

Abstract
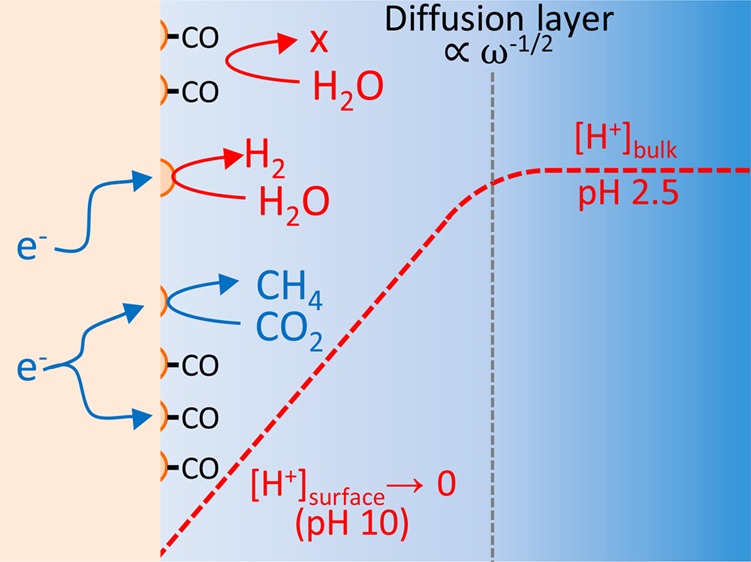
Understanding the competition between hydrogen evolution and CO2 reduction is of fundamental importance to increase the faradaic efficiency for electrocatalytic CO2 reduction in aqueous electrolytes. Here, by using a copper rotating disc electrode, we find that the major hydrogen evolution pathway competing with CO2 reduction is water reduction, even in a relatively acidic electrolyte (pH 2.5). The mass-transport-limited reduction of protons takes place at potentials for which there is no significant competition with CO2 reduction. This selective inhibitory effect of CO2 on water reduction, as well as the difference in onset potential even after correction for local pH changes, highlights the importance of differentiating between water reduction and proton reduction pathways for hydrogen evolution. In-situ FTIR spectroscopy indicates that the adsorbed CO formed during CO2 reduction is the primary intermediate responsible for inhibiting the water reduction process, which may be one of the main mechanisms by which copper maintains a high faradaic efficiency for CO2 reduction in neutral media.
Introduction
The electrochemical reduction of CO2 has received much attention in recent years1−12 as a potential method to produce useful chemicals from an abundant carbon source and renewable electricity. Target products include hydrocarbons such as methane4,5,10 and ethylene,5,6,10 or chemical feedstock such as formate,7,8 aldehydes,8,9 and alcohols9 which may be used for further chemical synthesis. This would effectively shift the carbon source from fossil fuels to (atmospheric) CO2, which would greatly enhance the sustainability and carbon-neutrality of modern society. However, one major challenge for CO2 reduction in aqueous electrolytes is the loss of faradaic efficiency due to the simultaneous evolution of hydrogen (HER, hydrogen evolution reaction),13,14 which may occur either through the reduction of protons (2H++2e– →H2) or through the reduction of the solvent molecule itself (2H2O+2e– → H2 + 2OH–). It is therefore of primary interest to uncover how the faradaic efficiency of CO2 reduction can be increased with respect to HER.
Recent reports have attempted to rationalize the preferred pathways based on the calculation of onset potentials by density functional theory (DFT).15,16 By focusing on the reaction step with the most negative potential in a given pathway, the onset potential of HER and CO2 reduction can be calculated based on the binding energies of H and CO2 related intermediates. For example, Durand et al. have found Cu (211) facets to be favored compared to Cu (111) and (100) facets15 based on a combined effect of a smaller thermodynamic driving force necessary for CO2 reduction and a larger driving force necessary for HER.
While the thermodynamic model is a powerful approach, it does not currently account for the two pathways of HER. They are identical within this framework due to the assumption that proton/hydroxide transfer is always accompanied by concerted electron transfer, meaning that pH is not an explicit parameter in the model. In reality, however, water reduction is expected to dominate over proton reduction at higher pH where the proton concentration is low. This effect may take place even in relatively acidic electrolytes because the local proton concentration at the cathode decreases during HER18 and CO2 reduction due to slow mass transport.19,20 The onset potential and kinetics of the two pathways may also change with pH, as is, for instance, well-known for HER on platinum electrodes.21−23 In the remainder of this article, we will refer to “proton reduction” as hydrogen evolution from acidic media showing a direct dependence on proton concentration, and to “water reduction” as hydrogen evolution from neutral media showing no direct dependence on proton concentration. We realize that the actual state of the proton or proton donor may be more complex. The importance of the proton source in hydrogen evolution has been demonstrated recently in organic solvents by Jackson et al.,24 who showed that steric effects of the proton donor drastically affected the HER Tafel slope, even when the pKa of the proton donors were similar. Differentiating the proton source is especially important from the viewpoint of discussing the competition of HER and CO2 reduction because the presence of CO2 may affect the two HER pathways differently. Furthermore, CO2 may act as or form pH buffers,19,20 adsorbates,17 and proton donor/acceptors25 during the reaction, all of which may interact differently with the HER pathway. Therefore, it is important to study the effects of CO2 on the two pathways of HER by explicitly taking into account the effect of mass transport and local pH at the electrode.19,20
Here, we have studied the competition of CO2 reduction with the overall HER rate from the reduction of both water and protons on a copper electrode, which is the most extensively studied electrocatalyst for CO2 reduction. The usage of a rotating disc electrode (RDE) system has allowed us to systematically change the flux of protons to the electrode–electrolyte interface, which in turn allows estimations of the local pH. Our results show that CO2 reduction does not significantly interfere with proton reduction in acidic electrolytes. Instead, the HER pathway that competes with CO2 reduction is the reduction of water. FTIR measurements suggest that this selective inhibition takes place through adsorbed CO. In addition to the different interactions with CO2 reduction, the onset potential of the two HER pathways is different even after compensating for local pH changes. These results highlight the importance of explicitly differentiating between HER from proton reduction and water reduction, and provide new insight into how copper reduces CO2 efficiently by suppressing HER.
