||
| 3 | 3 (6) | 4 (5) | 0 (0) | 0 (0) | ||
| Mean, median | 1.120, 1 | 1.488, 1 | <0.001 | 0.662, 1 | 1.033, 1 | <0.001 |
| H. pylori density [no. (%)] | ||||||
| 0 | 50 (100) | 9 (10) | 65 (100) | 9 (10) | ||
| 1 | 0 (0) | 16 (19) | 0 (0) | 32 (35) | ||
| 2 | 0 (0) | 25 (29) | 0 (0) | 40 (43) | ||
| 3 | 0 (0) | 36 (42) | 0 (0) | 11 (12) | ||
| Mean, median | 0, 0 | 2.023, 2 | <0.001 | 0, 0 | 1.576, 2 | <0.001 |
The prevalence of H. pylori was 63% in Bhutanese and 59% in the Dominican Republic subjects. Among the 86 H. pylori infected gastric mucosa from Bhutanese subjects, 70 cases were successfully cultured and were examined for the cagA genotype. Sixty-eight samples (97%) showed East Asian-type cagA, and only 2 samples (3%) showed the Western-type cagA genotype. From the 92 H. pylori-infected gastric mucosa from the Dominican Republic, 61 samples could be cultured. Of these samples, 14 (30%) were cagA negative and 47 (70%) were Western-type cagA (Table 1).
Gene expression profiles of IL-17 family members in H. pylori infection.
Microarray analysis was used to examine mRNA expression profiles of IL-17 ligand (IL-17A, B, C, D, E and F) and receptor (IL-17RA, RB, RC, RD, and RE) members in H. pylori-infected gastric mucosa (Table 2). We selected 32 samples (16 from Bhutan and 16 from the Dominican Republic) based on typical pathological findings (4 from H. pylori-negative normal mucosa and 12 from H. pylori-positive gastritis mucosa from each country, as described previously [23]).
TABLE 2.
Microarray analysis of IL-17 family gene expression
| IL-17 designation | Probe ID | Value(s) for subjects froma: |
|||||
|---|---|---|---|---|---|---|---|
| Total |
Bhutan |
Dominican Republic |
|||||
| Fold change | P value | Fold change | P value | Fold change | P value | ||
| IL17A | A_23_P332820 | 0.870 | 0.406 | 1.006 | 0.979 | 0.752 | 0.247 |
| IL17B | A_23_P167479 | 1.031 | 0.887 | 1.127 | 0.747 | ND | ND |
| IL17C | A_33_P3339625 | 4.493 | <0.001 | 3.150 | 0.002 | 6.408 | <0.001 |
| IL17D | A_23_P345692 | 1.276 | 0.243 | 1.349 | 0.285 | 1.208 | 0.543 |
| IL17E | A_23_P117437 | ND | ND | ND | ND | ND | ND |
| IL17F | A_23_P167882 | 1.744 | 0.008 | 1.509 | 0.079 | 2.015 | 0.048 |
| IL17RA | A_33_P3540143 | 1.176 | 0.085 | 1.202 | 0.080 | 1.150 | 0.361 |
| IL17RB | A_24_P157370 | 1.315 | 0.118 | 1.439 | 0.084 | 1.202 | 0.429 |
| IL17RC | A_23_P166775 | 0.786 | 0.041 | 0.819 | 0.054 | 0.754 | 0.082 |
| IL17RD | A_32_P188860 | 1.039 | 0.846 | 1.480 | 0.118 | 0.729 | 0.260 |
| IL17RE | A_23_P500206 | 0.576 | <0.001 | 0.562 | 0.013 | 0.590 | 0.001 |
Fold change indicates mean fold change between H. pylori-positive subjects and H. pylori-negative subjects in two separate experiments. ND, not detected.
IL-17C mRNA was significantly upregulated in H. pylori-infected gastric mucosa: 3.2-fold (P = 0.002) for subjects from Bhutan and 6.4-fold (P < 0.001) for subjects from the Dominican Republic. Other IL-17 ligands and receptors mRNA levels in H. pylori positive samples did not significantly differ from H. pylori-negative samples.
Regulator effects network analysis using IPA.
The array chipset contained 50,599 total probe sets, and the expression levels of 2,354 (4.6%) probe sets changed more than 2-fold (P < 0.05) in the presence of H. pylori infection (1,721 genes were upregulated and 633 genes were downregulated). These 2,354 significantly altered genes were uploaded into Ingenuity Pathway Analysis (IPA). Regulator effects analysis was performed to predict how activated IL-17C might cause an increase or decrease in phenotypic or functional outcomes downstream (Fig. 1).
FIG 1.

Ingenuity Pathway Analysis (IPA) of IL-17C in the gastric mucosa. (A) Regulator effects network generated from IPA (published with permission from QIAGEN). Upstream regulators (IL-17C) are displayed in the top tier, while functions are displayed in the bottom tier. The data set genes that connect IL-17C and the lower functions are displayed in the middle tier. This analysis presumed that IL-17C activated 8 genes (including IL-17C itself) and 3 functions (consistency score, 7.236). (B) Fold change and P value of the genes that connect IL-17C and the lower functions. BCL2, B-cell lymphoma 2; CCL20, C-C motif chemokine ligand 20; CXCL8, C-X-C motif chemokine ligand 8; IFNG, gamma interferon; TNF, tumor necrosis factor; DEFB4A/4B, defensin beta 4A/4B; S100A8/A9, S100 calcium-binding protein A8/A9; LCN2, Lipocalin-2.
This analysis presumed that IL-17C activated 8 genes (including IL-17C itself) and 3 functions (consistency score, 7.236). IL-17C activated CXCL8, IFNG, TNF, IL-6, IL-23A, S100A9, and LCN2. These activated genes are involved in migration of mononuclear leukocytes, cell movement of lymphocytes, and antimicrobial response (Fig. 1A). CCL20 (fold change, 25.00) and DEFB4A/DEFB4B (fold change, 18.96) were not predicted to be activated, probably due to both proteins binding to the same receptor, CCR6.
IL-17C mRNA expression levels in antral gastric mucosa.
To confirm microarray results, we measured IL-17C mRNA levels using quantitative real-time PCR (qPCR). IL-17C mRNA levels in samples from H. pylori-positive subjects (n = 178) were higher than those in samples from H. pylori-negative subjects (n = 115) (H. pylori negative, median of 0.37 and range of 0 to 7.55; H. pylori positive, median of 2.39 and range of 0 to 25.5; P < 0.001) (Fig. 2A).
FIG 2.

IL-17C mRNA levels in the antral gastric mucosa. (A) IL-17C mRNA levels in samples from H. pylori (HP)-positive subjects (n = 178) were higher than those from H. pylori-negative subjects (n = 115). (B) H. pylori-negative (n = 50) and H. pylori-positive (n = 86) subjects from Bhutan. IL-17C mRNA levels in samples from H. pylori-positive subjects were higher than those from H. pylori-negative subjects. (C) H. pylori-negative (n = 65) and H. pylori-positive (n = 92) subjects from the Dominican Republic. IL-17C mRNA levels in samples from H. pylori-positive subjects were higher than those from H. pylori-negative subjects. Beta-actin (ACTB) was used as the endogenous control for data normalization. Data were expressed by box plotting.
In biopsy specimens from Bhutan, the IL-17C mRNA levels in H. pylori-positive specimens (n = 86) were significantly higher than those in H. pylori-negative specimens (n = 50) (H. pylori negative, median of 0.51 and range of 0 to 5.26; H. pylori positive, median of 2.68 and range of 0 to 25.5; P < 0.001) (Fig. 2B). IL-17C mRNA was detected in 88% of all specimens (98% of H. pylori-positive versus 72% of H. pylori-negative specimens [P < 0.001]).
