

ABSTRACT
Tumor progression locus 2 (Tpl2) is a serine-threonine kinase that regulates Th1 differentiation, secretion of the inflammatory cytokine gamma interferon (IFN-γ), and host defense against the intracellular pathogens Toxoplasma gondii, Listeria monocytogenes, and Mycobacterium tuberculosis. However, relatively little is known about the contribution of Tpl2 to Th17 differentiation and immune cell function during infection with an extracellular pathogen. The goal of this study was to determine whether Tpl2 influences the immune response generated to the extracellular bacterium Citrobacter rodentium, which induces a mixed Th1 and Th17 response. During peak infection with C. rodentium, Tpl2−/− mice experienced greater bacterial burdens with evidence of dissemination to the liver and spleen but ultimately cleared the bacteria within 3 weeks postinfection, similar to the findings for wild-type mice. Tpl2−/− mice also recruited fewer neutrophils and monocytes to the colon during peak infection, which correlated with increased bacterial burdens. In mixed bone marrow chimeras, Tpl2 was shown to play a T cell-intrinsic role in promoting both IFN-γ and interleukin-17A production during infection with C. rodentium. However, upon CD4 T cell transfer into Rag−/− mice, Tpl2−/− CD4 T cells were as protective as wild-type CD4 T cells against the dissemination of bacteria and mortality. These data indicate that the enhanced bacterial burdens in Tpl2−/− mice are not caused primarily by impairments in CD4 T cell function but result from defects in innate immune cell recruitment and function.
KEYWORDS: Citrobacter, T helper cells, gastrointestinal infection, intestinal immunity, neutrophils
INTRODUCTION
Citrobacter rodentium is a nonmotile Gram-negative rod that is a natural mouse and gerbil pathogen (1, 2). Upon infection, C. rodentium colonizes the large intestine, primarily the cecum and distal portion of the colon (3), and forms a close association with the epithelium and lamina propria that results in attaching and effacing lesions in the large intestine (4, 5). However, C. rodentium can disseminate out of the intestines and be found in the nasopharynx, lung, heart, liver, and spleen (6). Early innate responses to C. rodentium are associated with recruitment and the antimicrobial functions of neutrophils, macrophages, NK cells, and innate lymphoid cells (7–12). Neutrophils secrete interleukin-17A (IL-17A) and IL-22, promote the production of antimicrobial defensins by epithelial cells, and protect against the development of diarrhea (11, 13). The bacterial association with the lamina propria of the large intestine subsequently induces a mixed Th1 and Th17 response associated with IL-12, gamma interferon (IFN-γ), tumor necrosis factor (TNF), IL-17A, and IL-22 expression (14–17). Clearance of the bacteria occurs within 3 weeks in a wild-type host and is dependent upon both CD4 T cell and B cell functions (18, 19).
Tumor progression locus 2 (Tpl2; also known as MAP3K8) is a serine-threonine protein kinase that is expressed in both innate and adaptive immune cells. The role of Tpl2 in promoting an inflammatory immune response has been extensively studied in macrophages and dendritic cells (20, 21). Tpl2 has been shown to promote Th1 cell differentiation and the production of IFN-γ (22). Therefore, Tpl2−/− mice experience greater susceptibility and infectious burden in response to the protozoan parasite Toxoplasma gondii (22) or the intracellular bacteria Listeria monocytogenes (21) and Mycobacterium tuberculosis (23) than wild-type mice. However, Tpl2−/− mice are resistant to endotoxin-induced septic shock due to the reduced production of TNF (20). Because Tpl2 promotes TNF processing and secretion (20, 24), it is being investigated as a therapeutic target for treating autoimmune diseases, especially those exacerbated by TNF, such as rheumatoid arthritis (25–27).
Th17 cells are a distinct lineage of CD4+ T cells that produce IL-17A, IL-17F, IL-21, and IL-22 (28–33), with one of their main downstream functions being recruitment of neutrophils to assist with the clearance of microbes (34–36). Together, Th17 effector cytokines are required for the clearance of extracellular bacterial and fungal infections, including those caused by Citrobacter rodentium, Klebsiella pneumoniae, Staphylococcus aureus, and Candida albicans, while they also contribute to the inflammation associated with autoimmune diseases (reviewed in reference 37). We have recently shown that Tpl2 promotes Th17 cell differentiation and the secretion of IL-17A but not the secretion of IL-22 in vitro (38). However, Tpl2 has little impact on Th17 cell production of IL-17A in vivo during myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis (39) or in a T cell transfer model of colitis (38). It has yet to be investigated whether Tpl2 influences Th17 cell differentiation, IL-17A production, or neutrophil accumulation during extracellular bacterial or fungal infections. Understanding how Tpl2 regulates Th17 responses during infection may provide valuable information about the range of potential benefits or risks associated with Tpl2 inhibition in various disease settings.
Upon infection with C. rodentium, Tpl2−/− mice experienced greater bacterial burdens during peak infection than wild-type mice but were capable of clearing the bacteria within 3 weeks, similar to the findings for wild-type mice. Infection in Tpl2−/− mice was not confined to the intestines and was also detected in the liver and spleen of infected mice. At 11 days postinfection (dpi), lymphocytes in the lamina propria expressed IL-17A, IL-22, and IFN-γ, with Tpl2−/− CD4 T cells trending toward reduced proportions of IL-17A-, IL-22-, and IFN-γ-positive cells. This defect was confirmed to be intrinsic to T cells, as Tpl2−/− CD4 T cells in mixed bone marrow chimeras were less likely to differentiate into Th1 and Th17 cells expressing IL-17A and IFN-γ, respectively. Despite this T cell-intrinsic defect, Tpl2−/− CD4 T cells transferred into Rag−/− mice were as protective as wild-type CD4 T cells in preventing bacterial dissemination and mortality, suggesting critical T cell-extrinsic functions for Tpl2 in protection against C. rodentium infection. Interestingly, the colons of Tpl2−/− mice had reduced inflammation relative to those of wild-type mice, indicating that impaired neutrophil recruitment and function may contribute to enhanced bacterial burdens in Tpl2−/− mice. Overall, our findings confirm the importance of Tpl2 in driving the development of the proinflammatory Th1 lineage as well as promoting IL-17A expression and neutrophil recruitment during infection with extracellular bacteria.
RESULTS
Tpl2−/− mice have greater bacterial burdens and dissemination than wild-type mice during peak infection with Citrobacter rodentium.
Wild-type and Tpl2−/− mice were infected with 2 × 109 CFU of Citrobacter rodentium ICC180. Bioluminescent images from the gastrointestinal region were collected throughout infection until clearance of the bacteria at 21 days postinfection (dpi). Infection was confirmed by measuring the fecal burdens of C. rodentium. Beginning at 7 dpi, Tpl2−/− mice had significantly greater bacterial burdens than wild-type mice, as detected by changes in luminescence (Fig. 1A and B). This trend continued until approximately 12 to 14 dpi, at which point both wild-type and Tpl2−/− mice similarly cleared the bacteria (Fig. 1).
FIG 1.