Experimental Section
All electrolytes were prepared using HPLC (= 99.0%) grade sodium perchlorate monohydrate (Fluka, 7791–07–3) and 60 wt % perchloric acid (Merck, 7601–90–3), and were used without further purification. A phosphate stock solution (pH 2.5; total concentration of phosphate ions, 10 M) was made from NaH2PO4·H2O (Merck, 10049-21-5) and H3PO4 (Merck,7664-38-2), and was diluted as necessary with the perchlorate solution to yield an electrolyte with the desired buffer concentration. The HCHO used (Sigma-Aldrich, 50-00-0) contained ca. 10–15 wt % CH3OH as a stabilizing agent. All electrolytes were bubbled with the appropriate gas (Ar, CO, or CO2) 30 min prior to the electrochemical measurement. The pH change due to switching from Ar atmosphere to CO2 is negligible, based on pH meter readings (ΔpH < 0.05) and the identical height of the diffusion plateau of HER during the CVs. The pH value of 2.5 was intentionally chosen because the proton concentration is high enough to ensure the pH does not change upon CO2 bubbling but low enough that the diffusion plateau for proton reduction can be observed at low current densities. The copper rotating disc electrode was polished to a mirror finish using alumina paste (1.0 μm, 0.3 μm, and 0.05 μm) and then sonicated before use. The copper thin film used for the FTIR measurements, which were performed with a Bruker Vertex 80 V IR spectrophotometer (with an MCT detector and p-polarized light, in the external reflection configuration), was deposited on the silicon prism via sputtering and was used without further cleaning. No electropolishing was conducted in any of the experiments. The spectra correspond to an average of 100 scans with 8 cm–1 resolution. The reference and counter electrodes were a commercial RHE (Gaskatel Hydroflex) and platinum wire, respectively, with the exception of the FTIR measurements where we employed a Ag/AgCl sat. reference electrode due to the cell configuration. All potentials are reported versus the RHE. Ohmic resistance was compensated during the measurements using the IVIUM potentiostat software (Ivium Soft) for the CVs and after the measurements for the FTIR measurements.
Results and Discussion
Figure 1 shows a
cyclic voltammogram (CV) of a polycrystalline copper rotating disc
electrode (RDE) in an Ar-saturated 0.1 M NaClO4 electrolyte
(pH 2.5). In the case of an unbuffered electrolyte (black lines),
the reduction current at ca. −0.5 V vs RHE (Reversible Hydrogen
Electrode) leads to a plateau current which correlates with the square
root of the rotation rate, indicating mass transport limitation.27 We ascribe this current to hydrogen evolution
from proton reduction,23 where the notation
“proton” refers collectively to all chemical species
which can be considered solvated forms of a hydrogen/proton nucleus.
The current which increases from ca. −1.2 V vs RHE, on the
other hand, is independent from proton mass transport. As the only
other proton (hydrogen nucleus) donor in this system is the water
molecule itself (the pKa of perchlorate
is −15, indicating that it is deprotonated under our experimental
conditions26), we assign this current to
HER from water reduction. The mass transport limitation of protons
leads to a discrepancy between the pH at the surface of the electrode
and the bulk electrolyte, which makes accurate determination of the
actual overpotential for water reduction difficult. Regardless of
the true overpotential, however, the marked difference in the apparent
onset potential between proton reduction and water reduction is an
observation which highlights the importance of explicitly distinguishing
between the two pathways. The RDE configuration allows precise control
over the diffusion layer thickness and the mass transport flux of
protons. Therefore, the surface pH relative to the bulk electrolyte
at a specific potential can be estimated based on the ratio of the
total current density at the potential to the plateau current density
(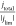 ; see modeling section for details). For
example, the local pH at −0.8 V is estimated to be c.a. 4.5
because the current density of proton reduction at −0.8 V corresponds
to 99% of the limiting current. As the current of proton reduction
is defined by mass transport, the proton concentration at the surface
of the electrode can be estimated to be 1% of that in the bulk electrolyte
(pH 4.5). CVs with an RHE potential scale which has been corrected
for local pH changes will be presented later in this study.
; see modeling section for details). For
example, the local pH at −0.8 V is estimated to be c.a. 4.5
because the current density of proton reduction at −0.8 V corresponds
to 99% of the limiting current. As the current of proton reduction
is defined by mass transport, the proton concentration at the surface
of the electrode can be estimated to be 1% of that in the bulk electrolyte
(pH 4.5). CVs with an RHE potential scale which has been corrected
for local pH changes will be presented later in this study.
Figure 1.
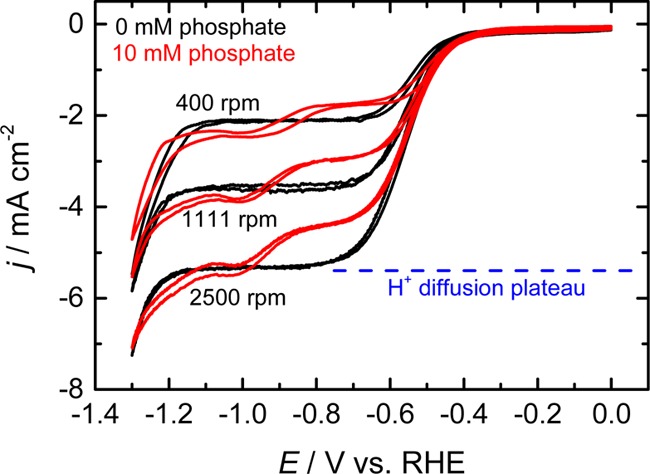
CVs of a polycrystalline copper RDE with (red) and without (black) 10 mM phosphate in a 0.1 M NaClO4 solution (pH 2.5) under Ar-saturated conditions. Scan rate: 50 mV/s.
