

Abstract
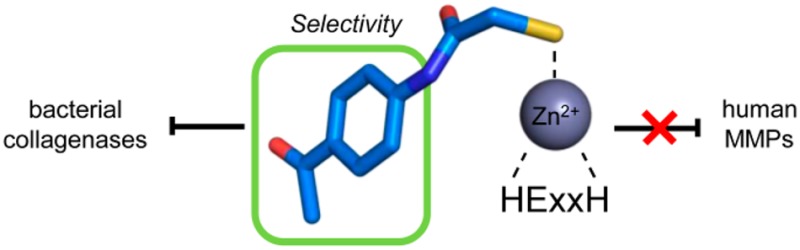
Secreted virulence factors like bacterial collagenases are conceptually attractive targets for fighting microbial infections. However, previous attempts to develop potent compounds against these metalloproteases failed to achieve selectivity against human matrix metalloproteinases (MMPs). Using a surface plasmon resonance-based screening complemented with enzyme inhibition assays, we discovered an N-aryl mercaptoacetamide-based inhibitor scaffold that showed sub-micromolar affinities toward collagenase H (ColH) from the human pathogen Clostridium histolyticum. Moreover, these inhibitors also efficiently blocked the homologous bacterial collagenases, ColG from C. histolyticum, ColT from C. tetani, and ColQ1 from the Bacillus cereus strain Q1, while showing negligible activity toward human MMPs-1, -2, -3, -7, -8, and -14. The most active compound displayed a more than 1000-fold selectivity over human MMPs. This selectivity can be rationalized by the crystal structure of ColH with this compound, revealing a distinct non-primed binding mode to the active site. The non-primed binding mode presented here paves the way for the development of selective broad-spectrum bacterial collagenase inhibitors with potential therapeutic application in humans.
Introduction
Clostridia represent a family of ubiquitously occurring Gram-positive bacteria comprising perilous pathogens that cause diseases such as botulism (Clostridium botulinum), gas gangrene (C. perfringens), tetanus (C. tetani), or pseudomembranous colitis (C. difficile).1,2 These toxigenic clostridia still represent a threat to public health, as tetanus and clostridial myonecrosis have maintained high mortality rates and pseudomembranous colitis is a known severe complication of antibiotic therapy.3−6 Furthermore, substantial amounts of pathogenic clostridia were cultured in the past 60 years for use as bioweapons.7 Consequently, massive efforts have been aimed at unraveling the molecular basis of these life-threatening infections. Nevertheless, such infections remain a major challenge, as this knowledge did not yet lead to satisfactory treatment options.
The high lethality of these bacteria is related to collagenases which are crucial for clostridial virulence, given their critical role in colonization and evasion of host immune defense, acquisition of nutrients, facilitation of dissemination, or tissue damage during infection. Additionally, they might potentiate clostridial histotoxicity by facilitating toxin diffusion.2,8,9
The physiological substrate of clostridial collagenases is collagen, the main component of the extracellular matrix in mammals (up to 90%).10,11 Its defining characteristic is the collagen triple-helix, which is perpetuated by the triplet repeat Gly-X-Y (X and Y positions are mostly occupied by proline (28%) and hydroxyproline (38%)).12 The natively folded triple helix is highly resistant to proteolysis.13,14 Even the most prominent human collagenases, the matrix metalloproteinases MMP-1, -2, -8, -13, -14, and -18, can cleave the triple helix only at a single site.15,16 In contrast to that, clostridial collagenases can process collagen triple helices at multiple sites, as the active site displays a remarkable selectivity for the Gly-Pro-Y triplets,17 and they can decompose collagen completely into small peptides.18,19
The inhibition of these extracellular collagenases is conceptually attractive, as it does not attack the pathogen directly but rather blocks the colonization and infiltration of the host by the clostridia. Thereby reducing the Darwinian selection pressure, targeting bacterial virulence is considered a promising approach to combat the emerging threat of drug-resistant bacteria.20−22 To date, several anti-virulence targets have been validated, demonstrating the potential of this approach.23−28 Kassegne et al. showed, for example, that a collagenase knock-out strain from Leptospira interrogans displayed reduced virulence in an in vivo model.28
Targeting extracellular enzymes provides a substantial benefit because inhibitors do not need to cross the bacterial cell wall, which has turned out to be challenging in many cases.29−31 Consequently, bacterial collagenases represent prime targets for an effective therapy against clostridial and bacillary infections.6,9,32,33
Clostridial collagenases are zinc metalloproteinases of ∼115 kDa with a multi-domain organization, homologues of which are also found in many bacilli. The mature protein harbors an N-terminal collagenase unit of ∼78 kDa, which is the minimal collagenolytic entity, followed by a varying composition of two to three accessory domains, which are thought to be involved in collagen swelling and binding to fibrillar collagen.34−38 The collagenase unit is composed of the activator domain and the peptidase domain.34 The peptidase domain harbors the catalytic zinc ion, which is coordinated by the two histidines of the canonical zinc-binding HEXXH motif, and a downstream glutamate.4,34,35,39−41 The glutamate residue in the HEXXH motif acts as the general acid/base, which polarizes the catalytic water essential for catalysis. This polarized water molecule performs the nucleophilic attack, while the zinc ion serves as an oxyanion hole to the carbonyl oxygen of the scissile peptide bond.42
Several groups have been working on the development of clostridial collagenase inhibitors in the past, focusing on the collagenases G (ColG) and H (ColH) from C. histolyticum. In this context, besides the identification of active compounds from Viola yedoensis,43 inhibitors based on sulfonylated derivatives of l-valine hydroxamate44 have been synthesized as well as sulfonyl aminoacyl hydroxamates.45 Furthermore, compounds incorporating 5-amino-2-mercapto-1,3,4-thiadiazole zinc binding functions,46 arylsulfonyl-ureido and 5-dibenzo-suberenyl/suberyl,47 or succinyl hydroxamate and iminodiacetic acid hydroxamate moieties48 have been described. These inhibitors follow the classic architecture of metalloprotease inhibitors with a backbone that mimics the natural substrate, which is connected via a linker to a zinc-binding group that chelates the catalytic zinc ion and, thereby, expels the essential catalytic water molecule from the active site.49,50 These inhibitors were developed as substrate analogues and/or designed on the basis of inhibitors for other metalloproteases that share the HEXXH motif,51 like thermolysin or MMPs.44,47,52−58 Unfortunately, the synthetic clostridial collagenase inhibitors are not selective, inhibiting clostridial collagenases and MMPs alike.44,45,47,48,55−57,59−61 Therefore, they are not suitable for antibacterial therapy in humans. Consequently, novel and more effective drug candidates are urgently needed.