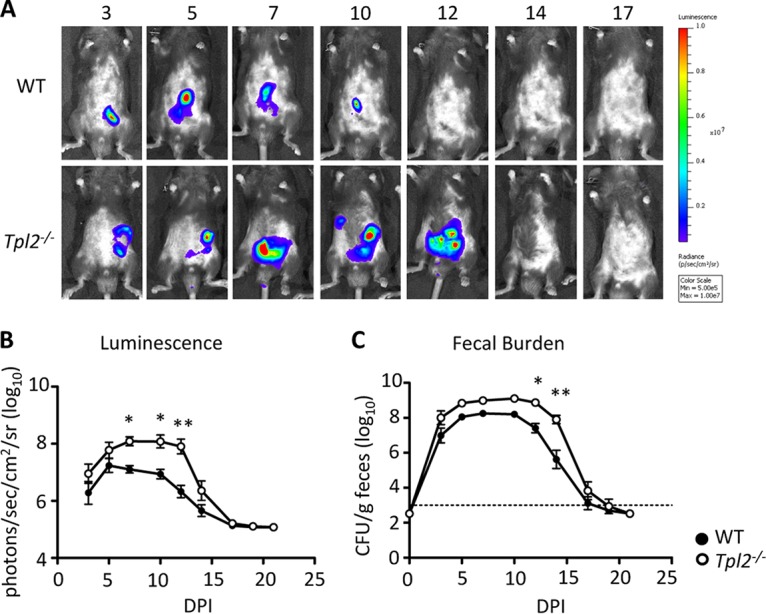
Tpl2−/− mice have a greater bacterial burden during peak infection. Wild-type (WT) and Tpl2−/− mice were gavaged with 2 × 109 CFU of Citrobacter rodentium (ICC180). Bioluminescent images from the gastrointestinal region are displayed as pseudocolor images, with variations in color representing the light intensity at a given location. Red represents the most intense light emission, while purple corresponds to the weakest signal. (A and B) Representative (A) and pooled (B) luminescence data for wild-type and Tpl2−/− mice from 3 to 21 dpi are shown. (C) The fecal burden was quantified at 0 to 21 dpi. Dashed line, limit of detection. When no fecal burden was detected, a value of 3 × 102 CFU/g was assigned. Data from two independent experiments were pooled (n = 8 mice). Error bars represent SEMs. P values were determined by two-way ANOVA. *, P < 0.05; **, P < 0.005.
C. rodentium is known to disseminate out of the intestines (6). Because Tpl2−/− mice showed greater luminescence than wild-type mice at 7 to 12 dpi (Fig. 1B) but no differences in fecal burdens between Tpl2−/− and wild-type mice were seen until 12 dpi (Fig. 1C), we hypothesized that greater bacterial dissemination was occurring in Tpl2−/− mice. As expected, Tpl2−/− mice had greater bacterial burdens in the liver and spleen than wild-type mice at 8 and 11 dpi (Fig. 2C and D). Pinpoint lesions were also more frequently observed on the livers of Tpl2−/− mice than on those of wild-type mice (Fig. 2A and B), although differential aerobic culture of pinpoint lesions did not result in bacterial growth (data not shown). Combined, Tpl2−/− mice not only showed greater bacterial burdens in the intestines than wild-type mice (Fig. 1) but also showed greater dissemination to other organs, including the liver and spleen (Fig. 2). Increased C. rodentium dissemination is consistent with the findings in our recent report describing the dissemination of the commensal bacterium Staphylococcus xylosus, normally found on the skin of healthy mice, into the lungs and lymph nodes of Rag−/− Tpl2−/− mice (40).
FIG 2.
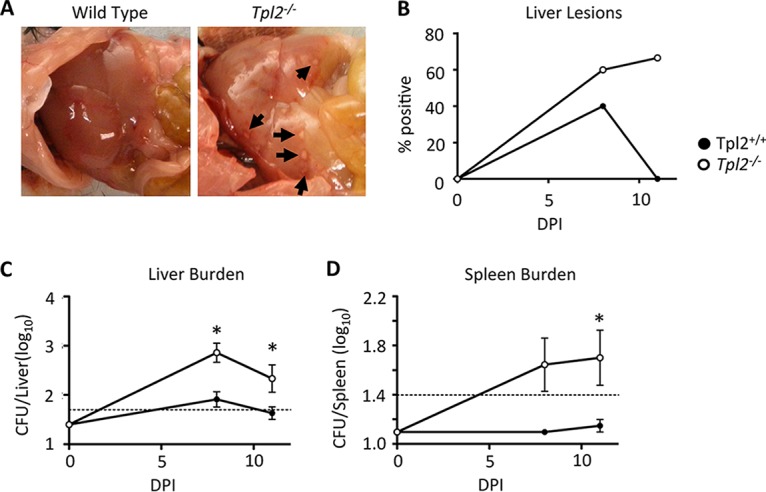
Tpl2−/− mice have greater bacterial dissemination during peak infection. Wild-type and Tpl2−/− mice were gavaged with 2 × 109 CFU Citrobacter rodentium (ICC180). Mice were euthanized at 8 or 11 dpi. Livers and spleens were collected, assessed for lesions (A and B), and homogenized to measure the bacterial burden (C and D). Dashed lines, limit of detection. Data from two or more independent experiments per time point were pooled (n ≥ 4 mice). *, P < 0.05 by two-way ANOVA.
The greater dissemination of C. rodentium indicates that Tpl2 may regulate intestinal permeability. Claudins aid in maintenance of the integrity of the epithelial barrier through the formation of tight junctions. Because Tpl2 signals upstream of the MEK/extracellular signal-regulated kinase (ERK) pathway (41) and the expression of claudins 2 and 5 on epithelial cell lines requires signaling through the MEK/ERK pathway (42, 43), we hypothesized that the expression of these claudins may be reduced by Tpl2 deficiency. Tpl2−/− mice showed a significant reduction in claudin 2 expression compared to wild-type mice at 8 dpi (Fig. 3A). A similar trend was also seen for claudin 5, although this was not statistically significant. By 11 dpi, both wild-type and Tpl2−/− mice showed a further reduction in the levels of expression of claudin 2 and claudin 5 (Fig. 3A). We hypothesized that an early reduction in the level of claudin expression during infection of Tpl2−/− mice may impair their intestinal barrier function and contribute to the greater bacterial dissemination seen in these mice (Fig. 2). To evaluate intestinal permeability directly, wild-type and Tpl2−/− mice were orally gavaged with fluorescein isothiocyanate (FITC)-dextran at 8 dpi. As expected, mice infected with C. rodentium had greater intestinal permeability with higher concentrations of circulating FITC-dextran (Fig. 3B). However, despite the reduced level of claudin expression in Tpl2−/− mice, no corresponding increase in intestinal permeability was detected in Tpl2−/− mice compared to wild-type mice. We therefore conclude that the greater bacterial burdens within the intestines of Tpl2−/− mice are likely sufficient to promote greater dissemination without a measurable change in barrier function per se.
FIG 3.

Tpl2 regulates claudin expression during C. rodentium infection. (A) Wild-type and Tpl2−/− mice were gavaged with 2 × 109 CFU Citrobacter rodentium (ICC180). Mice were euthanized at 8 or 11 dpi. The relative levels of expression of claudin 2 (Cldn2) and claudin 5 (Cldn5) in the colon were measured. Data from two or more independent experiments per time point were pooled (n ≥ 8 mice). *, P < 0.05 by two-way ANOVA. (B) Wild-type and Tpl2−/− mice were gavaged with 2 × 109 CFU of Citrobacter rodentium (ICC180) followed by FITC-dextran at 8 dpi. The concentrations of FITC-dextran in serum were quantified at 4 h posttreatment. Outliers were excluded from analysis using Grubb's test. **, P < 0.005 compared to control mice using one-way ANOVA.
Tpl2 enhances Th1 and Th17 responses to C. rodentium via a T cell-intrinsic mechanism.
The elevated bacterial burdens in Tpl2−/− mice at 8 to 12 dpi (Fig. 1B), as detected by luminescence, indicated a Tpl2-dependent defect in the adaptive immune response. As expected, lymphocytes in the lamina propria expressed IL-17A, IL-22, and IFN-γ (14–17). Compared to wild-type mice, Tpl2−/− mice consistently trended toward reduced levels of induction of Th1 and Th17 cells expressing IL-17A, IL-22, and IFN-γ (Fig. 4A and B). IL-6, IL-23, and transforming growth factor β are known to promote Th17 cell differentiation (44–47). Despite the elevated levels of cytokine production within the lamina propria lymphocytes (LPLs) at 11 dpi compared to sham-infected controls (Fig. 4B), the mRNA expression levels of Il23a and Tgfb were reduced to similar levels in the colon tissue of C. rodentium-infected wild-type and Tpl2−/− mice compared to sham-infected mice, whereas Il6 was more highly expressed in the colons of Tpl2−/−-infected mice than those of wild-type mice (see Fig. S1 in the supplemental material).