It should be noted that the change in local pH is also manifested in a buffered solution, as shown by the red lines in Figure 1. The presence of a plateau current can be seen even after the addition of 10 mM phosphate, indicating that the redox active species (protons) are depleted at the electrode surface (the plateau current at ca. −0.8 V is smaller than that in the unbuffered case due to the slightly higher pH in the buffered solution). The gradual increase of the pH at the vicinity of the electrode can also be observed from the increase in the plateau current at c.a. −1.0 V. When the pH at the electrode surface becomes more alkaline than the pKa2 of phosphate (7.2), the phosphate species at the electrode are deprotonated to form HPO4– species. This allows the transport of H2PO4– from the bulk electrolyte to make an additional contribution to the total proton flux. Such an effect is not unique to phosphate but may occur with other buffer molecules such as bicarbonate species, if the concentration and buffering capacity is sufficiently high, as will be discussed later in the article based on a quantitative model.
The transition of the HER pathway from proton reduction to water reduction in Figure 1 occurs at approximately −5.6 mA/cm2 at 2500 rpm, where the mass transport of protons becomes insufficient. However, the limiting current is linearly proportional to the concentration of protons, and therefore, the water reduction pathway will become increasingly important in more alkaline conditions. The limiting current of protons at pH 7 is approximately 0.2 μA/cm2 even at 2500 rpm, indicating the majority of the hydrogen detected in studies using stationary electrodes in mildly alkaline electrolytes is likely to be derived from water reduction.
Figure 2 shows the CVs measured in a CO2-saturated electrolyte for different values of the disc rotation rate. Compared to the Ar-saturated solution (black lines), the onset of the proton reduction current in CO2-saturated solution is slightly shifted to a more negative potential. In contrast, a more pronounced suppression is observed for the water reduction current at potentials more negative than −1.2 V, suggesting that CO2 interferes more strongly with the water reduction process than with proton reduction. This suppression in activity is not due to impurities in the electrolyte, as it does not occur in an Ar atmosphere. Buffering effects are also minimal, as no increase in proton flux could be observed, even when the pH at the electrode surface becomes more alkaline than the pKa1 of H2CO3 (3.6). This is most likely due to the low buffer capacity arising from the low solubility of CO2 in acidic solutions.
Figure 2.
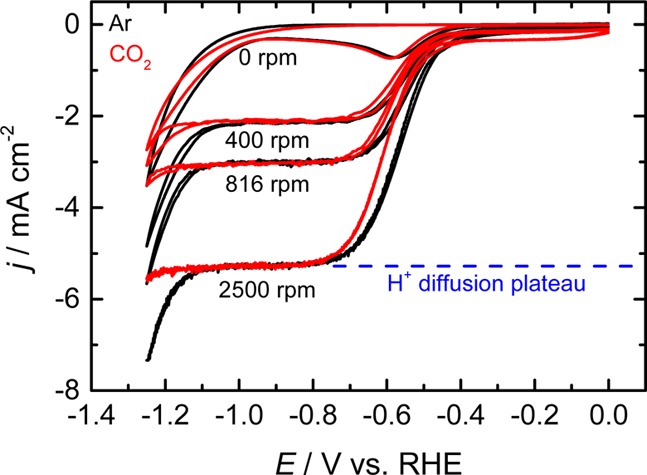
CVs of a polycrystalline copper RDE at various rotation rates in 0.1 M NaClO4 solution (pH 2.5) saturated with Ar (black lines) and CO2 (red lines). No bulk pH change was observed upon CO2 saturation as evidenced by the identical height of the limiting current. Scan rate: 50 mV/s.
In order to gain insight into the origin of this selective inhibitory behavior, CVs were measured in an electrolyte solution saturated with CO (Figure 3A). CO is the two-electron reduction product of CO228,29 and has been proposed to be the inhibitor of HER due to site-blocking effects.30,31 However, similar to the case with CO2, we find that the proton reduction process is not inhibited by the presence of CO as much as the water reduction process. The different interactions between the two pathways of HER with CO2 reduction further emphasizes the importance of explicitly distinguishing the substrate molecule for HER. The inhibitory effect was not observed when further reduced forms of CO2 such as HCHO and CH3OH were present in the electrolyte (Figure 3B). Although a portion of HCHO is known to hydrate to methanediol in water, these results nonetheless show that excessively reduced adsorbates cannot reproduce the inhibitory effects of CO2 or CO toward water reduction. These observations suggest the origin of the inhibitory effect comes primarily from CO adsorbed on the surface of copper.
Figure 3.
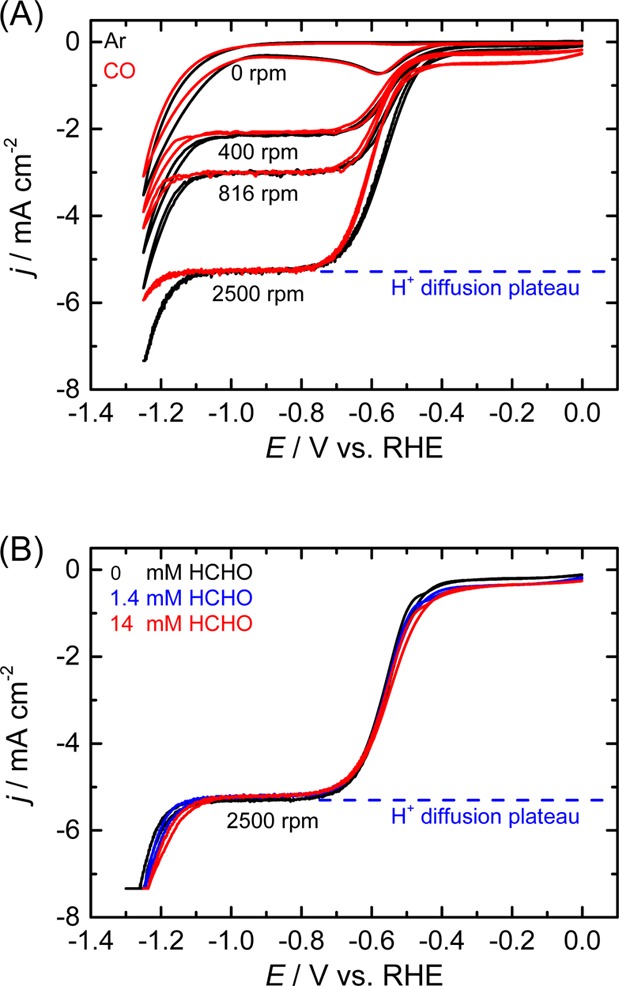
CVs of a polycrystalline copper electrode before (black) and after (red and blue) the addition of reduced forms of CO2 to a 0.1 M NaClO4 electrolyte (pH 2.5). (A) Ar-saturated solution resaturated with CO. (B) HCHO addition in Ar atmosphere. Scan rate: 50 mV/s.