In specimens from the Dominican Republic, the IL-17C mRNA levels in H. pylori-positive samples (n = 92) were also significantly greater than those in H. pylori-negative samples (n = 65) (H. pylori negative, median of 0.23 and range of 0 to 7.55; H. pylori positive, median of 2.24 and range of 0 to 618.8; P < 0.001) (Fig. 2C). IL-17C mRNA was detected in 82% of all samples. It was present in 95% of the H. pylori-positive versus 63% of the H. pylori-negative samples (P < 0.001). In both countries, the IL-17C mRNA levels in H. pylori-infected mucosa were significantly higher than those in uninfected mucosa even when subthreshold expression samples were excluded (P < 0.001).
Immunohistochemistry.
Although increased IL-17C mRNA levels were found in H. pylori-positive patients, the cellular source of IL-17C in the human gastric mucosa is unknown. We analyzed IL-17C protein expression in human gastric mucosa by immunohistochemistry (Fig. 3). IL-17C staining was observed primarily in the epithelial cells of H. pylori-infected mucosa. In addition, some infiltrating cells were also stained (Fig. 3C to F). In contrast, IL-17C-stained cells were rare in the uninfected gastric mucosa (Fig. 3A and B). Semiquantitative analysis of IL-17C staining intensity in gastric epithelial cells (H. pylori negative, n = 6; mild gastritis, n = 6; severe gastritis, n = 6; intestinal metaplasia [IM], n = 6) demonstrated significantly increased IL-17C in the mucosa with severe gastritis and IM compared with no infection and mild gastritis (Fig. 3G).
FIG 3.
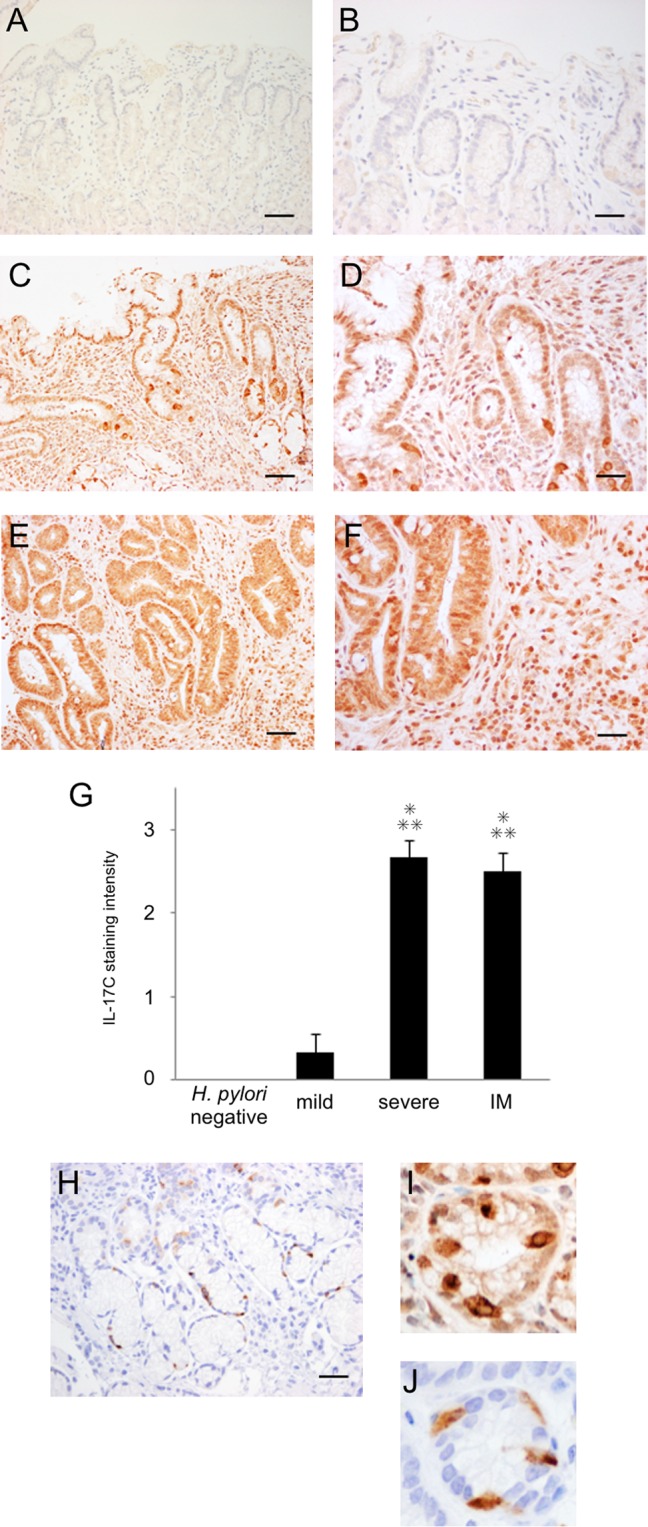
Immunohistochemistry of IL-17C (A to F and I) and chromogranin A (H and J) in the antral gastric mucosa. (A and B) Subject without H. pylori infection. (C, D, and H to J) Subject with H. pylori infection and severe gastritis. (E and F) Subjects with H. pylori infection and intestinal metaplasia. (G) Semiquantitative analysis of IL-17C. IL-17C staining was observed mainly in the epithelial cells of H. pylori-infected mucosa. Semiquantitative analysis of IL-17C in the gastric epithelial cells of H. pylori-negative and -positive subjects (no infection, n = 6; mild gastritis, n = 6; severe gastritis, n = 6; intestinal metaplasia [IM], n = 6) used the following scores: 0, no staining; 1, weak staining; 2, moderate staining; 3, strong staining (means ± standard errors [SE]; *, P < 0.001 compared with H. pylori-negative samples; **, P < 0.001 compared with mild gastritis). (I and J) Certain single cells in glandular epithelial cells were strongly stained, and these patterns resemble the staining of chromogranin A-positive enteroendocrine cells. Magnification, ×100 (scale bar, 250 μm) (A, C, E, and H) or ×400 (scale bar, 100 μm) (B, D, F, I, and J).
Within the glandular epithelium, certain cells were strongly stained in H. pylori-positive mucosa. These strongly stained IL-17C patterns were similar to the staining pattern of chromogranin A (CgA)-positive enteroendocrine cells (Fig. 3H to J). These findings suggested that IL-17C expression in H. pylori-infected gastric biopsy specimens was localized in both epithelial cells and in CgA-positive endocrine cells, and staining in CgA-positive cells was greater than that in epithelial cells.
IL-17C mRNA expression in relation to cagA genotype.
It is thought that IL-8, a CXC chemokine specific for neutrophil granulocyte chemotaxis, plays a major role in the H. pylori-associated acute inflammatory response (24, 25). We previously showed that gastric IL-8 mRNA levels were significantly higher in H. pylori-infected subjects from Bhutan (East Asian-type cagA) than from the Dominican Republic (in both Western-type cagA and cagA-negative infections) (22). The NF-κB signaling pathway is thought to be involved in the induction IL-8 and IL-17C (26). We hypothesized that IL-17C mRNA levels were also affected by cagA status (negative or positive) and genotype (East Asian type or Western type). The present study showed that the IL-17C mRNA levels were significantly higher in East Asian-type cagA samples (n = 68) from Bhutan than for cagA-negative samples (n = 14) from the Dominican Republic (East Asian-type cagA, median of 2.81 and range of 0 to 25.5; cagA negative, median of 1.60 and range of 0 to 5.11; P = 0.012) (Fig. 4). IL-17C mRNA expression levels were also higher for Western-type cagA samples (n = 47) than for cagA-negative specimens, but the difference was not statistically significant (Western-type cagA, median of 2.29 and range of 0.01 to 18.8; P = 0.073) (Fig. 4).
FIG 4.

IL-17C mRNA levels in relation to cagA genotype. IL-17C mRNA levels in East Asian-type cagA samples (n = 68) were significantly greater than those from cagA-negative samples (n = 14). IL-17C mRNA levels were higher for Western-type cagA samples (n = 47) than for cagA-negative specimens, but the difference was not statistically significant. Beta-actin (ACTB) was used as the endogenous control for data normalization. Data were expressed by box plotting.