FIG 4.
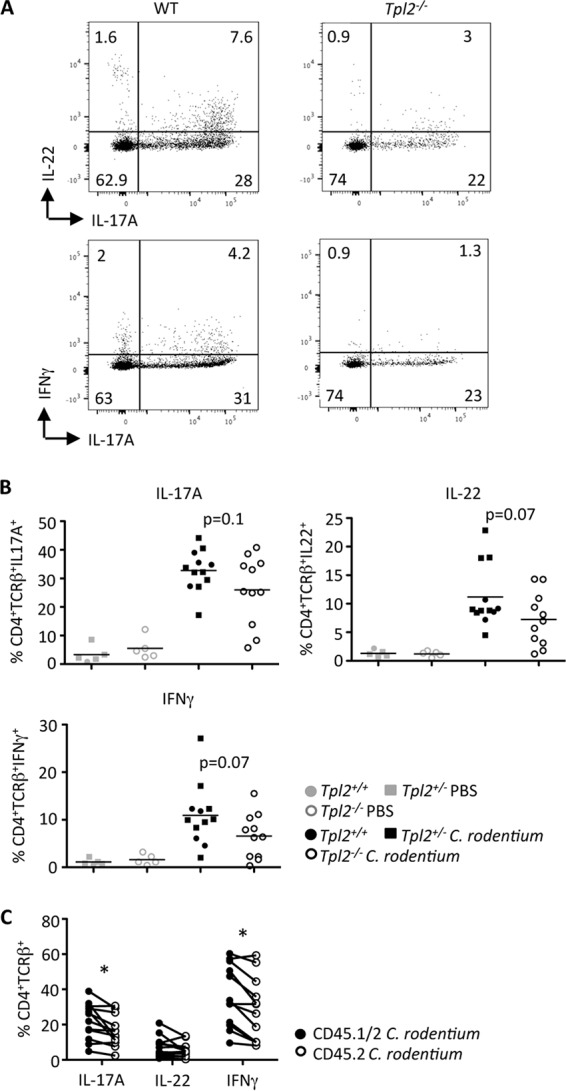
Tpl2 promotes a Th1/Th17 response. Wild-type (Tpl2+/+ and Tpl2+/−) and Tpl2−/− mice (A and B) or mixed bone marrow chimeras (C) were gavaged with 2 × 109 CFU Citrobacter rodentium (ICC180). Representative plots of LPLs isolated at 11 dpi (A) and stained for wild-type or Tpl2−/− CD4+ TCRβ+ cells expressing IL-17A, IL-22, or IFN-γ (A and B) are shown. The values in panel A represent percentages. The lines in panel B represent means (n ≥ 5 mice). (C) Relative frequency of CD45.1+ CD45.2+ wild-type or CD45.1− CD45.2+ Tpl2−/− CD4+ TCRβ+ cells expressing IL-17A, IL-22, or IFN-γ. Connecting lines represent percent cytokine-positive CD4 T cells from within the same host. Data from two or more independent experiments were pooled (n = 12 mice). *, P < 0.05 by paired Student's t test.
To determine whether Tpl2 influences T helper cell polarization in vivo via a T cell-intrinsic mechanism, mixed bone marrow chimeras were generated and similarly infected with C. rodentium. In mixed bone marrow chimeras, a significantly lower proportion of Tpl2−/− T cells (CD45.2+) than wild-type T cells (CD45.1/2+) in the same host expressed IL-17A and IFN-γ (Fig. 4C). However, Tpl2−/− T cells were similarly able to express IL-22 as wild-type T cells (Fig. 4C). These data confirm an accessory role for Tpl2 in Th1 cell differentiation and IFN-γ expression as well as Th17 cell differentiation and expression of IL-17A but not expression of IL-22 in response to C. rodentium infection.
Tpl2−/− CD4 T cells adoptively transferred into Rag1−/− mice are protective against C. rodentium.
To evaluate whether defects in Th1 and Th17 cell differentiation in the large intestines influenced the disease outcome, wild-type or Tpl2−/− CD4 T cells were adoptively transferred into Rag1−/− mice. At approximately 2 to 3 weeks posttransfer, mice were infected with a low dose of C. rodentium. Because Rag−/− mice are deficient in T cells and B cells (48), they are incapable of clearing the bacteria and quickly succumb to infection (49). The recipients of either wild-type or Tpl2−/− CD4 T cells survived up to 21 dpi (Fig. 5A) and had elevated bacterial burdens similar to those in Rag1−/− mice not receiving wild-type or Tpl2−/− CD4 T cells (Fig. 5B). The localization of C. rodentium was visualized using luminescent images taken throughout the time course (Fig. 5C), and dissemination was quantified at the time of death. Notably, transfer of either wild-type (50) or Tpl2−/− CD4 T cells into Rag1−/− mice partially restored the intestinal barrier function to Rag1−/− mice. Rag1−/− mice that received either wild-type or Tpl2−/− CD4 T cells had similarly reduced levels of dissemination of C. rodentium into the blood, liver, and spleen (Fig. 5D). For most Rag1−/− mice that received CD4 T cells, C. rodentium was no longer detected in the circulation (Fig. 5D). These results indicate that Tpl2−/− CD4 T cells are as protective as wild-type CD4 T cells upon transfer into Rag1−/− mice and that Tpl2 primarily functions in a CD4 T cell-extrinsic manner to influence bacterial burdens and dissemination during C. rodentium infection.
FIG 5.

Tpl2−/− CD4 cells are as protective as wild-type CD4 cells once they are transferred into Rag−/− mice. Rag−/− mice receiving wild-type CD4 cells, Tpl2−/− CD4 cells, or no CD4 cells were infected with low-dose Citrobacter rodentium. (A) Percent survival is shown. Body weights were recorded, and mice exhibiting severe signs of disease, including more than 20% weight loss, were euthanized. (B and C) Pooled luminescence data and fecal burden (B) plus representative images (C) from 3 to 21 dpi are shown. Dashed lines, limit of detection. Error bars represent SEMs. (D) Dissemination into the blood, liver, and spleen was quantified at the time of death. Lines represents means. Dashed lines, limits of detection for blood (top), liver (middle), and spleen (bottom). Data from two independent experiments were pooled (n ≥ 5 mice). P values were determined by one-way ANOVA. *, P < 0.05 comparing Rag−/− mice with or without CD4 cells; **, P < 0.005 comparing Rag−/− mice with or without CD4 cells.
Tpl2 promotes neutrophil accumulation in the colon during C. rodentium infection.
Because Tpl2 expression by CD4 T cells did not influence the total bacterial burdens in Rag1−/− mice (Fig. 5B), we next investigated the innate immune responses generated during peak infection. C. rodentium infection induces neutrophil recruitment into the large intestines (11, 12). Neutrophils not only phagocytose and kill bacteria (11, 12) but also contribute to inflammation and pathology (51). A comparison of the pathology between wild-type and Tpl2−/− mice showed reduced total inflammation in the large intestines of Tpl2−/− mice at 8 and 11 dpi (Fig. 6A and B), despite the significantly increased bacterial burdens over the same time period (Fig. 1A and B). The histopathology of colons isolated from C. rodentium-infected Tpl2−/− mice also showed colonic glands that were missing or markedly dilated due to the accumulation of cellular debris and large numbers of bacteria (Fig. 6A). Accordingly, an evaluation of innate immune cell infiltrates showed that reduced proportions of neutrophils and monocytes (Fig. 6C) were recruited to the epithelial barrier of the colons of Tpl2−/− mice during infection. Consistent with this finding, a 1:1 ratio of neutrophils and monocytes was observed in colon sections of wild-type and Tpl2−/− mice infected with C. rodentium (data not shown). Tpl2 did not significantly impact the recruitment of dendritic cells into the epithelial layer (Fig. 6C). The reduced levels of neutrophil recruitment in Tpl2−/− mice are consistent with previous reports of impaired neutrophil recruitment in models of inflammation primarily due to factors extrinsic to neutrophils (52–55).