The inhibitory effects of CO and CO2 on water reduction behave similarly with respect to the rotation rate, as shown in Figure 4. As the proton reduction rate is represented by its limiting current density, subtracting this limiting current from the total reduction current in either Ar atmosphere or CO2/CO atmosphere is expected to express the sum of the reaction rates for water reduction and CO2 (or CO) reduction. Therefore, comparing this reduction current at a potential in the water reduction regime in CO or CO2 atmosphere to the reduction current in Ar atmosphere illustrates to which extent CO and CO2 inhibit water reduction at different mass transport rates. If the CO or CO2 reduction current is negligible, the y-axis represents the rate of water reduction in CO2 or CO atmosphere relative to that in Ar. In both cases, water reduction is inhibited drastically from 0 to 500 rpm, whereas higher rotation rates have a less significant effect. The strong inhibition at low rotation rates (ω < 500 rpm) should be due to the increased coverage of CO on the copper surface due to the efficient transport of CO and CO2. This effect apparently saturates at higher rotation rates presumably because the CO coverage reaches a constant value. The current density in the case of CO-saturated electrolyte is larger than that in the CO2-saturated electrolyte. This may be due to CO2 reduction generating other inhibitory adsorbates such as formate. Another possibility is the difference in the CO coverage in different atmospheres, as a result of the different solubilities of CO2 and CO in water. In any case, further experiments would be necessary to uncover the difference in inhibitory effects.
Figure 4.
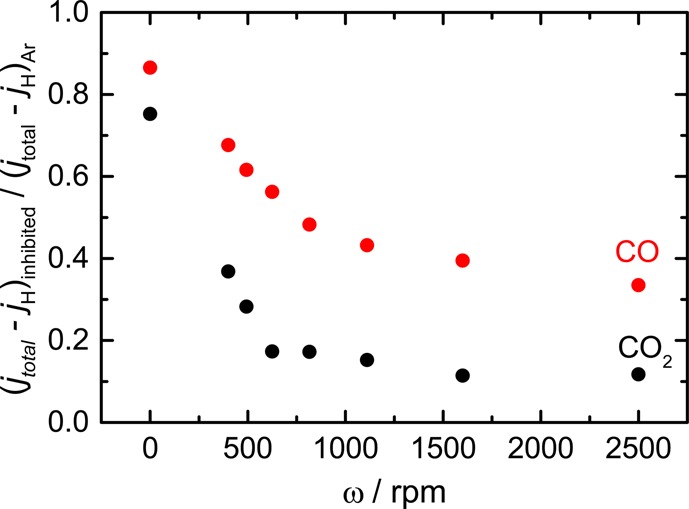
Inhibitory effects of CO2 (black) and CO (red) plotted with respect to the rotation rate of the copper RDE. The y-axis is the current density in each atmosphere normalized by that in an Ar-saturated atmosphere. A lower value of the ratio plotted on the y-axis implies a stronger inhibition of HER from water reduction. The limiting current density corresponding to the rate of proton reduction jH was subtracted to highlight the inhibition of the water reduction pathway. Ratios of current density values were calculated at −1.25 V vs RHE.
In order to confirm that the inhibitory effect of CO2 reduction on HER arises from adsorbed CO, in situ FTIR (Fourier transform infrared) spectra were measured in an ATR-IR (attenuated total reflection infrared) configuration (Figure 5). With the spectrum at 0 V as the reference, the IR band at ca. 2050 cm–1, which we assign to adsorbed CO,31 increases from −0.8 V during the negative potential step and remains until −1.2 V. Although CO is known to be further reduced at negative potentials,28,29 the constant CO coverage at water reduction potentials (E ∼ −1.2 V) indicates that an inhibitory interaction such as site blocking or a shift in the hydrogen binding energy32 is possible. The coverage of COads at potentials less negative than −0.8 V appears to be too low to have a significant inhibitory influence on proton reduction. The interaction between CO and copper is known to be favorable for CO2 reduction based on theoretical15,16 and experimental5,6,10 observations that CO can adsorb and desorb on copper efficiently. However, such an intermediate binding energy also allows the copper surface to maintain a high CO coverage during CO2 reduction, which enhances the faradaic efficiency with respect to HER. We have also observed the growth of vibrational bands 1435 and 1280 cm–1 at potentials more negative than −0.9 V (see Figure S1 in the Supporting Information). These bands resemble the IR bands of bicarbonate, in accord with the depletion of protons at the electrode surface.33
Figure 5.