Correlation between IL-17C and IL-8 mRNA levels.
Johnston et al. (27) reported that IL-17C appears to act upstream of proinflammatory cytokines, including IL-8, in the pathogenesis of psoriasis. Hence, we examined the correlation between IL-17C and IL-8 mRNA levels in the gastric mucosa. Significant but weak positive correlations were observed between IL-17C and IL-8 mRNA levels in H. pylori-positive samples (n = 178; correlation coefficient, 0.303; P < 0.001) and H. pylori-negative samples (n = 115; correlation coefficient, 0.364; P = 0.001) (Fig. 5). In addition, IL-17C mRNA levels correlated with IL-8 mRNA levels in cagA-positive samples (n = 115; correlation coefficient, 0.259; P = 0.005).
FIG 5.

Correlation coefficient for IL-17C and IL-8 mRNA levels in the gastric mucosa. Positive correlations were observed between IL-17C and IL-8 mRNA levels in H. pylori-positive samples (n = 178) and H. pylori-negative samples (n = 115).
Correlation between IL-17C mRNA levels and histological findings.
We analyzed the correlation between the IL-17C mRNA levels and histological findings and H. pylori density scores determined using the updated Sydney System. The IL-17C mRNA levels weakly correlated with the mononuclear cell (MNC) score in Bhutanese H. pylori-infected gastric specimens (correlation coefficient, 0.292; P = 0.006) (Table 3). In addition, there was a significant relationship between IL-17C mRNA levels and MNC score in Bhutanese samples (MNC score 1, median of 2.01 and range of 0 to 7.70; MNC score 2, median of 2.80 and range of 0 to 25.5; MNC score 3, median of 3.51 and range of 1.31 to 21.4; score 1 versus 2, P = 0.038; score 1 versus 3, P = 0.016;) (Fig. 6).
TABLE 3.
Correlation between IL-17C mRNA levels and histological findings
| Parameter | Bhutan |
Dominican Republic |
||
|---|---|---|---|---|
| Correlation coefficient | P value | Correlation coefficient | P value | |
| PMN | −0.019 | 0.863 | 0.038 | 0.722 |
| MNC | 0.292 | 0.006 | 0.051 | 0.627 |
| Atrophy | 0.210 | 0.053 | 0.034 | 0.747 |
| H. pylori density | 0.156 | 0.150 | 0.047 | 0.655 |
FIG 6.

IL-17C mRNA levels and mononuclear cell (MNC) infiltration in H. pylori-positive mucosa from Bhutanese subjects. IL-17C mRNA levels were increased in a step-like manner by MNC histologic grade scores using the updated Sydney System. Beta-actin (ACTB) was used as the endogenous control for data normalization. Data were expressed by box plotting.
In contrast, none of histological findings were significantly correlated with IL-17C mRNA levels from tissue from the Dominican Republic (polymorphonuclear leukocyte [PMN], P = 0.722; MNC, P = 0.627; atrophy, P = 0.747; H. pylori density, P = 0.655) (Table 3). However, the IL-17C mRNA levels correlated with the MNC score in cagA-positive samples (n = 115; correlation coefficient, 0.209; P = 0.023).
IL-17C expression in gastric epithelial cell lines in response to H. pylori infection.
We demonstrated that gastric mucosal IL-17C mRNA levels were increased in H. pylori-infected gastric mucosa compared to those in uninfected mucosa (Fig. 2), and immunohistochemistry examination revealed that IL-17C was produced in gastric epithelial cells (Fig. 3). To investigate how H. pylori regulates IL-17C mRNA and protein levels in gastric epithelial cells, we examined the effect of H. pylori infection using human gastric epithelial cancer cells. First, we looked for an alteration of IL-17C expression in response to H. pylori infection using qPCR and enzyme-linked immunosorbent assay (ELISA). H. pylori strain 26695 was used, and IL-17C mRNA levels were assessed 3 h after infection at a multiplicity of infection (MOI) of 100. IL-17C mRNA in H. pylori-infected cells was significantly upregulated compared to that of uninfected cells (Fig. 7A). Both H. pylori strain 26695 and TN2GF4 induced IL-17C from MKN28 cells, but the level of IL-17C protein was below the level of detection with both AGS and MKN45 cells (Fig. 7B). Of interest, even among gastric cancer cells we found differences in IL-17C levels. In a second step, MKN28 cells were infected with H. pylori for different time periods and at different MOIs (50, 100, and 200). In time course experiments, IL-17C mRNA levels quickly increased following H. pylori infection, reaching maximal levels within 3 h and then decreasing to baseline (Fig. 7C). IL-17C protein levels increased in a time-dependent manner after infection (Fig. 7D). IL-17C mRNA and protein levels were not dependent on H. pylori density (Fig. 7E and F).
FIG 7.

IL-17C mRNA and protein levels in gastric epithelial cell lines in response to H. pylori infection. (A) IL-17C mRNA levels in AGS, MKN28, and MKN45 cells. Cells were infected with H. pylori strain 26695 for 3 h at an MOI of 100. (B) IL-17C protein levels in AGS, MKN28, and MKN45 cells determined by ELISA. Cells were infected with H. pylori strain 26695 and TN2GF4 for 24 h at an MOI of 100. (C) IL-17C mRNA levels in MKN28 cells. Cells were infected with H. pylori strain 26695 and TN2GF4 for different time periods (0, 3, 6, and 24 h) at an MOI of 100. (D) IL-17C protein levels in MKN28 cells. Cells were infected with H. pylori strain 26695 and TN2GF4 for different time periods (0, 3, 6, and 24 h) at an MOI of 100. (E) IL-17C mRNA levels in MKN28 cells. Cells were infected with H. pylori strain 26695 and TN2GF4 for 3 h at different MOIs (50, 100, and 200). (F) IL-17C protein levels in MKN28 cells. Cells were infected with H. pylori strain 26695 and TN2GF4 for 24 h at different MOIs (50, 100, and 200). (G) IL-17C mRNA levels in MKN28 cells. Cells were infected with TN2GF4 wild type and cagA and cag PAI mutants for 3 h at an MOI of 100. (H) IL-17C protein levels in MKN28 cells. Cells were infected with TN2GF4 wild type and cagA and cag PAI mutants for 24 h at an MOI of 100. Error bars represent the standard deviations obtained from three experiments. Statistical differences between treated and control samples were analyzed using Student's t test. *, P < 0.05.
To investigate the effects of H. pylori virulence factors on IL-17C production, we cocultured cells with cagA mutants or with cag pathogenicity island (PAI) mutants (Fig. 7G and H). IL-17C expression was significantly decreased from MKN28 cocultured with either the cagA or cag PAI mutants. The results from in vitro coculturing experiments using gastric epithelial cells were identical to the data from in vivo measurement of IL-17C mRNA and protein levels using gastric biopsy specimens.
IL-17C is thought to be a cytokine produced by epithelial cells in an autocrine manner to induce a rapid innate immune response in the epithelium (18). IL-17C binds to IL-17 receptor A (RA) and IL-17RE complex on epithelial cells to trigger downstream signaling pathways. IL-17RE is considered a specific receptor for IL-17C (17, 18, 28). The protein expression of IL-17RE in MKN28 cells was confirmed by immunocytochemistry (Fig. 8). The cytoplasm of MKN28 cells was positively immunostained with an anti-human IL-17RE antibody.
FIG 8.