FIG 6.

Tpl2−/− mice have reduced neutrophil recruitment to the colon during the adaptive immune response. Wild-type and Tpl2−/− mice were gavaged with 2 × 109 CFU Citrobacter rodentium (ICC180). Mice were euthanized at 8 or 11 dpi. (A) Representative images are given. The inflammatory cells in the colons at 11 dpi are mostly histiocytes (red dashed arrows) and lymphocytes (black solid arrows), with some neutrophils (red arrowheads) being seen. Asterisks denote areas where the intestinal glands are missing. The solid red arrow points to a colonic gland that is markedly dilated due to the accumulation of cellular debris and large numbers of bacteria in the lumen of the colonic gland. Magnifications, ×200. (B and C) Colons were isolated and scored for pathology (B), and the proportion of myeloid cells in the epithelial layer was quantified by gating on CD45+ events (C). Lines represent means (n ≥ 3 mice). (B) Data from three independent experiments were pooled. (C) Data are representative of those from one (monocytes and dendritic cells) or four (neutrophils) independent experiments. P values were determined by unpaired Student's t test (B) and one-way ANOVA (C). *, P < 0.05.
DISCUSSION
Understanding how Tpl2 regulates Th17 responses during infection may provide valuable information about the range of potential benefits or risks associated with Tpl2 inhibition in various disease settings. In this study, we investigated the role of Tpl2 during host defense against the Gram-negative extracellular bacterium Citrobacter rodentium, which induces a mixed Th1 and Th17 response for protection. During peak infection with C. rodentium, Tpl2−/− mice had greater bacterial burdens than wild-type mice, as detected by luminescence and fecal burdens. Interestingly, the colons of Tpl2−/− mice had reduced levels of inflammation and recruitment of neutrophils and monocytes relative to wild-type mice, indicating that impaired innate immune cell recruitment and function may contribute to enhanced bacterial burdens in Tpl2−/− mice. Even with greater bacterial burdens and dissemination, Tpl2−/− mice were as capable as wild-type mice of clearing the bacteria within 3 weeks. Evaluation of Tpl2−/− CD4 T cells in the lamina propria of mixed bone marrow chimeras indicated a T cell-intrinsic role for Tpl2 in IL-17A and IFN-γ expression by CD4 T cells. However, Tpl2−/− CD4 T cells transferred into Rag−/− mice were as protective as wild-type CD4 T cells against bacterial dissemination and mortality, suggesting critical T cell-extrinsic functions for Tpl2 in protection against C. rodentium infection.
Tpl2 has previously been shown to promote Th1 cell differentiation and the production of IFN-γ in vitro and in vivo (22), as well as Th17 cell differentiation and the secretion of IL-17A but not the secretion of IL-22 in vitro (38). Accordingly, Tpl2−/− mice are more susceptible than wild-type mice to the Th1-inducing intracellular pathogens Toxoplasma gondii (22), Listeria monocytogenes (21), and Mycobacterium tuberculosis (23). M. tuberculosis is known to induce a mixed Th1/Th17 response (reviewed in reference 56), but whether Tpl2 impacted Th17 cell differentiation in this model was not investigated. Consistent with our previous reports of Tpl2 regulating Th1 and Th17 cell differentiation, Tpl2−/− CD4 T cells in the lamina propria were less likely to differentiate into Th1 and Th17 cells expressing IFN-γ and IL-17A, respectively, during infection with C. rodentium. In contrast, Tpl2 did not influence CD4 T cell expression of the Th17-associated cytokine IL-22 during infection. These findings are similar to those observed for Th17 cells cultured in vitro, in which IL-17A expression was significantly reduced but the expression of IL-22 or the Th17-associated transcription factors RORα, RORγt, and interferon regulatory factor 4 (IRF4) was unaffected (38), suggesting a specific defect in IL-17A expression rather than a global defect in Th17 cell differentiation.
The greater dissemination of C. rodentium into the organs of Tpl2−/− mice than into those of wild-type mice initially suggested that Tpl2 may regulate the permeability of the intestinal barrier. Lymphocytes in the lamina propria are known to assist with maintenance of the intestinal barrier by promoting epithelial cell differentiation (50); therefore, CD4 T cell- and B cell-deficient mice display a reduced ability to limit the dissemination of C. rodentium (57). However, because Rag1−/− mice that received either wild-type or Tpl2−/− CD4 T cells had similarly reduced levels of dissemination into the liver and spleen, Tpl2 expression by CD4 T cells did not appear to influence intestinal permeability. The absence of B cells and antibody production during infection with C. rodentium is associated with a significantly delayed clearance as well as an enhanced fecal burden over time (19, 58). B cell proliferation, activation, and secretion of antibodies are initialized through cross-linking of the CD40 expressed on the surface of B cells (reviewed in reference 59), which signals via Tpl2 to activate ERK and promote class switching to IgE (60). However, Tpl2 ablation does not impair B cell activation, proliferation, or secretion of IgG1 (60). Therefore, although B cells do not appear to influence bacterial clearance due to the ability of Tpl2−/− mice to clear infection similarly to wild-type mice, we cannot exclude the possibility that Tpl2-deficient B cells contribute to greater bacterial burdens and higher levels of dissemination at 2 weeks postinfection.
Prior to induction of an adaptive immune response, innate cells in the intestines respond to the pathogen-associated molecular patterns expressed by C. rodentium, including lipopolysaccharide (LPS) and lipoproteins that activate Toll-like receptor 2 (TLR2) (61) and TLR4 (62), leading to downstream signaling through MyD88. TLR2-, TLR4-, and MyD88-deficient mice infected with C. rodentium have significant mortality associated with epithelial barrier damage and reduced barrier function (12, 51, 61, 62). Because Tpl2 is activated downstream of TLR2 and TLR4, among others (20, 21, 63, 64), we would expect a severe pathology and reduced barrier function in Tpl2−/− mice. Contrary to this expectation, Tpl2−/− mice did not have a pathology more severe than that in wild-type mice but did have enhanced bacterial dissemination. Claudins are known to play a significant role in maintaining intestinal integrity through the formation of tight junctions. Claudin 2 and claudin 5 require signaling through the MEK/ERK pathway for protein expression on epithelial cell lines (42, 43), and expression of the genes for claudin 1 and 2 can be upregulated by IL-17A stimulation (42). Because Tpl2 signals upstream of the MEK/ERK pathway (41) and contributes to IL-17A production in vitro (38) and in vivo (this study), we tested the possibility that Tpl2 regulates intestinal barrier integrity by regulating the expression of claudin 2 and/or 5 on the surface of epithelial cells. Indeed, the expression of claudin 2 was significantly reduced and the expression of claudin 5 trended toward a reduction in Tpl2−/− mouse colon tissue at 8 dpi. Despite the reduction in claudin expression, intestinal permeability was similarly enhanced by infection in both wild-type and Tpl2−/− mice. It is possible that differences in intestinal permeability may exist at earlier time points or with different particle sizes that were not examined in the present study. However, it is likely that the greater bacterial burdens within the intestines of Tpl2−/− mice are sufficient to promote increased dissemination without a measurable change in barrier function per se.