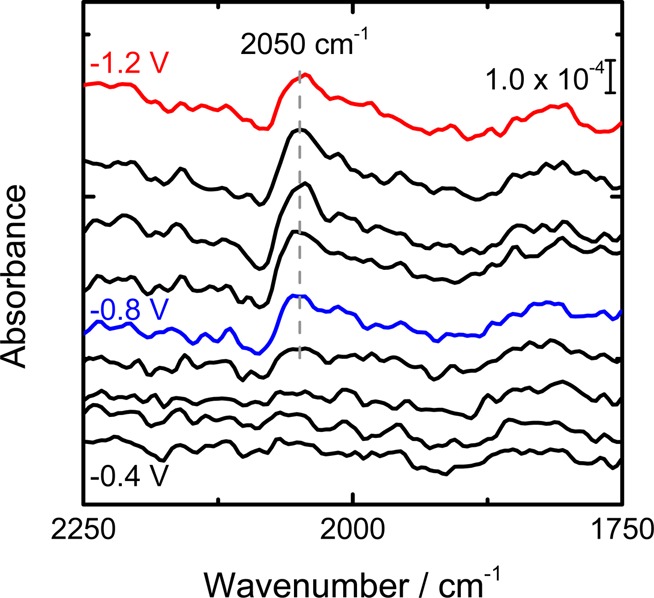
FTIR spectra of a polycrystalline copper electrode in 0.1 M NaClO4 electrolyte (pH 2.5). The copper film was deposited on the silicon prism by sputtering. The solution was saturated with CO2 for 30 min, after which the reference spectrum was measured at 0 V. The spectra were measured from −0.4 V to −1.2 V in 0.1 V intervals.
The idea of HER being inhibited by adsorbed CO has been proposed in the past.17,30 However, it should be noted that previous literature has not explicitly differentiated between the two HER pathways, which show marked differences in terms of apparent onset potentials (Figures 1–3) and interaction with adsorbed CO (Figure 5). As the pH change at the surface of the electrode would lead to an overestimation of the difference in overpotential, it is important to compensate for the change of pH for a better comparison. By modeling the local pH,18−20 we can estimate the true onset potential of the water reduction reaction and also show that CO2 buffering effects should indeed be negligible under our conditions.
As Auinger et al. have shown,18 the amount of protons consumed and hydroxide ions generated at the electrode due to HER corresponds to the mass transport flux of each ion under steady-state conditions, which should apply to our experiments. Therefore, the total current density (jtotal) can be expressed using the mass transport fluxes of H+ (JH) and OH– (JOH) as follows:
| 1 |
| 2 |
where x is the distance from the electrode, F is the faraday constant, [X] is the concentration of X, DX is the diffusion coefficient, and δX is the diffusion layer thickness. Subscripts b and s indicate bulk and surface values, and subscripts H and OH indicate protons and hydroxyl ions, respectively. δH and δOH depend on the rotation rate of the RDE following the Levich equation. Normalizing jtotal by the absolute value of the limiting current density23
| 3 |
generates a rotation-rate independent parameter.
| 4 |
Finally, assuming that water hydrolysis is in equilibrium allows for the substitution of hydroxide ion concentrations using the proton concentrations and the water dissociation constant Kw. The local proton concentration at the electrode can therefore be expressed as
| 5 |
where
| 6 |
This equation allows for the estimation
of
the surface pH based on the normalized current density, assuming that
water dissociation is the only solution equilibrium that protons or
hydroxide ions are involved in. An important implication from these
derivations is that the surface pH is a function of 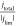 , not the raw rotation rate. This is because
although a faster rotation rate generates a larger proton mass transport
flux, the local pH is dictated based on the balance with the consumption
rate which is represented by the current density. Therefore,
, not the raw rotation rate. This is because
although a faster rotation rate generates a larger proton mass transport
flux, the local pH is dictated based on the balance with the consumption
rate which is represented by the current density. Therefore, 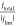 can be interpreted as a normalized rotation
rate, which shows the ratio of proton supply and consumption.
can be interpreted as a normalized rotation
rate, which shows the ratio of proton supply and consumption.
Figure 6 shows the results of the modeling based on eq 4. It can be seen that when the current density approaches the limiting current, the surface pH exhibits a sharp change from pH 4 to 10 in a narrow current range. The increase of pH at the electrode surface at current densities greater than the limiting current is sufficiently high for carbonic acid to deprotonate and generate bicarbonate and carbonate ions. This suggests that a pH buffering effect may be present even in unbuffered electrolytes if the mass transport of protons is slow compared to the rate of HER. Therefore, in a CO2 saturated electrolyte, it becomes necessary to model the electrode pH assuming the presence of a buffer molecule.
Figure 6.
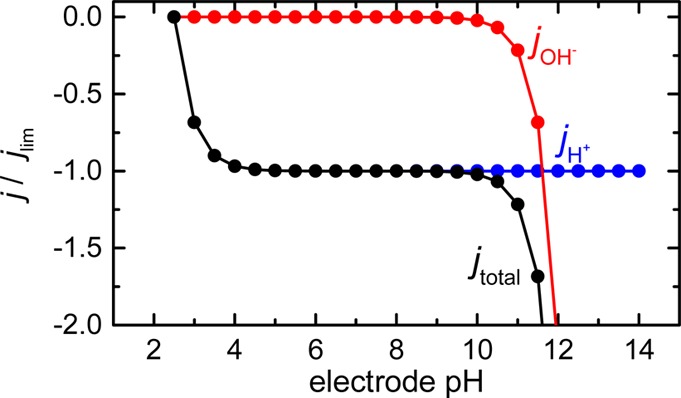
Estimation of the normalized current density based on the pH near the electrode surface (“electrode pH”) (eq 4). The first and second terms of eq 4 are shown in blue and red, and their total is shown in black. The diffusion layer thicknesses were calculated from the Levich equation using a kinematic viscosity of 1 cm2/s, DH = 9.31 × 10–5 [cm2/s], and DOH = 5.27 × 10–5 [cm2/s].18 The rotation rate plays no role due to the normalization with the limiting current.