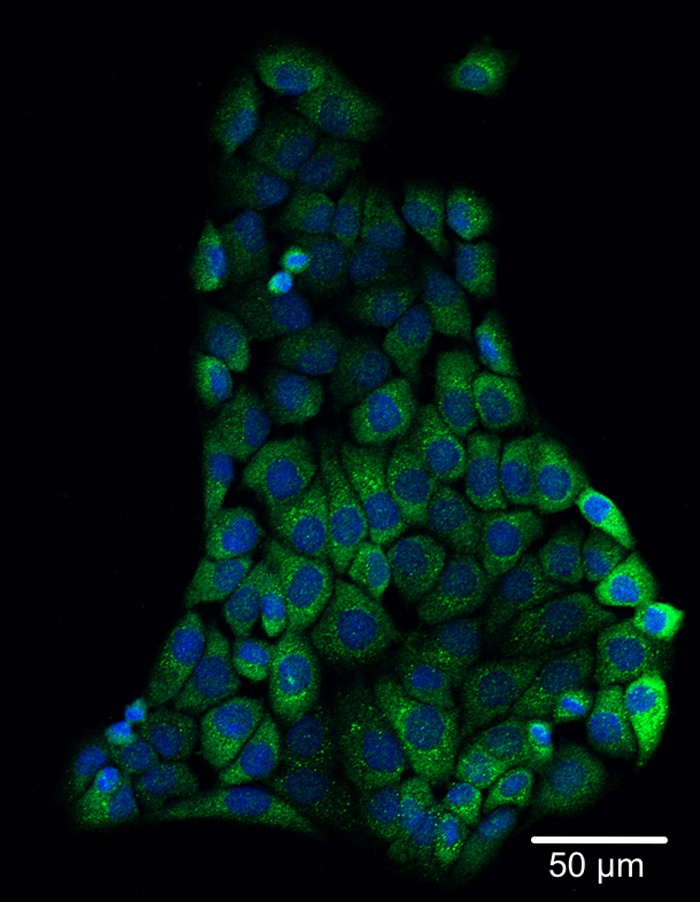
Immunocytochemical detection of IL-17RE in MKN28 cells. MKN28 cells were plated on glass slides and cultured overnight. After fixation as described in Materials and Methods, the cells were immunocytochemically stained with anti-human IL-17RE antibody and analyzed by confocal microscopy. The cytoplasm of MKN28 cells was positively immunostained with anti-human IL-17RE antibody. Nuclei were counterstained with DAPI. The scale bar represents 50 μm.
Currently, there are no reports investigating signal transduction pathways leading to IL-17C release from human gastric epithelial cells. We therefore aimed to identify signaling mediators crucial for H. pylori-induced IL-17C production in MKN28 cells. H. pylori is known to activate mitogen-activated protein kinases (MAPK) as well as NF-κB (29). We preincubated MKN28 cells with chemical inhibitors or dimethyl sulfoxide (DMSO) (for the infected control) for 1 h, followed by infection with wild-type H. pylori. Chemical inhibitors of IκB kinase (IKK) (SC-514; 20 μM), mitogen-activated protein extracellular signal-regulated kinase (MEK) (U0126; 10 μM), and Jun N-terminal kinase (JNK) (SP600125; 10 μM) inhibited the induction of both IL-17C and IL-8 proteins (Fig. 9A and B). p38 inhibitor (SB203580; 10 μM) did not affect IL-17C protein secretion despite reduced IL-8 protein.
FIG 9.

Effect of inhibitors of signal transduction on IL-17C induction following H. pylori infection. MKN28 cells were incubated with SC-514 (20 μM; IKK-2 inhibitor), U0126 (10 μM; MEK1/2 inhibitor), SP600125 (10 μM; JNK inhibitor), or SB203580 (10 μM; p38 inhibitor) for 1 h and subsequently cocultured with wild-type H. pylori strains for 24 h at an MOI of 100. IL-17C (A) and IL-8 (B) supernatant levels were determined using ELISA. IL-8 protein expression induced by H. pylori infection was significantly reduced in cells preincubated with p38 inhibitor but IL-17C was not reduced. Error bars represent the standard deviations obtained from three experiments. Statistical differences between treated and control samples were analyzed using Student's t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001 compared with an H. pylori-infected control without inhibitor.
DISCUSSION
Accumulating evidence has suggested that the epithelial expression of IL-17C can be induced directly by bacteria or indirectly through inflammatory cytokines (7, 8). The rapid kinetics of IL-17C expression following these stimuli positions IL-17C as an early player in the epithelial response to bacterial challenge (7). IL-17RE, a specific receptor for IL-17C, is located mainly on epithelial cells, keratinocytes, or Th17 cells. Gene expression profiling of various mouse tissues showed that the higher expression of IL-17RE was in mucosal organs, including the colon, lungs, and stomach (18). IL-17C activates the NF-κB pathway in normal human bronchial epithelial cells (30) and mouse primary intestinal epithelial cells (17), and it induces the expression of inflammatory cytokines, chemokines, and antibacterial peptides.
To the best of our knowledge, this is the first report that the expression of IL-17C in H. pylori-infected gastric mucosa was significantly higher than those in uninfected mucosa. Prior studies have shown that IL-17A was significantly increased in the gastric mucosa of H. pylori-infected subjects (14, 31, 32). In contrast, IL-17A mRNA levels in our study were not altered by H. pylori infection. We also performed real-time reverse transcription-PCR (RT-PCR); however, IL-17A mRNA levels were independent of H. pylori infection (data not shown). The reason for this difference is unclear and may be due to differences in the degree of lymphocyte infiltration, as IL-17A is produced mainly by Th17 cells, or differences in the method of IL-17A measurement.
Several immunohistochemical studies using clinical samples have indicated that IL-17C is expressed mainly in the epithelial cells, including for inflammatory bowel disease (IBD) (33, 34), chronic obstructive pulmonary disease (20), chronic rhinosinusitis with nasal polyposis (35), and recurrent aphthous ulcer (36). Similar to other tissues, our results revealed that IL-17C was present predominantly in the epithelial cells of H. pylori-infected gastric biopsy specimens as well as in chromogranin A-positive endocrine cells (mainly G cells). van Den Brink et al. (37) showed that active NF-κB was detected primarily in the cells deeper in the gastric glands, many of which are G cells. These findings suggest that IL-17C expression from G cells is higher than that from other epithelial cells due to differences in NF-κB activity.
Moreover, to confirm the expression of IL-17C in gastric epithelial cells, we performed in vitro infection with H. pylori using gastric epithelial cancer cell lines. IL-17C mRNA levels in H. pylori-infected cells were upregulated compared to those of uninfected cells (Fig. 7A). Our in vitro study also demonstrated that IL-17C is involved in the rapid response to H. pylori infection (Fig. 7C). Although AGS and MKN45 cells are responsive to H. pylori, they did not produce detectable IL-17C protein (Fig. 7A and B). Our previous study on IL-6 (2) reported that IL-6 levels were below the level of detection with MKN45, MKN74, KATOIII, AGS, SNU-1, SNU-16, and SNU-638 cells irrespective of whether or not H. pylori was present. H. pylori induced IL-6 from MKN1, MKN7, MKN28, SNU-668, and HeLa cells. Even among gastric cancer cells, we found differences in IL-6 levels. We believe it is likely that gene mutations in cancer cells influenced the production of cytokines. This is a flaw with all in vitro work.
Recently, a new gastric cell culture system from normal human gastric glands, gastric organoids, has been established as an advanced model for studies on infection with H. pylori in vitro (38, 39). IL-17C mRNA expression using microarray analysis was also found to be upregulated in H. pylori-infected organoids in the three-dimensional (3D) model (fold change, 2.61) (39) and 2D model (fold change, 9.26) (38).
The H. pylori virulence factor CagA is delivered into gastric epithelial cells by a type IV secretion system and is associated with severe gastritis and gastric carcinogenesis (40, 41). Brandt et al. (42) found that CagA could activate NF-κB and induce downstream IL-8 release via the MEK/ERK signaling pathway. cagA-positive strains have been subdivided into East Asian and Western types according to the sequence located in the 3′ region of cagA (43, 44). In vitro experiments have shown that East Asian-type CagA has a higher binding ability for the cytoplasmic Src homology 2-containing protein tyrosine phosphatase (SHP-2) and a greater ability to induce morphological change than Western-type CagA (45). In this study, we analyzed the association between IL-17C expression and cagA genotype in vivo (Fig. 3) and in vitro (Fig. 6G and H). Our findings suggested that cagA genotype influences IL-17C levels in human gastric mucosa.