Even with greater bacterial burdens, Tpl2−/− mice had reduced pathology and neutrophil recruitment to the large intestine compared with those in wild-type mice. These results are consistent with the finding that Tpl2 regulates the expression of inflammatory cytokines and chemokines and the recruitment of neutrophils to sites of inflammation (52–55, 65). The reduced intestinal pathology in Tpl2−/− mice may also be due to greater dissemination, which would induce a more diffuse and less localized inflammatory response in the host. Because a reduction in intestinal pathology was observed during the adaptive phase of the immune response, defects in neutrophil recruitment in Tpl2−/− mice may be an indirect result of reduced TNF production and/or reduced signaling in Tpl2−/− mice in response to LPS (20), as well as reduced IL-17A production. IL-17A and TNF in combination enhance neutrophil recruitment through elevated secretion of CXCL1, CXCL2, CXCL8, granulocyte-macrophage colony-stimulating factor, and granulocyte colony-stimulating factor (36, 66, 67). Similarly to Tpl2−/− mice, Cxcr2−/− mice, which are deficient in neutrophil recruitment, showed elevated fecal burdens at 2 weeks postinfection with C. rodentium as well as greater dissemination of the bacteria into the liver and spleen (11). These data suggest that the observed defects in neutrophil recruitment into the large intestine of Tpl2−/− mice may explain both the greater bacterial burdens and the increased dissemination in Tpl2−/− mice.
Overall, our findings underscore that Tpl2 is a contributor to the development of the proinflammatory Th1 lineage as well as promotes IL-17A expression. These results also confirm the importance of Tpl2 in promoting neutrophil recruitment and the development of pathology during inflammation. Furthermore, they highlight the important role of Tpl2 in the prevention of bacterial dissemination.
MATERIALS AND METHODS
Mice.
Wild-type C57BL/6 and Rag1−/− mice were obtained from The Jackson Laboratory. B6-Ly5.1/Cr (CD45.1+) mice were obtained from Charles River Laboratories and were intercrossed with wild-type mice to generate heterozygous CD45.1/CD45.2 mice at the University of Georgia (UGA). Tpl2-deficient mice backcrossed more than 10 generations onto the C57BL/6 genetic background were kindly provided by Philip Tsichlis and Thomas Jefferson University. Tpl2+/− matings generated the Tpl2+/+ and Tpl2−/− mice used for infections. Animals were used at 6 to 12 weeks of age and were age and sex matched for individual experiments. All experiments involving mice were performed according to the University of Georgia guidelines for laboratory animals and were approved by the UGA Institutional Animal Care and Use Committee.
Adoptive transfer and bone marrow chimeras.
Cells from the spleens and lymph nodes from wild-type or Tpl2−/− mice were disaggregated by pressing them through a 70-μm-pore-size filter, and CD4 cells were purified by negative selection using a CD4 isolation kit (Stemcell Technologies, Vancouver, Canada) according to the manufacturer's guidelines. CD4 cells (2 × 106 to 3 × 106) were transferred into Rag1−/− mice intravenously (i.v.). At 2 to 3 weeks posttransfer, the reconstitution of CD4 cells was measured by tail bleed. CD45.1+ mice were lethally irradiated with a dose of 1,100 rads. On the following day, the mice were reconstituted with bone marrow from CD45.1+ CD45.2+ and Tpl2−/− mice. Bone marrow was isolated from the femurs and tibiae of naive mice. T cells were depleted using CD3-biotin (clone number 145-2C11; eBioscience) and antibiotin (Miltenyi Biotech, Auburn, CA) microbeads. The negative fraction, devoid of T cells, was collected using an AutoMACS separator (Miltenyi Biotech) according to the manufacturer's guidelines. Cells were counted and mixed, and 3 × 106 to 4 × 106 mixed bone marrow cells were injected into CD45.1+ mice i.v.
Citrobacter rodentium infection and burden quantification.
The C. rodentium strain used was a luminescent strain (ICC180) kindly provided by Gad Frankel at Imperial College, London, United Kingdom (68). Mice were inoculated with a low dose (1 × 107 to 2 × 107 CFU) or a high dose (1 × 109 to 2 × 109 CFU) in a total volume of 200 μl via a gastric gavage. The dose was confirmed by retrospective plating on LB agar plates. For quantification of the bacterial burden, feces were diluted in 100 μl phosphate-buffered saline (PBS) per 0.01 g feces, spleens were homogenized in 1 ml PBS, livers were homogenized in 2 ml PBS, and 100 μl of blood was immediately diluted in 900 μl PBS followed by serial dilution and plating on LB agar in triplicate. The plates were imaged for luminescent colonies using an IVIS imager (PerkinElmer), and the bacteria were counted to determine the number of CFU per gram of feces, the number of CFU per spleen, the number of CFU per liver, and the number of CFU per milliliter of blood. The limit of detection was set at 103 CFU/g of feces, 500 CFU/ml blood, 50 CFU/liver, and 25 CFU/spleen. For imaging, mice were anesthetized using either tribromoethanol or isoflurane and imaged for 1 min using an IVIS Lumina imager (PerkinElmer) to collect luminescence data.
Intraepithelial cells and LPLs.
Lamina propria lymphocytes (LPLs) were purified from the colons of mice as previously described (69, 70). For isolation of intraepithelial cells, the colons were cut into fragments and washed 3 times with RPMI 1640 medium containing 5% fetal calf serum (FCS; Invitrogen) and 5 mM EDTA (Fisher Scientific) for 15 min at 37°C in a shaking incubator. For LPLs, tissue was further subjected to two digestions with 0.5 mg/ml collagenase (Sigma-Aldrich) and 0.1 mg/ml DNase (Roche) in RPMI 1640 medium containing 5% FCS and 15 mM HEPES (Invitrogen) with continuous shaking at 37°C for 20 min. Supernatants from each digestion were passed through a 70-μm-pore-size cell strainer. Lymphocytes were enriched by Percoll (GE Healthcare) gradient purification using a 30-45-70% gradient and collection of the cells at the 45%-70% interface. Cells were stimulated for 4 h at 37°C with 50 ng/ml phorbol myristate acetate (Sigma-Aldrich), 0.5 μg/ml ionomycin (Sigma-Aldrich), and Golgi transport inhibitor (BD Biosciences) according to the manufacturers' specifications. The following anti-mouse immunoglobulin monoclonal antibodies used were from eBioscience: CD16/CD32 (antibody 93), CD45.1 (antibody A20), CD45.2 (antibody 104), CD4 (antibody RM4-5), TCRβ (antibody H57-597), Gr-1 (antibody RB6-8C5), CD11b (antibody M1/70), CD11c (antibody N418), Ly-6C (antibody HK1.4), IL-17A (antibody eBio17B7), IFN-γ (antibody XMG1.2), and IL-22 (antibody 1H8PWSR). Samples were run on a BD LSRII flow cytometer, and the results were analyzed using FlowJo software (Tree Star, Inc., Ashland, OR).
Pathology.
Colonic sections from mice were collected and fixed in 10% neutral buffered formalin for 24 h at room temperature. Complete cross sections of formalin-fixed intestinal tissue sections were placed in cassettes, embedded in paraffin, sectioned to a thickness of 4 μm, mounted on glass slides, and stained with hematoxylin and eosin. Histological sections were evaluated by a veterinary pathologist (T.N.) and scored according to the following criteria: for the distribution of inflammation, 0 indicated no inflammation, 1 indicated focal inflammation, 2 indicated multifocal inflammation, and 3 indicated diffuse inflammation; for the degree of inflammation, 0 indicated no inflammation, 1 indicated mild inflammation, 2 indicated moderate inflammation, and 3 indicated severe inflammation; and for the extent of erosion and/or ulceration, 0 indicated no erosion and/or ulceration, 1 indicated superficial erosion and/or ulceration (erosion and/or ulceration in the lamina propria only), 2 indicated moderate erosion and/or ulceration (erosion and/or ulceration extends to the submucosa), and 3 indicated severe erosion and/or ulceration (transmural erosion and/or ulceration). Scores were pooled to give a total pathology score.