Efforts to design selective inhibitors were hampered by the lack of high-resolution structural data on clostridial collagenases until 2011. The first crystal structures revealed that, although there is no significant sequence homology between the peptidolytic domains of clostridial collagenases and MMPs, their active sites share a similar catalytic zinc ion-binding geometry and the canonical non-prime-site substrate-recognition motif, the edge strand.17
In this study, we wanted to capitalize on the recent crystal structures of the peptidase domains of three clostridial collagenases34,41 with the aim to rationally develop small organic molecules targeting collagenase ColH from C. histolyticum. In the following we describe the discovery of inhibitors which are highly active and selective for clostridial collagenases over MMPs and have the potential to be further optimized for a future therapeutic application in humans. Their selectivity can be rationalized on the basis of a co-crystal structure of the peptidase domain of ColH in complex with an inhibitor, revealing a distinct non-primed mode of binding of the inhibitor to the active site.
Results and Discussion
Discovery of New Inhibitory Scaffold
To discover new low-molecular-weight inhibitory compounds, a focused protease inhibitor library was screened with a surface plasmon resonance (SPR)-based binding assay using the amine-coupled peptidase domain of ColH (ColH-PD) as ligand. To ensure the integrity of ColH-PD after immobilization, the collagenase-specific peptidic substrate N-(3-[2-furyl]acryloyl)-l-leucyl-glycyl-l-prolyl-l-alanine (FALGPA)62 was used as positive control (Figure S1a). A total of 1520 structurally diverse small molecules with an average molecular weight (MW) of 389 ± 78 Da were screened at 100 μM. Compounds showing a MW-normalized response higher than that of 500 μM FALGPA (i.e., 35 μ-refractive index units) were classified as hits. The SPR screen resulted in 202 primary hits. Nineteen compounds were excluded from the subsequent testing as known promiscuous inhibitors,63 resulting in a hit rate of 12.0% (Figure S2).
The secondary functional screening assessed the potential of the 183 SPR hits to inhibit the peptidolytic activity of ColH-PD using a custom-made fluorescence resonance energy transfer (FRET) substrate. Typically, the FALGPA62 and Wünsch64 assays are used to characterize the activity of clostridial collagenases due to their easy setup and commercial availability next to their specificity for these enzymes.62,65,66 However, the low binding affinity of these peptides together with their low signal-to-noise ratios severely limits the sensitivity of these assays. Consequently, substantial amounts of enzyme and substrate are needed in characterization studies (e.g., KM values for FALPGA are in the mM range62,67). To facilitate our screening process, we designed and synthesized a decapeptide (Mca-Ala-Gly-Pro-Pro-Gly-Pro-Dpa-Gly-Arg-NH2) to be used as a substrate for a FRET-based assay. Its sequence was based on the detailed profile of the primed and non-primed cleavage site specificity of clostridial collagenases as determined by Proteomic Identification of protease Cleavage Sites (PICS) recently.17,68 The assay sensitivity was increased by several orders of magnitude compared to the FALGPA assay by the application of the FRET technology.69,70 The KM value of this substrate is 62 ± 8 μM for ColH-PD. The 183 SPR binders were screened at a final concentration of 40 μM. The inhibitor isoamylphosphonyl-Gly-Pro-Ala (Figure S1b) was used as positive control in the assay.58 In sum, the SPR-based and activity-based screenings led to six functional hits (>25% inhibition) with MWs ranging from ∼210 to ∼385 Da (Figure S3). The two most active inhibitors in this assay were mercaptoacetamides 1 and 2 (Table 1). Compound 2 led to an inhibition of ColH-PD in vitro similar to that of isoamylphosphonyl-Gly-Pro-Ala, both at 40 μM, i.e., 82 ± 3% and 81 ± 1%, respectively.
Table 1. IC50 Values of Mercaptoacetamide Compounds for ColH-PD.

Dose–response studies revealed an IC50 value of 1.9 ± 0.3 μM for this compound, while the non-substituted aniline derivative 1 showed lower activity toward ColH-PD (IC50 = 25 ± 6 μM). Our further hits showed considerably weaker inhibition and, in one case, proved to be incompatible with the FRET assay at high concentration. The high potency of the N-aryl mercaptoacetamides combined with their relatively low molecular weight encouraged us to investigate this promising compound class further in order to improve the inhibitory activity.
Characterization of Mercaptoacetamide Hits
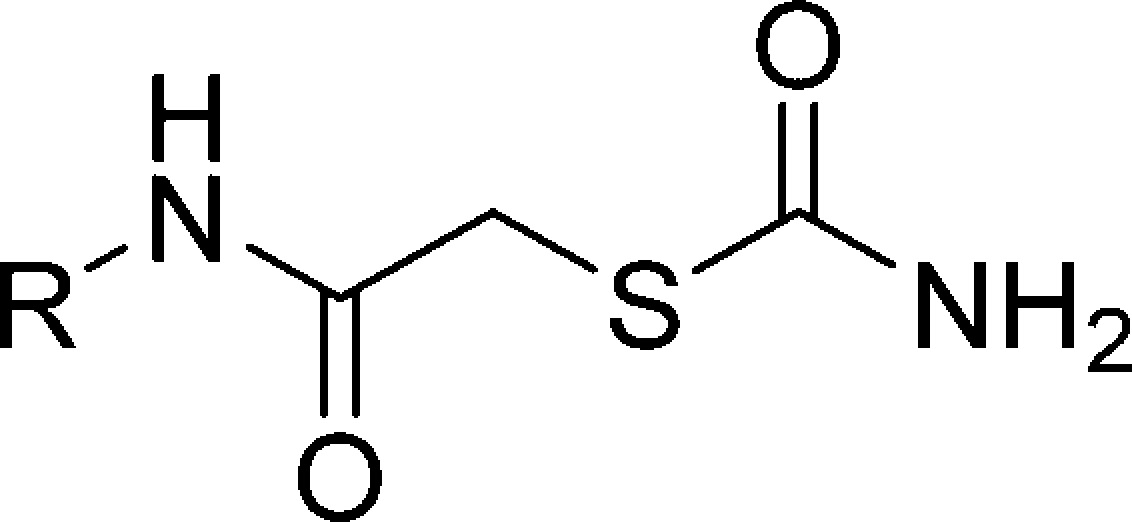
A total of 36 derivatives of this compound class were purchased (see Tables 1, 3, and S1). Six derivatives showed improved inhibition compared to 2 (3–8). Generally, the introduction of functional groups in para-position to the aniline turned out to be favorable, considering the striking loss of activity of ortho-methoxy-substituted compound 11 compared to its para-analogue 5 (ortho-effect), and ortho-chloro-substituted compound 12 compared to its para-analogue 7. The superior performance of para-derivatives was also evident regarding the 100-fold decrease in IC50 of 7 compared to its meta-chloro-substituted counterpart 9. In comparison to the unsubstituted compound 1, meta-substituted compounds 9 and 10 showed no significant improvement in IC50. Removal of the 3-methyl-group of compound 2 even led to a 16-fold decreased IC50 (compound 6), suggesting that the meta-substitution is not beneficial for ColH inhibition.