Recent investigations of IBD patients have demonstrated that IL-17C mRNA levels in inflamed colonic biopsy specimens were significantly higher than those in noninflamed specimens (19, 34). However, the etiology and pathogenesis of the IL-17C-induced mucosal inflammation have not been reported. Johnston et al. (27) suggested that IL-17C acts upstream of many proinflammatory cytokines critical in psoriasis pathogenesis, including IL-1β, IL-17A/F, IL-22, IL-6, CXCL8, and TNF. In the present study, the cause-and-effect analysis using IPA presumed that IL-17C activated MNC infiltration via the upregulation of H. pylori-related inflammatory cytokine genes (CXCL8 [1], IFNG [46], TNF [3], IL-6 [2], and CCL20 [47]) (Fig. 8). These findings suggest that IL-17C triggers many proinflammatory cytokines as a rapid host epithelial response to H. pylori infection. This is then followed by complicated proinflammatory cascades that may depend on the interactions among H. pylori virulence factors, host gastric mucosal factors, and the environment.
We also found no relationship between H. pylori density and IL-17C mRNA expression in H. pylori-infected gastric mucosa (Table 3) or in vitro (Fig. 7E and F). IL-17C could provide an epithelial responsive mechanism for relatively weaker H. pylori challenges. More studies are needed to examine the precise functional roles of IL-17C in the pathogenesis of H. pylori-induced gastritis.
Our in vitro results using pharmacological inhibitors showed that H. pylori activated NF-κB, MEK, and JNK signaling, thus inducing IL-17C similarly to that already described for IL-8 (29). Previously, the p38 pathway was shown to be important in activation of IL-8 production in response to H. pylori (48). Our experiments confirmed that IL-8 was significantly reduced in cells treated with the p38 inhibitor, but IL-17C production was not reduced. The different mechanisms of IL-17C induction from IL-8 might give us new insights into H. pylori-associated gastroduodenal pathogenesis.
Several lines of evidence have indicated that H. pylori infection is etiologically related to gastric cancer (49). Song et al. (21) reported that IL-17C mRNA and protein expression were upregulated in human colorectal cancer specimens. They also found that microbiota-driven IL-17C promotes colon cancer development by protecting intestinal epithelial cells (IECs) from apoptosis. The removal of the gut microbiota by antibiotics blocked the upregulation of IL-17C in colon tumors and tumor-derived IECs. In an immunohistochemistry study, IL-17RE, a specific receptor for IL-17C, was highly expressed in human gastric cancer tissue (50). Microarray analysis using gastric biopsy specimens from Vietnamese gastric cancer patients suggested that the IL-17C mRNA levels in cancerous tissue were 4.3-fold higher than those from noncancerous tissue (unpublished data). Gastric cancer is a major cause of cancer death in the world, and further studies to investigate the etiological roles of IL-17C in gastric cancer are needed.
In conclusion, our study revealed that the expression of IL-17C in human gastric mucosa was increased during H. pylori infection. The role of IL-17C in the pathogenesis of H. pylori-induced gastritis and in gastric cancer remains to be determined.
MATERIALS AND METHODS
Gastric biopsy specimens from subjects.
We used gastric biopsy specimens obtained from subjects with mild dyspeptic symptoms living in two widely separated regions, Bhutan, in South Asia, and the Dominican Republic, a Caribbean country (22, 23). Written informed consent was obtained from all participants, and the protocols were approved by the ethics committee of Oita University Faculty of Medicine (Japan), Chulalongkorn University (Thailand), and Universidad Autonoma de Santo Domingo (Dominican Republic), as well by the local hospitals where we collected gastric mucosal specimens. The clinical diagnoses after endoscopy included gastritis, gastric ulcer, duodenal ulcer, and gastric cancer. Gastritis and peptic ulcers were identified by endoscopy. Gastric cancers were confirmed by histology. During endoscopy, four gastric specimens were obtained from the normal-appearing areas in the antrum: one each for rapid urease test, histological examination, H. pylori culture, and RNA examination. Rapid urease tests were performed at the time of endoscopy. Gastric biopsy specimens for histology were fixed in buffered formalin at room temperature and were sent to the Oita University Faculty of Medicine for sectioning and analyses. Biopsy specimens for culture and RNA analyses were immediately placed in a −20°C freezer and subsequently sent with dry ice by Express Mail to the Oita University Faculty of Medicine, Japan, where they were stored at −80°C. Total RNA from the gastric specimens placed in RNAlater (Ambion, Life Technologies, Carlsbad, CA) was isolated using commercially available kits (Ambion).
H. pylori culture and cagA genotyping.
H. pylori culture was performed using standard methods as described previously (51). H. pylori status was determined using a combination of rapid urease test, culture, and histology. Subjects were considered H. pylori negative if all three tests were negative and were defined as H. pylori positive when at least one of these examinations yielded positive results (88% of Bhutanese subjects and 92% of Dominican subjects had at least 2 positive tests). H. pylori DNA was extracted using a commercially available kit (DNeasy blood and tissue kit; Qiagen, Valencia, CA). The cagA status was defined by PCR for a conserved region of cagA and by direct sequencing (52). The cagA-negative status was confirmed by PCR for the cag empty site (53). The cagA genotype (East Asian type and Western type) was identified by sequencing the PCR products (54).
Histology and immunohistochemistry.
Biopsy specimens for histology were fixed in 10% neutral buffered formalin, embedded in paraffin wax, and stained with hematoxylin-eosin and Giemsa stains, followed by evaluation by an experienced pathologist (T. Uchida) blinded to the clinical features of subjects and the characteristics of the H. pylori strains. The degree of mononuclear cell infiltration (inflammation), polymorphonuclear leukocyte infiltration (activity), and H. pylori density were determined according to the updated Sydney System (55).
Immunohistochemistry was performed as described previously (56). Briefly, after antigen retrieval and inactivation of endogenous peroxidase activity, tissue sections were incubated with anti-IL-17C (Sigma-Aldrich Corp., St. Louis, MO), anti-chromogranin A (Dako, Copenhagen, Denmark), or rabbit polyclonal anti-H. pylori (Dako, Copenhagen, Denmark) antibody with diluting solution (Dako) overnight at 4°C.
Gene expression microarray analysis.
Gene expression levels from the gastric samples were analyzed with a gene expression microarray. cRNA was amplified, labeled, and hybridized to a Agilent 44K 60-mer oligonucleotide microarray according to the manufacturer's instructions. All hybridized microarray slides were scanned using an Agilent scanner, and relative hybridization intensities and background hybridization values were calculated using Agilent Feature Extraction software (9.5.1.1).
RT-PCR and quantitative real-time PCR.
Expression levels of IL-17C mRNA from the gastric samples were analyzed by qPCR. RT-PCR was performed using SuperScript III reverse transcriptase (Invitrogen). qPCR was performed using TaqMan Universal PCR master mix (Applied Biosystems, Life Technologies, Carlsbad, CA) according to the manufacturer's instructions. Beta-actin (ACTB) was used as the endogenous control for data normalization. Predesigned TaqMan gene expression assays, including the primer set and TaqMan probe (IL-17C, Hs00171163_m1; ACTB, Hs01060665_g1) were purchased from Applied Biosystems. The mRNA levels in gastric mucosa were quantified using the ABI Prism 7300 sequence detection system (Applied Biosystems). The samples were placed in the analyzer, and PCR was conducted according to the manufacturer's instructions. The ratio change in target gene relative to the endogenous control gene was determined by the 2−ΔΔCT method. We used an uninfected sample as a calibrator. The expression of IL-8 mRNA was also analyzed by qPCR as described previously (22).
Gastric epithelial cell culture and H. pylori culture used for in vitro studies.
Human gastric epithelial cancer cell lines MKN28 and MKN45 were obtained from the Riken Cell Bank (Tsukuba, Japan), and AGS cells were purchased from the American Type Culture Collection (Manassas, Virginia). These three cell lines were routinely maintained in Roswell Park Memorial Institute 1640 medium (Lonza, Walkersville, MD) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Invitrogen, Life Technologies, Carlsbad, CA) and 1% penicillin-streptomycin at 37°C and 5% CO2.