The effect of buffer ions in the solution and their effect on local pH can be modeled using an approach similar to the one above. Assuming a buffer ion with the acid–base equilibrium (HA ⇆ H+ + A–, equilibrium constant Ka), the difference between bulk pH and surface pH can be suppressed because the flux of buffer molecules makes an additional contribution to the net proton flux. In steady state, the following equation should hold:
| 7 |
which is equivalent to
| 8 |
Given the acid–base equilibrium between HA and A–, this leads to the following solution for [A]s:
| 9 |
where
| 10 |
The current density in a buffered solution is given by
| 11 |
so that the normalized current density can be expressed as
| 12 |
The numerical solution to this equation
at various buffer concentrations
is shown in Figure 7, modeled for a bulk pH of 2.5 and using the diffusion constants
of CO2 and HCO3– as the protonated
and deprotonated forms of buffer molecule. When the buffer concentration
is low compared to the proton concentration (panels A and B), the
voltammogram resembles the unbuffered case. However, when the proton
concentration and buffer concentration are within an order of magnitude
(panels B and C), the buffer molecule makes a noticeable contribution
at surface pH > pKa. This can be understood
by comparing the first and third terms (proton flux and buffer flux,
respectively) of eq 12. As maximum flux is attained when [H]b ≫ [H]s and [HA]b ≫
[HA]s, the third term
becomes significant when 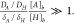
Figure 7.
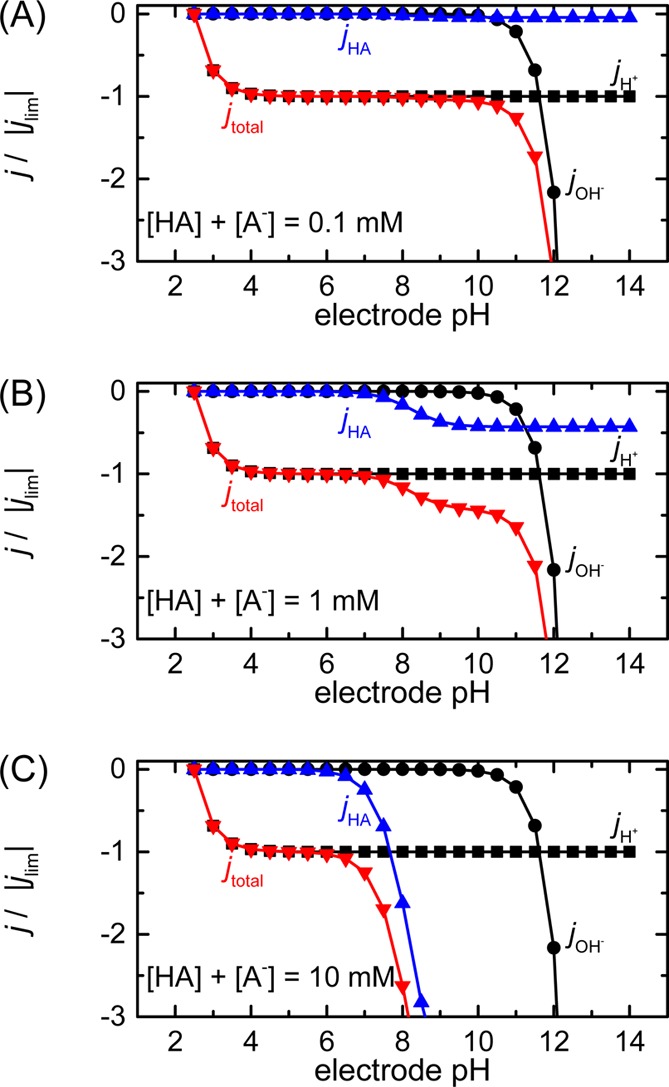
Estimation of the normalized current density based on the pH near the electrode surface (“electrode pH”) in a buffered solution with three different total buffer concentrations (A, 0.1 mM; B, 1 mM; and C, 10 mM). The contributions of the proton and hydroxide ion flux (black squares and circles, respectively) are the same as those in the unbuffered case. The total current density and the current density due to the flux of protons transported by HA are shown in red and blue triangles, respectively. Diffusion constants of CO2 and HCO3– were used as the values for the protonated and deprotonated forms of the buffer molecule (DHA = 1.47 × 10–8 [cm2/s] and DA = 7.02 × 10–9 [cm2/s]).34 All other parameters are the same as those in Figure 6.
In the case of an electrolyte in equilibrium with
1 atm of CO2, the concentration of carbonic acid (H2CO3) is around 6 × 10–5 M,
based on the
hydration equilibrium constant35 [H2CO3]/[CO2] = 1.7 × 10–3 and the solubility of CO2 (1.5 g/L).36 On the basis of the pKa of
carbonic acid (pKa = 3.6) and the diffusion
coefficient of bicarbonate 7.02 × 10–9 [cm2/s],34 this would lead to 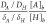 = 0.00125 ≪ 1, indicating that the
buffer capacity of CO2 is too small to play a role as a
pH buffer in our results. This is in accord with the identical limiting
current in the CVs before and after CO2 was introduced
into the electrochemical cell (Figure 2). However, changing the electrolyte pH has marked
consequences, as [A]b will increase simultaneously with
the decrease of [H]b. For example,
= 0.00125 ≪ 1, indicating that the
buffer capacity of CO2 is too small to play a role as a
pH buffer in our results. This is in accord with the identical limiting
current in the CVs before and after CO2 was introduced
into the electrochemical cell (Figure 2). However, changing the electrolyte pH has marked
consequences, as [A]b will increase simultaneously with
the decrease of [H]b. For example, 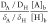 = 1.25 for a CO2-saturated
solution
at pH 4, indicating that buffering effects from bicarbonate play a
role at pH > 4.
= 1.25 for a CO2-saturated
solution
at pH 4, indicating that buffering effects from bicarbonate play a
role at pH > 4.
On a more qualitative note, the results in Figure 7 show that in the presence of a 10 mM buffer, the proton reduction current wave develops two plateaus, in agreement with the experimental results in Figure 1.