Table 3. Structure and Activity of Dithiocarbamates.


Regarding electronic properties of our hits it becomes apparent that the best compounds 3–5, displaying IC50 values in the two-digit nanomolar range, bear oxygen-containing groups with hydrogen bond accepting properties.
Selectivity against MMPs and Broad-Spectrum Inhibition of Other Bacterial Collagenases
To determine the selectivity of our compounds toward clostridial and bacillial collagenases on the one hand, and MMPs on the other, selected compounds (3 and 7,Figure 1) were tested using in vitro inhibition assays with ColH-PD and the peptidase domains of ColT (ColT-PD), the collagenase units of ColG (ColG-CU) and of ColQ1 (ColQ1-CU), as well as the catalytic domains of MMP-1, -2, -3, -7, -8, and -14. The hydroxamate-based peptidomimetic batimastat (Figure S1c) is a highly potent and unselective inhibitor of MMPs71 and was used as a positive control. MMPs are highly similar to each other in their active-site topology, which has made the development of selective active-site directed MMP inhibitors a challenging task.72,73 The S1′ binding site is the major specificity determinant in MMPs. Based on the S1′ site, the MMPs are typically divided into deep, intermediate and shallow S1′ binding pocket groups (e.g., deep: MMP-3, -12, and -14; intermediate: MMP-2, -8, and -9; shallow: MMP-1 and -7).74 Therefore, we chose a panel of MMPs to investigate the binding of our compounds to all three S1′ pocket types. In line with published results,71 batimastat displayed IC50 values below 10 nM for all of these MMPs (Table S2). As expected from this broad-spectrum zinc metalloproteinase inhibitor, batimastat also inhibited ColH-PD, ColT-PD, ColG-CU and ColQ1-CU (Figure S4). Intriguingly, compounds 3 and 7 resulted in no or negligible inhibition of the tested MMPs (Figure 1 and Figure S5). Only in case of MMP-2, we observed 25% inhibition at 100 μM compound 3, while ColH-PD was efficiently inhibited, showing less than 10% residual activity. Thus, we observed a more than 1000-fold selectivity of these two compounds for ColH over MMPs. Strikingly, the clostridial collagenase homologues ColG and ColT, and the bacillial collagenase ColQ1, were even more efficiently inhibited, showing 5% or less residual activity when treated with 100 μM compound 3 or 7. A similar compound scaffold had been reported by Zhu et al. to inhibit LasB, an extracellular elastase from Pseudomonas aeruginosa.75 In sum, these findings showed that the N-aryl mercaptoacetamide-based inhibitors are not only selective against MMPs, but are also potent broad-spectrum inhibitors of bacterial collagenases.
Figure 1.
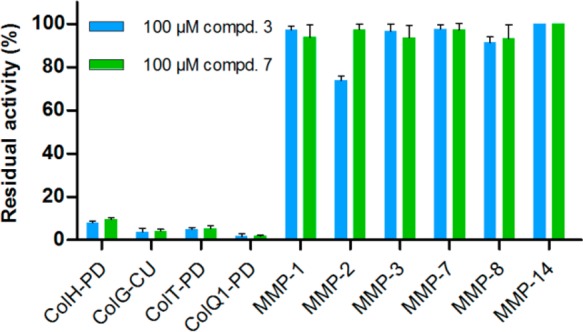
Inhibition of selected MMPs and bacterial collagenases by N-aryl mercaptoacetamide compounds 3 and 7.
Crystal Structure of the Peptidase Domain of ColH in Complex with Compound 3
To rationalize the binding mode of the N-aryl mercaptoacetamide-based inhibitors, we aimed to solve the crystal structure of ColH-PD in complex with compound 3. The structure was determined at 1.87 Å resolution with all residues being defined in the electron density at excellent geometric and crystallographic parameters (Table S3). The overall topology of the peptidase domain showed the expected thermolysin-like fold. The average root-mean-square displacements (RMSDs) of backbone atoms between the structure of the apo-peptidase domain and the peptidase domain in complex with isoamylphosphonyl-Gly-Pro-Ala were 0.133 and 0.123 Å, respectively. The peptidase domain of ColH is divided horizontally by the active-site cleft into an upper N-terminal and a lower C-terminal subdomain (CSD). Substrates can bind to the active-site cleft from the left (non-primed side) to the right (primed side) when viewed in standard orientation.76 Central elements of the N-terminal subdomain (NSD) are the active-site helix and a mixed five-strand β-sheet. The zinc-binding motif HEXXH, which provides the two zinc-coordinating histidines and the general acid/base glutamate, is located in the active-site helix (Figure 2). The lowermost β-strand of the mixed β-sheet shapes the upper perimeter of the active-site cleft, the edge strand. The edge strand interacts in an antiparallel manner with the substrate predominantly on the non-primed side34,77,78 The third zinc ligand is a glutamate residue, located on the glutamate helix of the CSD. The insertion of 30 residues between the HEXXH motif and this glutamate residue shapes (i) the non-primed side of the active-site cleft and (ii) a calcium-binding site crucial for enzymatic activity.34,41,77
Figure 2.
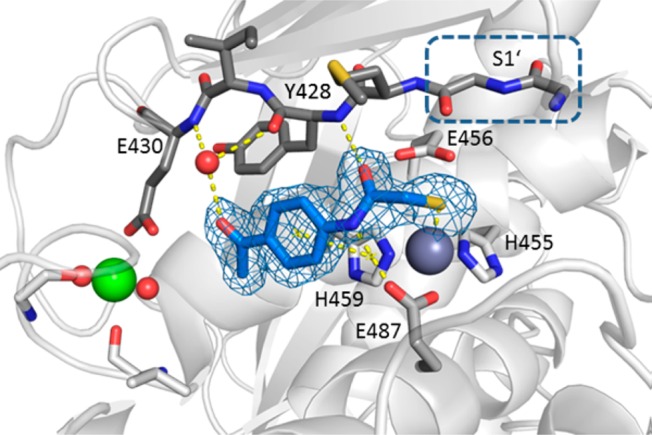
Peptidase domain of ColH in complex with the hydrolysis product of compound 3. Close-up view of the active site in ball-and-stick representation. The inhibitor (blue) is shown in sticks with the maximum likelihood weighted 2Fo – Fc electron density map contoured at 1σ. The catalytic zinc ion (dark gray), calcium ion (green), and water molecule (red) are shown as spheres. The S1′ site formed by Gly425 and Gly426 in the edge strand (shown in dark gray sticks) is indicated.