H. pylori strain 26695 and TN2GF4 were used from our stocks. TN2GF4 isogenic cagA-negative and cag PAI-negative mutants were constructed as described previously (2). For constructing the whole cag PAI-deleted mutants, regions upstream (hp0518-hp0519; bp 545, 254 to 547, and 164; locus numbers and locations are from H. pylori strain 26695, GenBank accession number AE000511) and downstream (hp0549-hp0550; bp 584, 570 to 586, and 563) of the cag PAI were amplified to delete the entire cag PAI from the H. pylori chromosome. These fragments, separated by a chloramphenicol resistance cassette (a gift from D. E. Taylor, University of Alberta, Edmonton, Canada), were cloned into the T7Blue vector (Novagen, Madison, WI). All plasmids (1 to 2 g) were used for inactivation of chromosomal genes by natural transformation. H. pylori was grown on brain heart infusion (BHI; Becton, Dickinson, and Company, Sparks, MD) agar plates containing 7% defibrinated horse blood for 72 h. Before infection, bacteria were inoculated into BHI broth with 10% FBS and grown under microaerophilic conditions at 37°C overnight with shaking. Bacterial density was determined from the optical density at 600 nm (OD600). Cultured gastric epithelial cells were infected with H. pylori at MOIs ranging from 50 to 200. Total RNA from gastric cell lines was extracted using commercially available kits (Ambion).
Immunocytochemistry.
Immunocytochemistry was performed as described previously (56). MKN28 cells were plated and grown on 25-mm coverslips overnight and then fixed with 4% paraformaldehyde for 10 min at room temperature. After being permeabilized with 0.5% Triton X-100 in 1× phosphate-buffered saline (PBS) for 10 min at room temperature, the cells were incubated with a rabbit anti-human IL-17RE antibody (1:100; HPA019011; Atlas Antibodies AB, Stockholm, Sweden) for 1 h at room temperature. After washing, the cells were incubated for 1 h with an Alexa Fluor 488-conjugated goat anti-rabbit IgG (H+L) (1:200; A11008; Invitrogen) in 0.1% Triton X-100 in 1× PBS at room temperature. The nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). The mounted coverslips were then observed using a Carl Zeiss LSM710 confocal microscope (Carl Zeiss, Inc., Jena, Germany).
Pharmacological inhibitor experiments.
Cells were pretreated (60 min before H. pylori infection) with the IKK-2 inhibitor SC-514 or a MAPK inhibitor (U0126, SB203580, or SP600125). U0126 is a specific inhibitor of MEK1/2 (MAPK/extracellular signal-regulated kinase 1/2 [ERK1/2]), which is located upstream of ERK1/2. SB203580 is a specific inhibitor of p38, and SP600125 is a specific inhibitor of the Jun N-terminal kinase (JNK). We used each inhibitor according to our previous determination of optimal concentrations (2) as well as our preliminary experiments (data not shown). All inhibitors were purchased from ENZO Life Sciences (Farmingdale, NY).
ELISA.
The IL-17C protein levels in human gastric cancer cell lines were measured using an IL-17C Duoset ELISA kit (R&D Systems, Minneapolis, MN). The ELISA was carried out according to the manufacturer's protocol. The final result was determined by a microplate spectrophotometer (Epoch; BioTek, Winooski, VT) at 450 nm. All measurements were performed in duplicate.
Regulator effects in IPA.
Qiagen's Ingenuity Pathway Analysis (IPA; Qiagen, Redwood City, CA) is an all-in-one web-based application used to examine molecular interactions. IPA's causal analytics are made possible by the Ingenuity Knowledge Base, a large structured collection of observations in various experimental contexts manually curated from the biomedical literature or integrated from third-party databases (57). Regulator effects analytics in IPA provide insight into the causes and effects of differentially expressed genes in a data set. Regulator effects explain how predicted activated/inhibited upstream regulators cause increases/decreases in functional outcomes downstream. The consistency score is a measurement used to decide rank or to prioritize the most useful networks. The details are available at www.qiagen.com/ingenuity.
We uploaded our microarray data containing gene identifiers, their corresponding fold changes, and P values. Focus genes were selected by fold change of expression values of <0.5 and >2.0, and the P value was < 0.05 using the t test. The activation state of each upstream regulator from the experimental data set was determined by calculating the Z score (>2, activated; <−2, inhibited).
Statistics.
All statistical analyses were performed by JMP 10.0 software (SAS, Cary, NC). Clinical samples were analyzed using the χ2 test to compare discrete variables and the Mann-Whitney U test to compare continuous variables. In vitro samples were analyzed using Student's t test. Correlation coefficients were calculated by Spearman's rank correlation coefficient. All statistical tests were two-sided, and a P value of <0.05 was considered statistically significant.
Accession number(s).
The microarray data were registered in the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/info/linking.html) under accession number GSE60427.
ACKNOWLEDGMENTS
This work was supported in part by grants from the National Institutes of Health (DK62813 to Y.Y. and DK56338, which funds the Texas Medical Center Digestive Diseases Center), Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan (15H02657, 16H0591, and 26640114 to Y.Y.), the Strategic Young Researcher Overseas Visits Program for Accelerating Brain Circulation from the Japan Society for the Promotion of Science (JSPS), and the Strategic Funds for the Promotion of Science and Technology from the Japan Science and Technology Agency (JST). It was also supported in part by a grant from The National Fund for Innovation and Development of Science and Technology (FONDOCYT) from the Ministry of Higher Education Science and Technology (MESCyT) of the Dominican Republic (2012-2013-2A1-65) (M.C.).
D.Y.G. is a consultant for RedHill Biopharma regarding novel H. pylori therapies, has received research support for culture of Helicobacter pylori, and is the principle investigator of an international study of the use of antimycobacterial therapy for Crohn's disease. He is also a consultant for BioGaia in relation to probiotic therapy for H. pylori infection and for Takeda in relation to H. pylori therapies. The other authors have no financial conflicts of interest.