FITC-dextran assay.
Food and water were withdrawn, and mice were gavaged with 150 μl the permeability tracer FITC-dextran (Sigma-Aldrich) at a concentration of 80 mg/ml. Blood was collected via cardiac puncture or the tail vein at 4 h posttreatment, and the FITC-dextran concentration in serum was measured with a fluorescence spectrophotometer using emission and excitation wavelengths of 485 nm and 535 nm, respectively. Concentrations were calculated from standard curves generated by serial dilution of FITC-dextran.
RNA isolation and reverse transcription-PCR.
RNA was isolated from colon tissue using an EZ-RNA extraction kit (Omega Bio-Tek, Norcross, GA) and converted to cDNA using a high-capacity cDNA reverse transcription kit (Life Technologies). The relative levels of Il6, Il22, Il23a, Il12b, Il23r, Tgfb, Cldn2, and Cldn5 expression were measured using a SensiFAST Probe Hi-ROX kit (Bioline, Taunton, MA) and predesigned TaqMan probe and primer sets (Applied Biosystems, Grand Island, NY). Samples were run on a StepOnePlus quantitative PCR machine (Applied Biosystems). The results given are relative to those for wild-type sham-infected controls and the actin housekeeping gene using the ΔΔCT threshold cycle (CT) method.
Statistics.
P values were derived by paired or unpaired Student's t test, the log-rank test, one-way analysis of variance (ANOVA), or two-way ANOVA, as indicated in the figure legends, using Prism software. Differences were considered statistically significant if P was ≤0.05.
Supplementary Material
ACKNOWLEDGMENTS
We thank Monica LaGatta for excellent technical support and maintenance of our mouse colony as well as UGA's Veterinary Medicine Central Animal Facility for animal care.
The research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number R01AI099058 to W.T.W.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00193-17.
REFERENCES
- 1.Schauer DB, Zabel BA, Pedraza IF, O'Hara CM, Steigerwalt AG, Brenner DJ. 1995. Genetic and biochemical characterization of Citrobacter rodentium sp. nov. J Clin Microbiol 33:2064–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de la Puente-Redondo VA, Gutiérrez-Martín CB, Pérez-Martínez C, del Blanco NG, García-Iglesias MJ, Pérez-García CC, Rodríguez-Ferri EF. 1999. Epidemic infection caused by Citrobacter rodentium in a gerbil colony. Vet Rec 145:400–403. doi: 10.1136/vr.145.14.400. [DOI] [PubMed] [Google Scholar]
- 3.Wiles S, Clare S, Harker J, Huett A, Young D, Dougan G, Frankel G. 2004. Organ specificity, colonization and clearance dynamics in vivo following oral challenges with the murine pathogen Citrobacter rodentium. Cell Microbiol 6:963–972. doi: 10.1111/j.1462-5822.2004.00414.x. [DOI] [PubMed] [Google Scholar]
- 4.Schauer DB, Falkow S. 1993. Attaching and effacing locus of a Citrobacter freundii biotype that causes transmissible murine colonic hyperplasia. Infect Immun 61:2486–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schauer DB, Falkow S. 1993. The eae gene of Citrobacter freundii biotype 4280 is necessary for colonization in transmissible murine colonic hyperplasia. Infect Immun 61:4654–4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barthold SW, Coleman GL, Bhatt PN, Osbaldiston GW, Jonas AM. 1976. The etiology of transmissible murine colonic hyperplasia. Lab Anim Sci 26(6 Pt 1):889–894. [PubMed] [Google Scholar]
- 7.Schreiber HA, Loschko J, Karssemeijer RA, Escolano A, Meredith MM, Mucida D, Guermonprez P, Nussenzweig MC. 2013. Intestinal monocytes and macrophages are required for T cell polarization in response to Citrobacter rodentium. J Exp Med 210:2025–2039. doi: 10.1084/jem.20130903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hall LJ, Murphy CT, Hurley G, Quinlan A, Shanahan F, Nally K, Melgar S. 2013. Natural killer cells protect against mucosal and systemic infection with the enteric pathogen Citrobacter rodentium. Infect Immun 81:460–469. doi: 10.1128/IAI.00953-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kum WWS, Lo BC, Deng W, Ziltener HJ, Finlay BB. 2010. Impaired innate immune response and enhanced pathology during Citrobacter rodentium infection in mice lacking functional P-selectin. Cell Microbiol 12:1250–1271. doi: 10.1111/j.1462-5822.2010.01466.x. [DOI] [PubMed] [Google Scholar]
- 10.Geddes K, Rubino SJ, Magalhaes JG, Streutker C, Le Bourhis L, Cho JH, Robertson SJ, Kim CJ, Kaul R, Philpott DJ, Girardin SE. 2011. Identification of an innate T helper type 17 response to intestinal bacterial pathogens. Nat Med 17:837–844. doi: 10.1038/nm.2391. [DOI] [PubMed] [Google Scholar]
- 11.Spehlmann ME, Dann SM, Hruz P, Hanson E, McCole DF, Eckmann L. 2009. CXCR2-dependent mucosal neutrophil influx protects against colitis-associated diarrhea caused by an attaching/effacing lesion-forming bacterial pathogen. J Immunol 183:3332–3343. doi: 10.4049/jimmunol.0900600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lebeis SL, Bommarius B, Parkos CA, Sherman MA, Kalman D. 2007. TLR signaling mediated by MyD88 is required for a protective innate immune response by neutrophils to Citrobacter rodentium. J Immunol 179:566–577. doi: 10.4049/jimmunol.179.1.566. [DOI] [PubMed] [Google Scholar]
- 13.Zindl CL, Lai J-F, Lee YK, Maynard CL, Harbour SN, Ouyang W, Chaplin DD, Weaver CT. 2013. IL-22-producing neutrophils contribute to antimicrobial defense and restitution of colonic epithelial integrity during colitis. Proc Natl Acad Sci U S A 110:12768–12773. doi: 10.1073/pnas.1300318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, Fujikado N, Tanahashi Y, Akitsu A, Kotaki H, Sudo K, Nakae S, Sasakawa C, Iwakura Y. 2009. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 30:108–119. doi: 10.1016/j.immuni.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 15.Higgins LM, Frankel G, Douce G, Dougan G, MacDonald TT. 1999. Citrobacter rodentium infection in mice elicits a mucosal Th1 cytokine response and lesions similar to those in murine inflammatory bowel disease. Infect Immun 67:3031–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, Ouyang W. 2008. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med 14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 17.Basu R, O'Quinn DB, Silberger DJ, Schoeb TR, Fouser L, Ouyang W, Hatton RD, Weaver CT. 2012. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity 37:1061–1075. doi: 10.1016/j.immuni.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simmons CP, Clare S, Ghaem-Maghami M, Uren TK, Rankin J, Huett A, Goldin R, Lewis DJ, MacDonald TT, Strugnell RA, Frankel G, Dougan G. 2003. Central role for B lymphocytes and CD4+ T cells in immunity to infection by the attaching and effacing pathogen Citrobacter rodentium. Infect Immun 71:5077–5086. doi: 10.1128/IAI.71.9.5077-5086.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maaser C, Housley MP, Iimura M, Smith JR, Vallance BA, Finlay BB, Schreiber JR, Varki NM, Kagnoff MF, Eckmann L. 2004. Clearance of Citrobacter rodentium requires B cells but not secretory immunoglobulin A (IgA) or IgM antibodies. Infect Immun 72:3315–3324. doi: 10.1128/IAI.72.6.3315-3324.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dumitru CD, Ceci JD, Tsatsanis C, Kontoyiannis D, Stamatakis K, Lin J-H, Patriotis C, Jenkins NA, Copeland NG, Kollias G, Tsichlis PN. 2000. TNF-α induction by LPS is regulated posttranscriptionally via a Tpl2/ERK-dependent pathway. Cell 103:1071–1083. doi: 10.1016/S0092-8674(00)00210-5. [DOI] [PubMed] [Google Scholar]