A well-defined electron density was observed for the ligand bound in the active site. The structure of compound 3 could be clearly modeled into the density (Figure 2), except for the carbamoyl unit of the thiocarbamate moiety. Instead, the electron density showed the sulfur atom coordinating the catalytic zinc ion, suggesting that the thioester group had been hydrolyzed in the co-crystallization process. This result prompted an investigation of our newly discovered class of inhibitors with particular emphasis on the stability of the thiocarbamate function in aqueous buffers such as the buffer system of the functional assay and the crystallization buffer.
Stability of the Thiocarbamate Unit
Two inhibitors with major differences in potency (7, 12) were selected and the hydrolytic formation of the corresponding free thiol was analyzed by liquid chromatography–mass spectrometry (LC-MS). Free thiols were synthesized as references for the stability assay. The conversions of compounds 7 and 12 into compounds 14 and 15, respectively, proceeded rapidly in 10 mM HEPES, pH 7.5 at 22.5 °C, with thiocarbamate half-lives of 26.8 ± 1.4 min (7) and 20.6 ± 0.9 min (12, Figure 3). These results corroborated that the inhibition of thiocarbamates 1–12 was predominantly due to the respective free thiols. Considering the preparation time and the pre-incubation time of 1 h for each compound with ColH-PD before the functional assay was started by addition of the substrate the thiocarbamates were quantitatively converted within the time frame of the experiment. Thiol formation was also demonstrated at pH 6.4, corresponding to the buffer used for co-crystallization (Figure S6).
Figure 3.
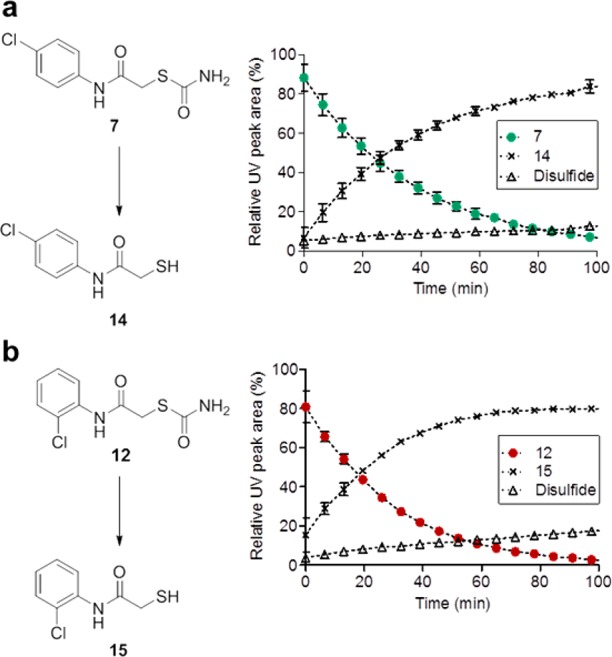
Conversion of thiocarbamates 7 and 12 into the respective corresponding free thiols: (a) compound 7 into 14 and (b) compound 12 into 15. Time course of hydrolysis in 10 mM Hepes, pH 7.4 (10% methanol), at 22.5 °C was monitored by LC-MS, showing conversion into corresponding thiol and to minor extent into another compound which is most likely the disulfide oxidation product.79
Confirmation of Thiol as Active Compound
To further substantiate these findings, we followed two different strategies. First, we studied the inhibitory activities of the free thiols 13–15. Thus, we determined the IC50 values of the free thiols 13–15 with ColH-PD in the presence of the reducing agent TCEP. The resulting IC50 values of 0.017, 0.21, and 40 μM corresponded well with 0.010, 0.19, and 31 μM of the thiocarbamate analogues (Tables 1 and 2).
Table 2. Inhibition of ColH-PD by Thiol Compounds in the Presence of 5 mM TCEP.


The results of the MMP and bacterial collagenase inhibition assays could also be reproduced using the thiol compounds, with 13 and 14 demonstrating a similarly high selectivity against MMPs and a broad-spectrum inhibition of bacterial collagenases (Figure 4).
Figure 4.
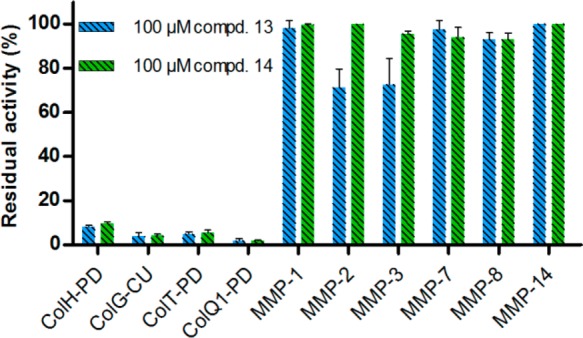
Inhibition of selected MMPs and bacterial collagenases by thiol compounds 13 and 14.
As a second strategy, we aimed to synthesize a structural analogue of compound 1 lacking the hydrolytically instable thioester motif. The formal replacement of the sulfur atom with a methylene group led to the carboxamide analogue 16, which was prepared and tested for its inhibitory activity toward ColH-PD (Figure 5). Compound 16 was devoid of any activity even at 1000 μM. This demonstrated that the carbamoyl moiety in 1 does not contribute to target binding, but is just part of a prodrug-like structure which furnishes the corresponding bioactive thiol by chemical hydrolysis.
Figure 5.

Structure and inhibitory activity of the non-hydrolyzable carboxamide analogue 16.
Further in line with our findings, dithiocarbamates 17–21 (Table 3) were inactive toward ColH-PD. LC-MS experiments with the dithiocarbamate analogue of our best hit 3 showed no formation of free thiol 13 within the time frame of our assay, explaining the inactivity of these derivatives by stability toward hydrolysis (Figure S7).
In addition, the thermodynamic profile of the interaction between compounds 7 and 14 and ColH-PD was determined. As expected, isothermal titration calorimetry (ITC) measurements resulted in very similar affinities and free energy values, resulting from the hydrolysis of 7 to furnish thiol 14 (Table 4). Compound binding to ColH-PD turned out to be enthalpy-driven. In sum, the findings from the stability assay, the in vitro assay and the ITC data confirmed the thiols as active compounds in our enzyme inhibition assay.