REFERENCES
- 1.Crabtree JE, Farmery SM, Lindley IJ, Figura N, Peichl P, Tompkins DS. 1994. CagA/cytotoxic strains of Helicobacter pylori and interleukin-8 in gastric epithelial cell lines. J Clin Pathol 47:945–950. doi: 10.1136/jcp.47.10.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu H, Wu J-Y, Kudo T, Ohno T, Graham DY, Yamaoka Y. 2005. Regulation of interleukin-6 promoter activation in gastric epithelial cells infected with Helicobacter pylori. Mol Biol Cell 16:4954–4966. doi: 10.1091/mbc.E05-05-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tanahashi T, Kita M, Kodama T, Yamaoka Y, Sawai N, Ohno T, Mitsufuji S, Wei YP, Kashima K, Imanishi J. 2000. Cytokine expression and production by purified Helicobacter pylori urease in human gastric epithelial cells. Infect Immun 68:664–671. doi: 10.1128/IAI.68.2.664-671.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yamaoka Y, Kita M, Kodama T, Sawai N, Imanishi J. 1996. Helicobacter pylori cagA gene and expression of cytokine messenger RNA in gastric mucosa. Gastroenterology 110:1744–1752. doi: 10.1053/gast.1996.v110.pm8964399. [DOI] [PubMed] [Google Scholar]
- 5.Yamaoka Y, Kita M, Kodama T, Sawai N, Kashima K, Imanishi J. 1997. Induction of various cytokines and development of severe mucosal inflammation by cagA gene positive Helicobacter pylori strains. Gut 41:442–451. doi: 10.1136/gut.41.4.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iwakura Y, Ishigame H, Saijo S, Nakae S. 2011. Functional specialization of interleukin-17 family members. Immunity 34:149–162. doi: 10.1016/j.immuni.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 7.Pappu R, Rutz S, Ouyang W. 2012. Regulation of epithelial immunity by IL-17 family cytokines. Trends Immunol 33:343–349. doi: 10.1016/j.it.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 8.Song X, Qian Y. 2013. IL-17 family cytokines mediated signaling in the pathogenesis of inflammatory diseases. Cell Signal 25:2335–2347. doi: 10.1016/j.cellsig.2013.07.021. [DOI] [PubMed] [Google Scholar]
- 9.Gu C, Wu L, Li X. 2013. IL-17 family: cytokines, receptors and signaling. Cytokine 64:477–485. doi: 10.1016/j.cyto.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. 2005. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 11.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang Y-H, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. 2005. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kabir S. 2011. The role of interleukin-17 in the Helicobacter pylori induced infection and immunity. Helicobacter 16:1–8. doi: 10.1111/j.1523-5378.2010.00812.x. [DOI] [PubMed] [Google Scholar]
- 13.Bagheri N, Azadegan-Dehkordi F, Shirzad H, Rafieian-Kopaei M, Rahimian G, Razavi A. 2015. The biological functions of IL-17 in different clinical expressions of Helicobacter pylori-infection. Microb Pathog 81:33–38. doi: 10.1016/j.micpath.2015.03.010. [DOI] [PubMed] [Google Scholar]
- 14.Caruso R, Fina D, Paoluzi OA, Del Vecchio Blanco G, Stolfi C, Rizzo A, Caprioli F, Sarra M, Andrei F, Fantini MC, MacDonald TT, Pallone F, Monteleone G. 2008. IL-23-mediated regulation of IL-17 production in Helicobacter pylori-infected gastric mucosa. Eur J Immunol 38:470–478. doi: 10.1002/eji.200737635. [DOI] [PubMed] [Google Scholar]
- 15.Shi Y, Liu X-F, Zhuang Y, Zhang J-Y, Liu T, Yin Z, Wu C, Mao XH, Jia K-R, Wang F-J, Guo H, Flavell RA, Zhao Z, Liu K-Y, Xiao B, Guo Y, Zhang W-J, Zhou W-Y, Guo G, Zou QM. 2010. Helicobacter pylori-induced Th17 responses modulate Th1 cell responses, benefit bacterial growth, and contribute to pathology in mice. J Immunol 184:5121–5129. doi: 10.4049/jimmunol.0901115. [DOI] [PubMed] [Google Scholar]
- 16.Serrano C, Wright SW, Bimczok D, Shaffer CL, Cover TL, Venegas A, Salazar MG, Smythies LE, Harris PR, Smith PD. 2013. Downregulated Th17 responses are associated with reduced gastritis in Helicobacter pylori-infected children. Mucosal Immunol 5:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song X, Zhu S, Shi P, Liu Y, Shi Y, Levin SD, Qian Y. 2011. IL-17RE is the functional receptor for IL-17C and mediates mucosal immunity to infection with intestinal pathogens. Nat Immunol 12:1151–1158. doi: 10.1038/ni.2155. [DOI] [PubMed] [Google Scholar]
- 18.Ramirez-Carrozzi V, Sambandam A, Luis E, Lin Z, Jeet S, Lesch J, Hackney J, Kim J, Zhou M, Lai J, Modrusan Z, Sai T, Lee W, Xu M, Caplazi P, Diehl L, de Voss J, Balazs M, Gonzalez L, Singh H, Ouyang W, Pappu R. 2011. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat Immunol 12:1159–1166. doi: 10.1038/ni.2156. [DOI] [PubMed] [Google Scholar]
- 19.Im E, Jung J, Rhee SH. 2012. Toll-like receptor 5 engagement induces interleukin-17C expression in intestinal epithelial cells. J Interferon Cytokine Res 32:583–591. doi: 10.1089/jir.2012.0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pfeifer P, Voss M, Wonnenberg B, Hellberg J, Seiler F, Lepper PM, Bischoff M, Langer F, Schäfers H-J, Menger MD, Bals R, Beisswenger C. 2013. IL-17C is a mediator of respiratory epithelial innate immune response. Am J Respir Cell Mol Biol 48:415–421. doi: 10.1165/rcmb.2012-0232OC. [DOI] [PubMed] [Google Scholar]
- 21.Song X, Gao H, Lin Y, Yao Y, Zhu S, Wang J, Liu Y, Yao X, Meng G, Shen N, Shi Y, Iwakura Y, Qian Y. 2014. Alterations in the microbiota drive interleukin-17C production from intestinal epithelial cells to promote tumorigenesis. Immunity 40:140–152. doi: 10.1016/j.immuni.2013.11.018. [DOI] [PubMed] [Google Scholar]
- 22.Nagashima H, Iwatani S, Cruz M, Jiménez Abreu JA, Tronilo L, Rodríguez E, Disla M, Terao H, Uchida T, Mahachai V, Vilaichone RK, Tshering L, Mitsui T, Shiota S, Graham DY, Yamaoka Y. 2015. Differences in interleukin 8 expression in Helicobacter pylori-infected gastric mucosa tissues from patients in Bhutan and the Dominican Republic. Hum Pathol 46:129–136. doi: 10.1016/j.humpath.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagashima H, Iwatani S, Cruz M, Jiménez Abreu JA, Uchida T, Mahachai V, Vilaichone R-K, Graham DY, Yamaoka Y. 2015. Toll-like receptor 10 in Helicobacter pylori infection. J Infect Dis 212:1666–1676. doi: 10.1093/infdis/jiv270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baggiolini M, Walz A, Kunkel SL. 1989. Neutrophil-activating peptide-1/interleukin 8, a novel cytokine that activates neutrophils. J Clin Investig 84:1045–1049. doi: 10.1172/JCI114265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crowe SE, Alvarez L, Dytoc M, Hunt RH, Muller M, Sherman P, Patel J, Jin Y, Ernst PB. 1995. Expression of interleukin 8 and CD54 by human gastric epithelium after Helicobacter pylori infection in vitro. Gastroenterology 108:65–74. doi: 10.1016/0016-5085(95)90009-8. [DOI] [PubMed] [Google Scholar]
- 26.Johansen C, Riis JL, Gedebjerg A, Kragballe K, Iversen L. 2011. Tumor necrosis factor α-mediated induction of interleukin 17C in human keratinocytes is controlled by nuclear factor κB. J Biol Chem 286:25487–25494. doi: 10.1074/jbc.M111.240671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnston A, Fritz Y, Dawes SM, Diaconu D, Al-Attar PM, Guzman AM, Chen CS, Fu W, Gudjonsson JE, McCormick TS, Ward NL. 2013. Keratinocyte overexpression of IL-17C promotes psoriasiform skin inflammation. J Immunol 190:2252–2262. doi: 10.4049/jimmunol.1201505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang SH, Reynolds JM, Pappu BP, Chen G, Martinez GJ, Dong C. 2011. Interleukin-17C promotes Th17 cell responses and autoimmune disease via interleukin-17 receptor E. Immunity 35:611–621. doi: 10.1016/j.immuni.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Backert S, Naumann M. 2010. What a disorder: proinflammatory signaling pathways induced by Helicobacter pylori. Trends Microbiol 18:479–486. doi: 10.1016/j.tim.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 30.Kusagaya H, Fujisawa T, Yamanaka K, Mori K, Hashimoto D, Enomoto N, Inui N, Nakamura Y, Wu R, Maekawa M, Suda T, Chida K. 2014. Toll-like receptor-mediated airway IL-17C enhances epithelial host defense in an autocrine/paracrine manner. Am J Respir Cell Mol Biol 50:30–39. [DOI] [PubMed] [Google Scholar]