- 21.Mielke LA, Elkins KL, Wei L, Starr R, Tsichlis PN, O'Shea JJ, Watford WT. 2009. Tumor progression locus 2 (Map3k8) is critical for host defense against Listeria monocytogenes and IL-1β production. J Immunol 183:7984–7993. doi: 10.4049/jimmunol.0901336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watford WT, Hissong BD, Durant LR, Yamane H, Muul LM, Kanno Y, Tato CM, Ramos HL, Berger AE, Mielke L, Pesu M, Solomon B, Frucht DM, Paul WE, Sher A, Jankovic D, Tsichlis PN, O'Shea JJ. 2008. Tpl2 kinase regulates T cell interferon-γ production and host resistance to Toxoplasma gondii. J Exp Med 205:2803–2812. doi: 10.1084/jem.20081461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McNab FW, Ewbank J, Rajsbaum R, Stavropoulos E, Martirosyan A, Redford PS, Wu X, Graham CM, Saraiva M, Tsichlis P, Chaussabel D, Ley SC, O'Garra A. 2013. TPL-2–ERK1/2 signaling promotes host resistance against intracellular bacterial infection by negative regulation of type I interferon production. J Immunol 191:1732–1743. doi: 10.4049/jimmunol.1300146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rousseau S, Papoutsopoulou M, Symons A, Cook D, Lucocq JM, Prescott AR, O'Garra A, Ley SC, Cohen P. 2008. TPL2-mediated activation of ERK1 and ERK2 regulates the processing of pre-TNFα in LPS-stimulated macrophages. J Cell Sci 121:149–154. doi: 10.1242/jcs.018671. [DOI] [PubMed] [Google Scholar]
- 25.George D, Salmeron A. 2009. Cot/Tpl-2 protein kinase as a target for the treatment of inflammatory disease. Curr Top Med Chem 9:611–622. doi: 10.2174/156802609789007345. [DOI] [PubMed] [Google Scholar]
- 26.Hall JP, Kurdi Y, Hsu S, Cuozzo J, Liu J, Telliez JB, Seidl KJ, Winkler A, Hu Y, Green N, Askew GR, Tam S, Clark JD, Lin LL. 2007. Pharmacologic inhibition of Tpl2 blocks inflammatory responses in primary human monocytes, synoviocytes, and blood. J Biol Chem 282:33295–33304. doi: 10.1074/jbc.M703694200. [DOI] [PubMed] [Google Scholar]
- 27.Green N, Hu Y, Janz K, Li H-Q, Kaila N, Guler S, Thomason J, Joseph-McCarthy D, Tam SY, Hotchandani R, Wu J, Huang A, Wang Q, Leung L, Pelker J, Marusic S, Hsu S, Telliez J-B, Hall JP, Cuozzo JW, Lin L-L. 2007. Inhibitors of tumor progression loci-2 (Tpl2) kinase and tumor necrosis factor α (TNF-α) production: selectivity and in vivo antiinflammatory activity of novel 8-substituted-4-anilino-6-aminoquinoline-3-carbonitriles. J Med Chem 50:4728–4745. doi: 10.1021/jm070436q. [DOI] [PubMed] [Google Scholar]
- 28.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. 2005. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 29.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang Y-H, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. 2005. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. 2005. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei L, Laurence A, Elias KM, O'Shea JJ. 2007. IL-21 is produced by Th17 cells and drives IL-17 production in a STAT3-dependent manner. J Biol Chem 282:34605–34610. doi: 10.1074/jbc.M705100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, Dong C. 2007. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature 448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 33.Liang SC, Tan X-Y, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA. 2006. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med 203:2271–2279. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roussel L, Houle F, Chan C, Yao Y, Bérubé J, Olivenstein R, Martin JG, Huot J, Hamid Q, Ferri L, Rousseau S. 2010. IL-17 promotes p38 MAPK-dependent endothelial activation enhancing neutrophil recruitment to sites of inflammation. J Immunol 184:4531–4537. doi: 10.4049/jimmunol.0903162. [DOI] [PubMed] [Google Scholar]
- 35.Miyamoto M, Prause O, Sjöstrand M, Laan M, Lötvall J, Lindén A. 2003. Endogenous IL-17 as a mediator of neutrophil recruitment caused by endotoxin exposure in mouse airways. J Immunol 170:4665–4672. doi: 10.4049/jimmunol.170.9.4665. [DOI] [PubMed] [Google Scholar]
- 36.Griffin GK, Newton G, Tarrio ML, Bu DX, Maganto-Garcia E, Azcutia V, Alcaide P, Grabie N, Luscinskas FW, Croce KJ, Lichtman AH. 2012. IL-17 and TNF-alpha sustain neutrophil recruitment during inflammation through synergistic effects on endothelial activation. J Immunol 188:6287–6299. doi: 10.4049/jimmunol.1200385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miossec P, Kolls JK. 2012. Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discov 11:763–776. doi: 10.1038/nrd3794. [DOI] [PubMed] [Google Scholar]
- 38.Acuff NV, Li X, Kirkland R, Nagy T, Watford WT. 2015. Tumor progression locus 2 differentially regulates IFNγ and IL-17 production by effector CD4+ T cells in a T cell transfer model of colitis. PLoS One 10:e0119885. doi: 10.1371/journal.pone.0119885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sriskantharajah S, Gückel E, Tsakiri N, Kierdorf K, Brender C, Ben-Addi A, Veldhoen M, Tsichlis PN, Stockinger B, O'Garra A, Prinz M, Kollias G, Ley SC. 2014. Regulation of experimental autoimmune encephalomyelitis by TPL-2 kinase. J Immunol 192:3518–3529. doi: 10.4049/jimmunol.1300172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Acuff NV, LaGatta M, Nagy T, Watford WT. 6 April 2017. Severe dermatitis associated with spontaneous Staphylococcus xylosus infection in Rag−/− Tpl2−/− mice. Comp Med. Epub ahead of print. [PMC free article] [PubMed] [Google Scholar]
- 41.Gantke T, Sriskantharajah S, Ley SC. 2011. Regulation and function of TPL-2, an IκB kinase-regulated MAP kinase kinase kinase. Cell Res 21:131–145. doi: 10.1038/cr.2010.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kinugasa T, Sakaguchi T, Gu X, Reinecker H-C. 2000. Claudins regulate the intestinal barrier in response to immune mediators. Gastroenterology 118:1001–1011. doi: 10.1016/S0016-5085(00)70351-9. [DOI] [PubMed] [Google Scholar]
- 43.D'Agnillo F, Williams Moayeri MC, Warfel JM. 2013. Anthrax lethal toxin downregulates claudin-5 expression in human endothelial tight junctions. PLoS One 8:e62576. doi: 10.1371/journal.pone.0062576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. 2007. TGF-β and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain TH-17 cell-mediated pathology. Nat Immunol 8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- 45.Qin H, Wang L, Feng T, Elson CO, Niyongere SA, Lee SJ, Reynolds SL, Weaver CT, Roarty K, Serra R, Benveniste EN, Cong Y. 2009. TGF-β promotes Th17 cell development through inhibition of SOCS3. J Immunol 183:97–105. doi: 10.4049/jimmunol.0801986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. 2006. Transforming growth factor-[beta] induces development of the TH17 lineage. Nature 441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 47.Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, Grainger JR, Chen Q, Kanno Y, Watford WT, Sun HW, Eberl G, Shevach EM, Belkaid Y, Cua DJ, Chen W, O'Shea JJ. 2010. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature 467:967–971. doi: 10.1038/nature09447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. 1992. RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68:869–877. doi: 10.1016/0092-8674(92)90030-G. [DOI] [PubMed] [Google Scholar]