Table 4. ITC and IC50 Results of the Thiocarbamate–Thiol Pair 7 and 14.
| 7 | 14 | |
|---|---|---|
| IC50 (μM)a | 0.19 ± 0.02 | 0.21 ± 0.01 |
| KD (μM)b | 0.309 ± 0.045 | 0.360 ± 0.038 |
| ΔG (kcal mol–1)b | –8.9 ± 0.1 | –8.8 ± 0.1 |
| ΔH (kcal mol–1)b | –12.7 ± 1.2 | –15.4 ± 0.3 |
| –TΔS(kcal mol–1)b | 3.8 ± 1.3 | 6.6 ± 0.4 |
| Nb,c | 0.54 ± 0.05 | 0.48 ± 0.03 |
IC50 refers to the functional FRET assay.
Results are from at least two independent measurements.
The low stoichiometry could be explained by incomplete zinc occupation of the active sites.41
Cytotoxicity Test
Regarding the potential therapeutic use of our compounds in humans, we investigated the cytotoxic properties of selected N-aryl mercaptoacetamides. Cytotoxicity tests using HEP G2 cells showed compounds 13 and 14 to display low cytotoxicity, comparable to that of the marketed antibiotic rifampicin (Table 5), while doxorubicin as control showed the expected cytotoxic effect. These findings underline the potential of our compounds for the development of novel anti-infectives.
Table 5. Cytotoxicity of 13, 14, and Three Reference Compounds in HEP G2 Cells.
| compound | concn (μM) | reduction of viability (%) |
|---|---|---|
| 13 | 100 | 17 ± 12 |
| 14 | 100 | 28 ± 12 |
| rifampicin | 100 | 29 ± 5 |
| doxorubicin | 1 | 50 ± 5 |
| batimastat | 100 | 13 ± 7 |
Zinc Coordination by a Thiolate
The identification of the thiol as active compound in our functional assays was in excellent agreement with the crystal structure analysis which demonstrated that only a sulfur atom, to be precise a thiolate, was coordinating the catalytic zinc ion. To validate our conclusions on the protonation state of the sulfur atom, we calculated the pKa values for the thiol group resulting from the hydrolysis of thiocarbamate 3 in solution and when bound to the active site using the Molecular Operating Environment (MOE) software.80 The pKa of the thiol group was strongly lowered from 9.0 in the solvent to 3.1 by the direct coordination to the zinc ion. This suggests that the thiol is fully ionized to the thiolate form in both the activity assay and the crystallization experiment upon binding to the active site.
Binding Mode of the N-Aryl Mercaptoacetamide Compound to the Active Site of ColH-PD
The hydrolysis product of compound 3 binds to the S3 to S1 substrate binding pockets (Figure 2). The thiolate coordinates the zinc ion with a sulfur-to-zinc distance of 2.27 Å. In the S1 pocket, the amide oxygen of compound 3 forms a hydrogen bond with the main-chain amide nitrogen of Tyr428 (3.11 Å) of the NSD, while the amide nitrogen of compound 3 hydrogen-bonds with the carbonyl oxygen (OE2) of Glu487 (2.97 Å) of the CSD. In addition, the benzene ring of the ligand is involved in a π–π-stacking interaction with the imidazole ring of His459 (centroid-centroid distance of 3.80 Å). The oxygen of the acetyl group of 3 interacts via a bridging water molecule (3.07 Å) with the main-chain oxygen of Tyr428 in S1 (3.11 Å) and with the main-chain nitrogen of Glu430 in S3 (2.84 Å). Thus, the inhibitor is well-braced in-between the NSD and the CSD of the peptidase domain.
Importantly, the binding mode of the inhibitor is not directed toward the primed substrate-binding sites, but toward the non-primed recognition sites in-between the calcium-binding site and the catalytic zinc ion. Thus, this complex of ColH-PD with the thiol derived from compound 3 is the first to describe non-primed interactions between a clostridial collagenase and an active site-directed ligand.
Identification of Selectivity Determinant
The thiol derived from compound 3 interacts with two central elements of the active site: (i) the zinc ion and its liganding sphere (His455, Glu456, His459, and Glu487), and (ii) the edge strand (Gly425-Glu430). These two central elements are also present in MMPs:17 A structurally, but not sequentially, homologous edge strand frames the upper rim of the active site in MMPs, and the zinc-liganding sphere composed of the HEXXH motif and a third proteinaceous ligand is nearly identical between the MMPs and the clostridial collagenases. The geometry of the zinc-liganding sphere is almost perfectly superimposable in clostridial collagenases and MMPs (RMSD = 0.060 Å between ColH and MMP-1). Only the third zinc-binding residue differs. While in MMPs, this position is occupied by a histidine; in clostridial collagenases, this ligand is a glutamate provided by the gluzincin-specific glutamate helix. Given this high similarity in the active site between clostridial collagenases and MMPs, this triggered the question of how we can rationalize the observed differences in selectivity of the N-aryl mercaptoacetamide compounds toward the two enzyme families. A first in silico structural analysis of the active sites of MMP-1, -2, -3, -8, -12, and -13 suggested that (i) these enzymes could accommodate the mercaptoacetamide compounds in their non-primed substrate pockets, and that (ii) the residues on the edge strand and the zinc ion are positioned as such as to allow productive interactions with the thiolate. Yet, the MMPs, lacking the zinc-binding glutamate, cannot provide the hydrogen-bonding partner for the amide nitrogen of the mercaptoacetamide inhibitor. Hence, is the interaction with the gluzincin-specific Glu487 crucial for selectivity? To test this hypothesis, we mimicked the zinc-liganding sphere of MMPs in ColH-PD by mutating Glu487 into a histidine. A comparison of the apparent inhibition constant Ki(app) of compound 11 toward wild-type ColH-PD and the mutant E487H, 92 ± 8 and 166 ± 23 μM, respectively, showed that the mutation did not result in a drastic change in the inhibitory potency. This suggests that the interaction with the edge strand on the non-primed site, mediated via main-chain contacts, is the main structural selectivity determinant. This hypothesis is further supported by a structural analysis of the edge-strand conformations in MMPs and clostridial collagenases in the ligand-bound state. Within each family, the ligand-bound edge-strand conformation is highly conserved (Figure 6). Compared to the clostridial situation, the edge strand in MMPs is tilted by 27–29°. This tilted orientation could explain the inefficient binding of the N-aryl mercaptoacetamide compounds to the MMPs, suggesting that the interactions with the non-primed edge strand (S1–S3) are the crucial selectivity determinants.
Figure 6.