The lack of pH buffering effects
from CO2 indicates
that equations without explicitly taking buffering effects into account
can be applied to CO2-saturated solutions. Specifically, eqs 56 can be used to calculate the local pH at the surface of the electrode
during the CVs. Compared to Figure 2, which shows the current density with respect to the
RHE at the bulk pH, the CVs in Figure 8 show the normalized current density with respect to
the RHE by correcting the potential (Ecorrected) using the pH existing at the electrode surface. The large horizontal
noise near 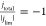 is due to the large pH change (4 < pH
< 10), which amplifies the noise within the current density data
from the original CV. The marked shift of the onset potential of water
reduction compared to the original voltammogram is noteworthy. In
the original CVs, proton reduction appeared to be favored over water
reduction due to the 600 mV smaller “apparent” overpotential.
A large part of this is due to the difference between the bulk pH
and the pH at the surface of the working electrode. However, there
is still a 200 mV difference in the onset potential even after correcting
for the local pH change, indicating the activation energy for water
reduction is larger than that of proton reduction. The larger activation
energy for water reduction is in accord with previous HER studies
on platinum in acid and alkaline electrolytes.22,23 Therefore, these studies highlight the importance of differentiating
between the two HER pathways for discussing the competition with CO2 reduction.
is due to the large pH change (4 < pH
< 10), which amplifies the noise within the current density data
from the original CV. The marked shift of the onset potential of water
reduction compared to the original voltammogram is noteworthy. In
the original CVs, proton reduction appeared to be favored over water
reduction due to the 600 mV smaller “apparent” overpotential.
A large part of this is due to the difference between the bulk pH
and the pH at the surface of the working electrode. However, there
is still a 200 mV difference in the onset potential even after correcting
for the local pH change, indicating the activation energy for water
reduction is larger than that of proton reduction. The larger activation
energy for water reduction is in accord with previous HER studies
on platinum in acid and alkaline electrolytes.22,23 Therefore, these studies highlight the importance of differentiating
between the two HER pathways for discussing the competition with CO2 reduction.
Figure 8.

CVs after correcting for the local pH change at the electrode based on eqs 5 and 6. Original experimental data are the same as those in Figure 2. Black lines, Ar atmosphere; red lines, CO2 atmosphere.
Summary and Conclusions
In conclusion, the main HER pathway competing with CO2 reduction on a copper electrode was found to be water reduction, even in relatively acidic (pH 2.5) electrolytes, where the proton reduction reaction is a diffusion-limited process hardly influenced by the presence of CO2 and CO. We find that the water reduction pathway is specifically inhibited by adsorbed CO and that this “CO poisoning” leads to a high faradaic efficiency for CO2 reduction on copper electrodes. The inhibition of the HER pathway becomes more pronounced when the mass transport of CO2 is improved. Water reduction appears to be a slower process than proton reduction even after correcting for the pH change at the electrode. The different interactions with adsorbed CO, along with the difference in overpotential, highlight the importance of differentiating between water reduction and proton reduction HER pathways.
Acknowledgments
We express our thanks to A. F. Beker and T. Mechielsen who fabricated the copper electrode, and to Dr. Ioannis Katsounaros for helpful discussions. H.O. received financial support from the Japan Society for the Promotion of Science (JSPS).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.langmuir.7b00696.
FTIR spectra showing the vibration region for bicarbonate and carbonate (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Qiao J.; Liu Y.; Hong F.; Hong F.; Zhang J. A Review of Catalysts for the Electroreduction of Carbon Dioxide to Produce Low-Carbon Fuels. Chem. Soc. Rev. 2014, 43, 631–675. 10.1039/C3CS60323G. [DOI] [PubMed] [Google Scholar]
- Loiudice A.; Lobaccaro P.; Kamali E. A.; Thao T.; Huang B. H.; Ager J. W.; Buonsati R. Tailoring Copper Nanocrystals towards C2 Products in Electrochemical CO2 Reduction. Angew. Chem., Int. Ed. 2016, 55, 5789–5792. 10.1002/anie.201601582. [DOI] [PubMed] [Google Scholar]
- Kortlever R.; Shen J.; Schouten K. J. P.; Calle-Vallejo F.; Koper M. T. M. Catalysts and Reaction Pathways for the Electrochemical Reduction of Carbon Dioxide. J. Phys. Chem. Lett. 2015, 6, 4073–4082. 10.1021/acs.jpclett.5b01559. [DOI] [PubMed] [Google Scholar]
- Kas R.; Kortlever R.; Milbrat A.; Koper M. T. M.; Mul G.; Baltrusaitis J. Electrochemical CO2 Reduction on Cu2O-derived Copper Nanoparticles: Controlling the Catalytic Selectivity of Hydrocarbons. Phys. Chem. Chem. Phys. 2014, 16, 12194–12201. 10.1039/c4cp01520g. [DOI] [PubMed] [Google Scholar]