- 49.Vallance BA, Deng W, Knodler LA, Finlay BB. 2002. Mice lacking T and B lymphocytes develop transient colitis and crypt hyperplasia yet suffer impaired bacterial clearance during Citrobacter rodentium infection. Infect Immun 70:2070–2081. doi: 10.1128/IAI.70.4.2070-2081.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dahan S, Rabinowitz KM, Martin AP, Berin MC, Unkeless JC, Mayer L. 2011. Notch-1 signaling regulates intestinal epithelial barrier function, through interaction with CD4+ T cells, in mice and humans. Gastroenterology 140:550–559. doi: 10.1053/j.gastro.2010.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gibson DL, Ma C, Bergstrom KSB, Huang JT, Man C, Vallance BA. 2008. MyD88 signalling plays a critical role in host defence by controlling pathogen burden and promoting epithelial cell homeostasis during Citrobacter rodentium-induced colitis. Cell Microbiol 10:618–631. doi: 10.1111/j.1462-5822.2007.01071.x. [DOI] [PubMed] [Google Scholar]
- 52.Soria-Castro I, Krzyzanowska A, Pelaéz ML, Regadera J, Ferrer G, Montoliu L, Rodríguez-Ramos R, Fernández M, Alemany S. 2010. Cot/Tpl2 (MAP3K8) mediates myeloperoxidase activity and hypernociception following peripheral inflammation. J Biol Chem 285:33805–33815. doi: 10.1074/jbc.M110.169409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sanz-Garcia C, Ferrer-Mayorga G, González-Rodríguez Á Valverde Á Martín-Duce MA, Velasco-Martín JP, Regadera J, Fernández M, Alemany S. 2013. Sterile inflammation in acetaminophen-induced liver injury is mediated by Cot/Tpl2. J Biol Chem 288:15342–15351. doi: 10.1074/jbc.M112.439547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Van Acker GJD, Perides G, Weiss ER, Das S, Tsichlis PN, Steer ML. 2007. Tumor progression locus-2 is a critical regulator of pancreatic and lung inflammation during acute pancreatitis. J Biol Chem 282:22140–22149. doi: 10.1074/jbc.M702225200. [DOI] [PubMed] [Google Scholar]
- 55.Acuff NV, Li X, Elmore J, Rada B, Watford WT. 2017. Tpl2 promotes neutrophil trafficking, oxidative burst, and bacterial killing. J Leukoc Biol 101:1325–1333. doi: 10.1189/jlb.3A0316-146R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Torrado E, Cooper AM. 2010. IL-17 and Th17 cells in tuberculosis. Cytokine Growth Factor Rev 21:455–462. doi: 10.1016/j.cytogfr.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bry L, Brenner MB. 2004. Critical role of T cell-dependent serum antibody, but not the gut-associated lymphoid tissue, for surviving acute mucosal infection with Citrobacter rodentium, an attaching and effacing pathogen. J Immunol 172:433–441. doi: 10.4049/jimmunol.172.1.433. [DOI] [PubMed] [Google Scholar]
- 58.Frankel G, Phillips AD, Novakova M, Field H, Candy DC, Schauer DB, Douce G, Dougan G. 1996. Intimin from enteropathogenic Escherichia coli restores murine virulence to a Citrobacter rodentium eaeA mutant: induction of an immunoglobulin A response to intimin and EspB. Infect Immun 64:5315–5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Banchereau J, Bazan F, Blanchard D, Briere F, Galizzi JP, van Kooten C, Liu YJ, Rousset F, Saeland S. 1994. The CD40 antigen and its ligand. Annu Rev Immunol 12:881–922. doi: 10.1146/annurev.iy.12.040194.004313. [DOI] [PubMed] [Google Scholar]
- 60.Eliopoulos AG, Wang CC, Dumitru CD, Tsichlis PN. 2003. Tpl2 transduces CD40 and TNF signals that activate ERK and regulates IgE induction by CD40. EMBO J 22:3855–3864. doi: 10.1093/emboj/cdg386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gibson DL, Ma C, Rosenberger CM, Bergstrom KSB, Valdez Y, Huang JT, Khan MA, Vallance BA. 2008. Toll-like receptor 2 plays a critical role in maintaining mucosal integrity during Citrobacter rodentium-induced colitis. Cell Microbiol 10:388–403. [DOI] [PubMed] [Google Scholar]
- 62.Khan MA, Ma C, Knodler LA, Valdez Y, Rosenberger CM, Deng W, Finlay BB, Vallance BA. 2006. Toll-like receptor 4 contributes to colitis development but not to host defense during Citrobacter rodentium infection in mice. Infect Immun 74:2522–2536. doi: 10.1128/IAI.74.5.2522-2536.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Banerjee A, Gugasyan R, McMahon M, Gerondakis S. 2006. Diverse Toll-like receptors utilize Tpl2 to activate extracellular signal-regulated kinase (ERK) in hemopoietic cells. Proc Natl Acad Sci U S A 103:3274–3279. doi: 10.1073/pnas.0511113103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kuriakose T, Rada B, Watford WT. 2014. Tumor progression locus 2-dependent oxidative burst drives phosphorylation of extracellular signal-regulated kinase during TLR3 and 9 signaling. J Biol Chem 289:36089–36100. doi: 10.1074/jbc.M114.587121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xiao Y, Jin J, Chang M, Nakaya M, Hu H, Zou Q, Zhou X, Brittain GC, Cheng X, Sun S-C. 2014. TPL2 mediates autoimmune inflammation through activation of the TAK1 axis of IL-17 signaling. J Exp Med 211:1689–1702. doi: 10.1084/jem.20132640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lemos HP, Grespan R, Vieira SM, Cunha TM, Verri WA, Fernandes KSS, Souto FO, McInnes IB, Ferreira SH, Liew FY, Cunha FQ. 2009. Prostaglandin mediates IL-23/IL-17-induced neutrophil migration in inflammation by inhibiting IL-12 and IFNγ production. Proc Natl Acad Sci U S A 106:5954–5959. doi: 10.1073/pnas.0812782106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Henness S, van Thoor E, Ge Q, Armour CL, Hughes JM, Ammit AJ. 2006. IL-17A acts via p38 MAPK to increase stability of TNF-α-induced IL-8 mRNA in human ASM. Am J Physiol Lung Cell Mol Physiol 290:L1283–L1290. doi: 10.1152/ajplung.00367.2005. [DOI] [PubMed] [Google Scholar]
- 68.Wiles S, Pickard KM, Peng K, MacDonald TT, Frankel G. 2006. In vivo bioluminescence imaging of the murine pathogen Citrobacter rodentium. Infect Immun 74:5391–5396. doi: 10.1128/IAI.00848-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pesu M, Watford WT, Wei L, Xu L, Fuss I, Strober W, Andersson J, Shevach EM, Quezado M, Bouladoux N, Roebroek A, Belkaid Y, Creemers J, O'Shea JJ. 2008. T-cell-expressed proprotein convertase furin is essential for maintenance of peripheral immune tolerance. Nature 455:246–250. doi: 10.1038/nature07210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, Knight ZA, Cobb BS, Cantrell D, O'Connor E, Shokat KM, Fisher AG, Merkenschlager M. 2008. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci U S A 105:7797–7802. doi: 10.1073/pnas.0800928105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.