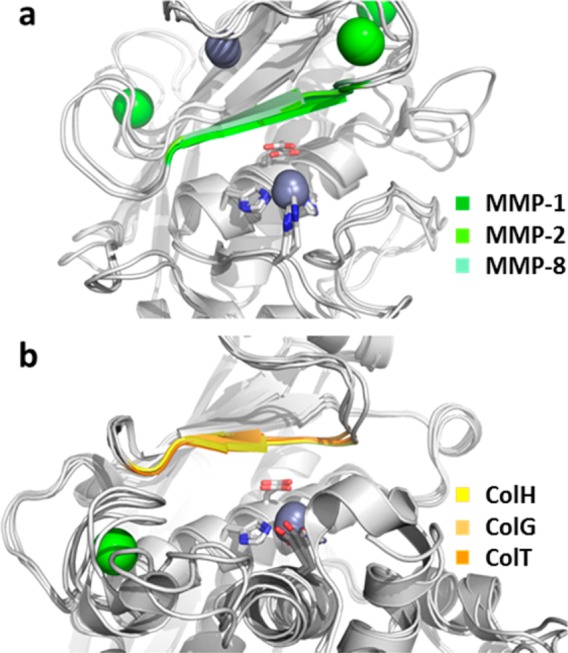
Close-up on the superpositioned active sites of three MMPs (a) and of three clostridial collagenases (b) in the ligand-bound state. The ligands have been removed for better visualization. The HEXXH motif is shown in sticks, and the zinc ions (gray) and calcium ions (green) are shown as spheres. The edge strand on top of the catalytic zinc is highlighted in color.
It was also interesting to see that related mercaptoacetamide derivatives were shown to inhibit LasB from P. aeruginosa.75 Analysis of our best compound 13 in an in vitro LasB inhibition assay revealed a more than 1000-fold lower activity compared to ColH (data not shown). This is likely due to the distinct binding mode of compound 13 to the non-primed binding site of ColH (Figure 2) in contrast to the proposed primed binding mode of the related compounds in LasB by Zhu et al.75
With regard to future inhibitor design, these findings suggest that by (i) amplification of the interactions with the edge strand and (ii) extension of the inhibitor scaffold in para, i.e., by developing the compound further into the non-primed substrate recognition pockets, even more potent and selective compounds could be developed. In compounds with optimized affinity to the edge strand and the non-primed substrate binding pockets, we plan to investigate the replacement of the thiol moiety by a less reactive ZBG. Such lead compounds hold the promise of higher efficacy and therefore higher safety in potential therapeutic applications in humans.
Conclusion
We identified a novel compound scaffold for the selective inhibition of clostridial collagenases. Starting with an SPR-based primary screening of a focused library, we validated the SPR-hits in a secondary enzyme inhibition assay using a custom-tailored FRET peptide substrate for clostridial collagenases. Two mercaptoacetamide derivatives were the most potent functional hits in this assay. Further derivatization of these initials hits, in particular the introduction of oxygen-containing groups in para-position to the aniline, led to the generation of highly potent N-aryl mercaptoacetamide-based clostridial collagenase inhibitors with IC50 values in the two-digit nanomolar range. These compounds showed unprecedented selectivity against MMPs, while at the same time they displayed a broad-spectrum inhibition of bacterial collagenases. The selectivity of these compounds could be rationalized on the basis of a co-crystal structure of ColH-PD with the most active compound, revealing a distinct non-primed binding mode of the inhibitor to the active site. The mercaptoacetamides were also shown to display no cytotoxicity toward human cells. These insights pave the way for the development of selective broad-spectrum bacterial collagenase inhibitors with potential therapeutic application in humans.
Acknowledgments
This work was supported by grants from the Austrian Science Fund (project W_01213 & M_1901). We thank Dr. Elfriede Dall for X-ray data collection, Dr. Werner Tegge for peptide synthesis, and Dr. Christine Maurer and Jeannine Jung for technical support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b06935.
Supporting Figures S1–S7 and Tables S1–S3, giving molecular structures of positive controls, SPR and functional screening hits, inhibition of the selected MMPs and bacterial collagenases by batimastat, MMP inhibition assay, LC-MS analyses of 3 and 21, structure and activity of additional thiocarbamates and related compounds, and data collection and refinement statistics (PDF)
Author Contributions
⊥ E.S. and A.M.K. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Cato E.; George W.; Finegold S.. Bergey’s Manual of Systematic Bacteriology; Williams & Wilkins: Baltimore, 1986; pp 1141–1200. [Google Scholar]
- Hatheway C. L. Clin. Microbiol. Rev. 1990, 3, 66–98. 10.1128/CMR.3.1.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke M. P.; Opeskin K. Am. J. Forensic Med. Pathol. 1999, 20, 158–162. 10.1097/00000433-199906000-00011. [DOI] [PubMed] [Google Scholar]
- Bruggemann H.; Baumer S.; Fricke W. F.; Wiezer A.; Liesegang H.; Decker I.; Herzberg C.; Martinez-Arias R.; Merkl R.; Henne A.; Gottschalk G. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 1316–1321. 10.1073/pnas.0335853100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubes G. Science 2008, 321, 360. 10.1126/science.321.5887.360. [DOI] [PubMed] [Google Scholar]
- Taubes G. Science 2008, 321, 356–361. 10.1126/science.321.5887.356. [DOI] [PubMed] [Google Scholar]
- Arnon S. S.; Schechter R.; Inglesby T. V.; Henderson D. A.; Bartlett J. G.; Ascher M. S.; Eitzen E.; Fine A. D.; Hauer J.; Layton M.; Lillibridge S.; Osterholm M. T.; O’Toole T.; Parker G.; Perl T. M.; Russell P. K.; Swerdlow D. L.; Tonat K.; JAMA 2001, 285, 1059–1070. 10.1001/jama.285.8.1059. [DOI] [PubMed] [Google Scholar]
- Matsushita O.; Okabe A. Toxicon 2001, 39, 1769–1780. 10.1016/S0041-0101(01)00163-5. [DOI] [PubMed] [Google Scholar]
- Popoff M. R.; Bouvet P. Future Microbiol. 2009, 4, 1021–1064. 10.2217/fmb.09.72. [DOI] [PubMed] [Google Scholar]