- 31.Luzza F, Parrello T, Monteleone G, Sebkova L, Romano M, Zarrilli R, Imeneo M, Pallone F. 2000. Up-regulation of IL-17 is associated with bioactive IL-8 expression in Helicobacter pylori-infected human gastric mucosa. J Immunol 165:5332–5337. doi: 10.4049/jimmunol.165.9.5332. [DOI] [PubMed] [Google Scholar]
- 32.Mizuno T, Ando T, Nobata K, Tsuzuki T, Maeda O, Watanabe O, Minami M, Ina K, Kusugami K, Peek RM, Goto H. 2005. Interleukin-17 levels in Helicobacter pylori-infected gastric mucosa and pathologic sequelae of colonization. World J Gastroenterol 11:6305–6311. doi: 10.3748/wjg.v11.i40.6305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harvey BS, Sia TC, Wattchow DA, Smid SD. 2014. Interleukin 17A evoked mucosal damage is attenuated by cannabidiol and anandamide in a human colonic explant model. Cytokine 65:236–244. doi: 10.1016/j.cyto.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 34.Friedrich M, Diegelmann J, Schauber J. 2015. Intestinal neuroendocrine cells and goblet cells are mediators of IL-17A-amplified epithelial IL-17C production in human inflammatory bowel disease. Mucosal Immunol 8:943–958. doi: 10.1038/mi.2014.124. [DOI] [PubMed] [Google Scholar]
- 35.Jin J, Rha K-S, Kim DW, Kim YM. 2014. IL-17C expression in nasal epithelial cells of chronic rhinosinusitis with nasal polyposis. Eur Arch Otorhinolaryngol 271:1097–1105. doi: 10.1007/s00405-013-2683-x. [DOI] [PubMed] [Google Scholar]
- 36.Al-Samadi A, Kouri V-P, Salem A, Ainola M, Kaivosoja E, Barreto G, Konttinen YT, Hietanen J, Häyrinen-Immonen R. 2014. IL-17C and its receptor IL-17RA/IL-17RE identify human oral epithelial cell as an inflammatory cell in recurrent aphthous ulcer. J Oral Pathol Med 43:117–124. doi: 10.1111/jop.12095. [DOI] [PubMed] [Google Scholar]
- 37.van Den Brink GR, ten Kate FJ, Ponsioen FJCY, Rive MM, Tytgat GN, van Deventer SJ, Peppelenbosch MP. 2000. Expression and activation of NF-kappa B in the antrum of the human stomach. J Immunol 164:3353–3359. doi: 10.4049/jimmunol.164.6.3353. [DOI] [PubMed] [Google Scholar]
- 38.Schlaermann P, Toelle B, Berger H, Schmidt SC, Glanemann M, Ordemann J, Bartfeld S, Mollenkopf HJ, Meyer TF. 2016. A novel human gastric primary cell culture system for modelling Helicobacter pylori infection in vitro. Gut 65:202–213. doi: 10.1136/gutjnl-2014-307949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bartfeld S, Bayram T, van de Wetering M, Huch M, Begthel H, Kujala P, Vries R, Peters PJ, Clevers H. 2015. In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology 148:126–136. doi: 10.1053/j.gastro.2014.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamaoka Y. 2010. Mechanisms of disease: Helicobacter pylori virulence factors. Nat Rev Gastroenterol Hepatol 7:629–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Backert S, Blaser MJ. 2016. The role of CagA in the gastric biology of Helicobacter pylori. Cancer Res 76:4028–4031. doi: 10.1158/0008-5472.CAN-16-1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brandt S, Kwok T, Hartig R, König W, Backert S. 2005. NF-kappaB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc Natl Acad Sci U S A 102:9300–9305. doi: 10.1073/pnas.0409873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamaoka Y, Kodama T, Kashima K, Graham DY, Sepulveda AR. 1998. Variants of the 3′ region of the cagA gene in Helicobacter pylori isolates from patients with different H. pylori-associated diseases. J Clin Microbiol 36:2258–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamaoka Y, El-Zimaity HM, Gutierrez O, Figura N, Kim JG, Kodama T, Kashima K, Graham DY, Kim JK. 1999. Relationship between the cagA 3′ repeat region of Helicobacter pylori, gastric histology, and susceptibility to low pH. Gastroenterology 117:342–349. doi: 10.1053/gast.1999.0029900342. [DOI] [PubMed] [Google Scholar]
- 45.Hatakeyama M. 2004. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer 4:688–694. doi: 10.1038/nrc1433. [DOI] [PubMed] [Google Scholar]
- 46.Sayi A, Kohler E, Hitzler I, Arnold I, Schwendener R, Rehrauer H, Müller A. 2009. The CD4+ T cell-mediated IFN-gamma response to Helicobacter infection is essential for clearance and determines gastric cancer risk. J Immunol 182:7085–7101. doi: 10.4049/jimmunol.0803293. [DOI] [PubMed] [Google Scholar]
- 47.Wu Y-Y, Tsai H-F, Lin W-C, Hsu P-I, Shun C-T, Wu M-S, Hsu P-N. 2007. Upregulation of CCL20 and recruitment of CCR6+ gastric infiltrating lymphocytes in Helicobacter pylori gastritis. Infect Immun 75:4357–4363. doi: 10.1128/IAI.01660-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Allison CC, Kufer TA, Kremmer E, Kaparakis M, Ferrero RL. 2009. Helicobacter pylori induces MAPK phosphorylation and AP-1 activation via a NOD1-dependent mechanism. J Immunol 183:8099–8109. doi: 10.4049/jimmunol.0900664. [DOI] [PubMed] [Google Scholar]
- 49.Graham DY. 2015. Helicobacter pylori update: gastric cancer, reliable therapy, and possible benefits. Gastroenterology 148:719–731. doi: 10.1053/j.gastro.2015.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, Zwahlen M, Kampf C, Wester K, Hober S, Wernerus H, Björling L, Ponten F. 2010. Towards a knowledge-based human protein atlas. Nat Biotech 28:1248–1250. doi: 10.1038/nbt1210-1248. [DOI] [PubMed] [Google Scholar]
- 51.Yamaoka Y, Kodama T, Kita M, Imanishi J, Kashima K, Graham DY. 1998. Relationship of vacA genotypes of Helicobacter pylori to cagA status, cytotoxin production, and clinical outcome. Helicobacter 3:241–253. doi: 10.1046/j.1523-5378.1998.08056.x. [DOI] [PubMed] [Google Scholar]
- 52.Yamaoka Y, Osato MS, Sepulveda AR, Gutierrez O, Figura N, Kim JG, Kodama T, Kashima K, Graham DY. 2000. Molecular epidemiology of Helicobacter pylori: separation of H. pylori from East Asian and non-Asian countries. Epidemiol Infect 124:91–96. doi: 10.1017/S0950268899003209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Akopyants NS, Clifton SW, Kersulyte D, Crabtree JE, Youree BE, Reece CA, Bukanov NO, Drazek ES, Roe BA, Berg DE. 1998. Analyses of the cag pathogenicity island of Helicobacter pylori. Mol Microbiol 28:37–53. doi: 10.1046/j.1365-2958.1998.00770.x. [DOI] [PubMed] [Google Scholar]
- 54.Xia Y, Yamaoka Y, Zhu Q, Matha I, Gao X. 2009. A comprehensive sequence and disease correlation analyses for the C-terminal region of CagA protein of Helicobacter pylori. PLoS One 4:e7736-8. doi: 10.1371/journal.pone.0007736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dixon MF, Genta RM, Yardley JH, Correa P. 1996. Classification and grading of gastritis. The updated Sydney System. International Workshop on the Histopathology of Gastritis, Houston 1994. Am J Surg Pathol 20:1161–1181. [DOI] [PubMed] [Google Scholar]
- 56.Uchida T, Kanada R, Tsukamoto Y, Hijiya N, Matsuura K, Yano S, Yokoyama S, Kishida T, Kodama M, Murakami K, Fujioka T, Moriyama M. 2007. Immunohistochemical diagnosis of the cagA-gene genotype of Helicobacter pylori with anti-East Asian CagA-specific antibody. Cancer Sci 98:521–528. doi: 10.1111/j.1349-7006.2007.00415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krämer A, Green J, Pollard J, Tugendreich S. 2014. Causal analysis approaches in Ingenuity Pathway Analysis (IPA). Bioinformatics 30:523–530. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]