- Burgeson R. E.; Nimni M. E. Clin. Orthop. Relat. Res. 1992, 282, 250–272. 10.1097/00003086-199209000-00033. [DOI] [PubMed] [Google Scholar]
- Brozek J.; Grande F.; Anderson J. T.; Keys A. Ann. N. Y. Acad. Sci. 1963, 110, 113–140. 10.1111/j.1749-6632.1963.tb17079.x. [DOI] [PubMed] [Google Scholar]
- Ramshaw J. A.; Shah N. K.; Brodsky B. J. Struct. Biol. 1998, 122, 86–91. 10.1006/jsbi.1998.3977. [DOI] [PubMed] [Google Scholar]
- Bruckner P.; Prockop D. J. Anal. Biochem. 1981, 110, 360–368. 10.1016/0003-2697(81)90204-9. [DOI] [PubMed] [Google Scholar]
- Bächinger H. P.; Bruckner P.; Timpl R.; Prockop D. J.; Engel J. Eur. J. Biochem. 1980, 106, 619–632. 10.1111/j.1432-1033.1980.tb04610.x. [DOI] [PubMed] [Google Scholar]
- Nagase H.; Visse R.; Murphy G. Cardiovasc. Res. 2006, 69, 562–573. 10.1016/j.cardiores.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Fields G. B. J. Biol. Chem. 2013, 288, 8785–8793. 10.1074/jbc.R113.451211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckhard U.; Huesgen P. F.; Brandstetter H.; Overall C. M. J. Proteomics 2014, 100, 102–114. 10.1016/j.jprot.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifter S.; Harper E. In The Enzymes; Boyer P. D.; Academic Press: New York, 1971; pp 649–697. [Google Scholar]
- Mookhtiar K. A.; Van Wart H. E. Matrix Suppl. 1992, 1, 116–126. [PubMed] [Google Scholar]
- Rasko D. A.; Sperandio V. Nat. Rev. Drug Discovery 2010, 9, 117–128. 10.1038/nrd3013. [DOI] [PubMed] [Google Scholar]
- Heras B.; Scanlon M. J.; Martin J. L. Br. J. Clin. Pharmacol. 2015, 79, 208–215. 10.1111/bcp.12356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clatworthy A. E.; Pierson E.; Hung D. T. Nat. Chem. Biol. 2007, 3, 541–548. 10.1038/nchembio.2007.24. [DOI] [PubMed] [Google Scholar]
- Storz M. P.; Maurer C. K.; Zimmer C.; Wagner N.; Brengel C.; de Jong C.; Lucas S.; Müsken M.; Häussler S.; Steinbach A.; Hartmann R. W. J. Am. Chem. Soc. 2012, 134, 16143–16146. 10.1021/ja3072397. [DOI] [PubMed] [Google Scholar]
- Lu C.; Maurer C. K.; Kirsch B.; Steinbach A.; Hartmann R. W. Angew. Chem., Int. Ed. 2014, 53, 1109–1112. 10.1002/anie.201307547. [DOI] [PubMed] [Google Scholar]
- Hung D. T.; Shakhnovich E. A.; Pierson E.; Mekalanos J. J. Science 2005, 310, 670–674. 10.1126/science.1116739. [DOI] [PubMed] [Google Scholar]
- Wagner S.; Sommer R.; Hinsberger S.; Lu C.; Hartmann R. W.; Empting M.; Titz A. J. Med. Chem. 2016, 59, 5929–5969. 10.1021/acs.jmedchem.5b01698. [DOI] [PubMed] [Google Scholar]
- Böttcher T.; Sieber S. A. J. Am. Chem. Soc. 2008, 130, 14400–14401. 10.1021/ja8051365. [DOI] [PubMed] [Google Scholar]
- Kassegne K.; Hu W.; Ojcius D. M.; Sun D.; Ge Y.; Zhao J.; Yang X. F.; Li L.; Yan J. J. Infect. Dis. 2014, 209, 1105–1115. 10.1093/infdis/jit659. [DOI] [PubMed] [Google Scholar]
- Lewis K. Nat. Rev. Drug Discovery 2013, 12, 371–387. 10.1038/nrd3975. [DOI] [PubMed] [Google Scholar]
- Payne D. J.; Gwynn M. N.; Holmes D. J.; Pompliano D. L. Nat. Rev. Drug Discovery 2007, 6, 29–40. 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- Graef F.; Vukosavljevic B.; Michel J.-P.; Wirth M.; Ries O.; De Rossi C.; Windbergs M.; Rosilio V.; Ducho C.; Gordon S.; Lehr C.-M. J. Controlled Release 2016, 243, 214–224. 10.1016/j.jconrel.2016.10.018. [DOI] [PubMed] [Google Scholar]
- Peterkofsky B. Methods Enzymol. 1982, 82, 453–471. 10.1016/0076-6879(82)82078-8. [DOI] [Google Scholar]
- Supuran C. T.; Scozzafava A.; Mastrolorenzo A. Expert Opin. Ther. Pat. 2001, 11, 221–259. 10.1517/13543776.11.2.221. [DOI] [Google Scholar]
- Eckhard U.; Schönauer E.; Nüss D.; Brandstetter H. Nat. Struct. Mol. Biol. 2011, 18, 1109–1114. 10.1038/nsmb.2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita O.; Jung C. M.; Katayama S.; Minami J.; Takahashi Y.; Okabe A. J. Bacteriol. 1999, 181, 923–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita O.; Jung C. M.; Minami J.; Katayama S.; Nishi N.; Okabe A. J. Biol. Chem. 1998, 273, 3643–3648. 10.1074/jbc.273.6.3643. [DOI] [PubMed] [Google Scholar]
- Matsushita O.; Koide T.; Kobayashi R.; Nagata K.; Okabe A. J. Biol. Chem. 2001, 276, 8761–8770. 10.1074/jbc.M003450200. [DOI] [PubMed] [Google Scholar]
- Wang Y.-K.; Zhao G.-Y.; Li Y.; Chen X.-L.; Xie B.-B.; Su H.-N.; Lv Y.-H.; He H.-L.; Liu H.; Hu J.; Zhou B.-C.; Zhang Y.-Z. J. Biol. Chem. 2010, 285, 14285–14291. 10.1074/jbc.M109.087023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung C. M.; Matsushita O.; Katayama S.; Minami J.; Sakurai J.; Okabe A. J. Bacteriol. 1999, 181, 2816–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond M. D.; Van Wart H. E. Biochemistry 1984, 23, 3085–3091. 10.1021/bi00308a036. [DOI] [PubMed] [Google Scholar]
- Eckhard U.; Schönauer E.; Brandstetter H. J. Biol. Chem. 2013, 288, 20184–20194. 10.1074/jbc.M112.448548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews B. W. Acc. Chem. Res. 1988, 21, 333–340. 10.1021/ar00153a003. [DOI] [Google Scholar]
- Oshima N.; Narukawa Y.; Takeda T.; Kiuchi F. J. Nat. Med. 2013, 67, 240–245. 10.1007/s11418-012-0665-8. [DOI] [PubMed] [Google Scholar]
- Supuran C. T.; Scozzafava A. Eur. J. Pharm. Sci. 2000, 10, 67–76. 10.1016/S0928-0987(99)00090-1. [DOI] [PubMed] [Google Scholar]
- Clare B. W.; Scozzafava A.; Supuran C. T. J. Med. Chem. 2001, 44, 2253–2258. 10.1021/jm010087e. [DOI] [PubMed] [Google Scholar]
- Scozzafava A.; Supuran C. T. Bioorg. Med. Chem. Lett. 2002, 12, 2667–2672. 10.1016/S0960-894X(02)00564-4. [DOI] [PubMed] [Google Scholar]
- Ilies M. A. M.; Banciu M. D.; Scozzafava A.; Ilies M. A. M.; Caproiu M. T.; Supuran C. T. Bioorg. Med. Chem. 2003, 11, 2227–2239. 10.1016/S0968-0896(03)00113-5. [DOI] [PubMed] [Google Scholar]
- Santos M. A.; Marques S.; Gil M.; Tegoni M.; Scozzafava A.; Supuran C. T. J. Enzyme Inhib. Med. Chem. 2003, 18, 233–242. 10.1080/1475636031000081134. [DOI] [PubMed] [Google Scholar]
- Jacobsen F. E.; Lewis J. A.; Cohen S. M. ChemMedChem 2007, 2, 152–171. 10.1002/cmdc.200600204. [DOI] [PubMed] [Google Scholar]
- Rouffet M.; Cohen S. M. Dalton Trans. 2011, 40, 3445–3454. 10.1039/c0dt01743d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper N. M. FEBS Lett. 1994, 354, 1–6. 10.1016/0014-5793(94)01079-X. [DOI] [PubMed] [Google Scholar]
- Grobelny D.; Galardy R. E. Biochemistry 1985, 24, 6145–6152. 10.1021/bi00343a017. [DOI] [PubMed] [Google Scholar]
- Vencill C. F.; Rasnick D.; Crumley K. V.; Nishino N.; Powers J. C. Biochemistry 1985, 24, 3149–3157. 10.1021/bi00334a012. [DOI] [PubMed] [Google Scholar]
- Dive V.; Yiotakis A.; Nicolaou A.; Toma F. Eur. J. Biochem. 1990, 191, 685–693. 10.1111/j.1432-1033.1990.tb19175.x. [DOI] [PubMed] [Google Scholar]
- Scozzafava A.; Supuran C. T. Bioorg. Med. Chem. 2000, 8, 637–645. 10.1016/S0968-0896(99)00316-8. [DOI] [PubMed] [Google Scholar]
- Scozzafava A.; Ilies M. A.; Manole G.; Supuran C. T. Eur. J. Pharm. Sci. 2000, 11, 69–79. 10.1016/S0928-0987(00)00089-0. [DOI] [PubMed] [Google Scholar]
- Supuran C. T.; Briganti F.; Mincione G.; Scozzafava A. J. Enzyme Inhib. 2000, 15, 111–128. 10.1080/14756360009030345. [DOI] [PubMed] [Google Scholar]
- Galardy R. E.; Grobelny D. Biochemistry 1983, 22, 4556–4561. 10.1021/bi00288a032. [DOI] [PubMed] [Google Scholar]
- Scozzafava A.; Supuran C. T. J. Med. Chem. 2000, 43, 3677–3687. 10.1021/jm000027t. [DOI] [PubMed] [Google Scholar]
- Scozzafava A.; Supuran C. T. Eur. J. Med. Chem. 2000, 35, 299–307. 10.1016/S0223-5234(00)00127-6. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. In Drug Design of Zinc-Enzyme Inhibitors; Supuran C. T., Winum J.-Y., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, 2009; pp 721–729. [Google Scholar]
- Van Wart H. E.; Steinbrink D. R. Anal. Biochem. 1981, 113, 356–365. 10.1016/0003-2697(81)90089-0. [DOI] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. J. Med. Chem. 2010, 53, 2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Wünsch E.; Heidrich H. H.-G. Hoppe-Seyler's Z. Physiol. Chem. 1963, 333, 149–151. 10.1515/bchm2.1963.333.1.149. [DOI] [PubMed] [Google Scholar]
- Komsa-Penkova R. S.; Rashap R. K.; Yomtova V. M. J. Biochem. Biophys. Methods 1997, 34, 237–249. 10.1016/S0165-022X(97)00016-X. [DOI] [PubMed] [Google Scholar]
- Watanabe K. Appl. Microbiol. Biotechnol. 2004, 63, 520–526. 10.1007/s00253-003-1442-0. [DOI] [PubMed] [Google Scholar]
- Eckhard U.; Schönauer E.; Ducka P.; Briza P.; Nüss D.; Brandstetter H. Biol. Chem. 2009, 390, 11–18. 10.1515/BC.2009.004. [DOI] [PubMed] [Google Scholar]
- Schilling O.; Overall C. M. Nat. Biotechnol. 2008, 26, 685–694. 10.1038/nbt1408. [DOI] [PubMed] [Google Scholar]
- Fields G. B. Methods Mol. Biol. 2010, 622, 393–433. 10.1007/978-1-60327-299-5_24. [DOI] [PubMed] [Google Scholar]
- Knight C. G. Methods Enzymol. 1995, 248, 18–34. 10.1016/0076-6879(95)48004-8. [DOI] [PubMed] [Google Scholar]
- Rasmussen H. S.; McCann P. P. Pharmacol. Ther. 1997, 75, 69–75. 10.1016/S0163-7258(97)00023-5. [DOI] [PubMed] [Google Scholar]
- Cathcart J.; Pulkoski-Gross A.; Cao J. Genes Dis. 2015, 2, 26–34. 10.1016/j.gendis.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overall C. M.; López-Otín C. Nat. Rev. Cancer 2002, 2, 657–672. 10.1038/nrc884. [DOI] [PubMed] [Google Scholar]
- Park H. I.; Jin Y.; Hurst D. R.; Monroe C. A.; Lee S.; Schwartz M. A.; Sang Q.-X. A. J. Biol. Chem. 2003, 278, 51646–51653. 10.1074/jbc.M310109200. [DOI] [PubMed] [Google Scholar]
- Zhu J.; Cai X.; Harris T. L.; Gooyit M.; Wood M.; Lardy M.; Janda K. D. Chem. Biol. 2015, 22, 483–491. 10.1016/j.chembiol.2015.03.012. [DOI] [PubMed] [Google Scholar]
- Gomis-Rüth F. X.; Botelho T. O.; Bode W. Biochim. Biophys. Acta, Proteins Proteomics 2012, 1824, 157–163. 10.1016/j.bbapap.2011.04.014. [DOI] [PubMed] [Google Scholar]
- Cerdà-Costa N.; Xavier Gomis-Rüth F. Protein Sci. 2014, 23, 123–144. 10.1002/pro.2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomis-Rüth F. X. J. Biol. Chem. 2009, 284, 15353–15357. 10.1074/jbc.R800069200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramar Y.; Das Gupta V.; Bethea C. J. Clin. Pharm. Ther. 1992, 17, 185–189. 10.1111/j.1365-2710.1992.tb01291.x. [DOI] [PubMed] [Google Scholar]
- Labute P. Proteins: Struct., Funct., Genet. 2009, 75, 187–205. 10.1002/prot.22234. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.