- Schouten K. J. P.; Gallent E. P.; Koper M. T. M. The Influence of pH on the Reduction of CO and CO2 to Hydrocarbons on Copper Electrodes. J. Electroanal. Chem. 2014, 716, 53–57. 10.1016/j.jelechem.2013.08.033. [DOI] [Google Scholar]
- Chen C. S.; Handoko A. D.; Wan J. H.; Ma L.; Ren D.; Yeo B. S. Stable and Selective Electrochemical Reduction of CO to Ethylene on Copper Mesocrystals. Catal. Sci. Technol. 2015, 5, 161–168. 10.1039/C4CY00906A. [DOI] [Google Scholar]
- Kortlever R.; Tan K. H.; Kwon Y.; Koper M. T. M. Electrochemical Carbon Dioxide and Bicarbonate Reduction on Copper in Weakly Alkaline Media. J. Solid State Electrochem. 2013, 17, 1843–1849. 10.1007/s10008-013-2100-9. [DOI] [Google Scholar]
- Kuhl K. P.; Cave E. R.; Abram D. N.; Jaramillo T. F. New Insights into the Electrochemical Reduction of Carbon Dioxide on Metallic Copper Surfaces. Energy Environ. Sci. 2012, 5, 7050–7059. 10.1039/c2ee21234j. [DOI] [Google Scholar]
- Kuhl K. P.; Hatsukade T.; Cave E. R.; Abram D. N.; Kibsgaard J.; Jaramillo T. F. Electrocatalytic Conversion of Carbon Dioxide to Methane and Methanol on Transition Metal Surfaces. J. Am. Chem. Soc. 2014, 136, 14107–14113. 10.1021/ja505791r. [DOI] [PubMed] [Google Scholar]
- Hori Y.; Kikuchi K.; Suzuki S. Production of CO and CH4 in Electrochemical Reduction of CO2 at Metal Electrodes in Aqueous Hydrogencarbonate Solution. Chem. Lett. 1985, 14, 1695–1698. 10.1246/cl.1985.1695. [DOI] [Google Scholar]
- Hori Y.; Kikuchi K.; Murata A.; Suzuki S. Production of Methane and Ethylene in Electrochemical Reduction of Carbon Dioxide at Copper Electrode in Aqueous Hydrogencarbonate Solution. Chem. Lett. 1986, 15, 897–898. 10.1246/cl.1986.897. [DOI] [Google Scholar]
- Lee C. H.; Kanan M. W. Controlling H+ vs CO2 Reduction Selectivity on Pb Electrodes. ACS Catal. 2015, 5, 465–469. 10.1021/cs5017672. [DOI] [Google Scholar]
- Ma M.; Djanashvili K.; Smith W. A. Controllable Hydrocarbon Formation from the Electrochemical Reduction of CO2 over Cu Nanowire Arrays. Angew. Chem., Int. Ed. 2016, 55, 6680–6684. 10.1002/anie.201601282. [DOI] [PubMed] [Google Scholar]
- Tang W.; Peterson A. A.; Varela A. S.; Jovanov Z. P.; Bech L.; Durand W. J.; Dahl S.; Norskov J. K.; Chorkendorff I. The Importance of Surface Morphology in Controlling the Selectivity of Polycrystalline Copper for CO2 Electroreduction. Phys. Chem. Chem. Phys. 2012, 14, 76–81. 10.1039/C1CP22700A. [DOI] [PubMed] [Google Scholar]
- Durand W. J.; Peterson A. A.; Studt F.; Abild-Pedersen F.; Norskov J. K. Structure Effects on the Energetics of the Electrochemical Reduction of CO2 by Copper Surfaces. Surf. Sci. 2011, 605, 1354–1359. 10.1016/j.susc.2011.04.028. [DOI] [Google Scholar]
- Peterson A. A.; Norskov J. K. Activity Descriptors for CO2 Electroreduction to Methane on Transition-Metal Catalysts. J. Phys. Chem. Lett. 2012, 3, 251–258. 10.1021/jz201461p. [DOI] [Google Scholar]
- Peterson A. A.; Abild-Pedersen F.; Studt F.; Rossmeisl J.; Norskov J. K. How Copper Catalyzes the Electroreduction of Carbon Dioxide into Hydrocarbon Fuels. Energy Environ. Sci. 2010, 3, 1311–1315. 10.1039/c0ee00071j. [DOI] [Google Scholar]
- Auinger M.; Katsounaros I.; Meier J. C.; Klemm S. O.; Biedermann P. U.; Topalov A. A.; Rohwerder M.; Mayrhofer K. J. J. Near-Surface Ion Distribution and Buffer Effects During Electrochemical Reactions. Phys. Chem. Chem. Phys. 2011, 13, 16384–16394. 10.1039/c1cp21717h. [DOI] [PubMed] [Google Scholar]
- Gupta N.; Gattrell M.; Bacdougall B. Calculation for the Cathode Surface Concentrations in the Electrochemical Reduction of CO2 in KHCO3 Solutions. J. Appl. Electrochem. 2006, 36, 161–172. 10.1007/s10800-005-9058-y. [DOI] [Google Scholar]
- Kas R.; Kortlever R.; Yilmaz H.; Koper M. T. M.; Mul G. Manipulating the Hydrocarbon Selectivity of Copper Nanoparticles in CO2 Electroreduction by Process Conditions. ChemElectroChem 2015, 2, 354–358. 10.1002/celc.201402373. [DOI] [Google Scholar]
- Sheng W.; Gasteiger H. A.; Shao-Horn Y. Hydrogen Oxidation and Evolution Reaction Kinetics on Platinum: Acid vs Alkaline Electrolytes. J. Electrochem. Soc. 2010, 157, B1529–B1536. 10.1149/1.3483106. [DOI] [Google Scholar]
- Ledezma-Yanez I.; Wallace W. D. Z.; Sebastian-Pascual P.; Climent V.; Feliu J. M.; Koper M. T. M. Interfacial Water Reorganization as a pH-Dependent Descriptor of the Hydrogen Evolution Rate on Platinum Electrodes. Nature Energy 2017, 2, 17031. 10.1038/nenergy.2017.31. [DOI] [Google Scholar]
- Strmcnik D.; Uchimura M.; Wang C.; Subbaraman R.; Danilovic N.; van der Vliet D.; Paulikas A. P.; Stamenkovic V. R.; Markovic N. M. Improving the Hydrogen Oxidation Reaction Rate by Promotion of Hydroxyl Adsorption. Nat. Chem. 2013, 5, 300–306. 10.1038/nchem.1574. [DOI] [PubMed] [Google Scholar]
- Jackson M. N.; Surendranath Y. Donor-Dependent Kinetics of Interfacial Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2016, 138, 3228–3234. 10.1021/jacs.6b00167. [DOI] [PubMed] [Google Scholar]
- Tu C.; Tripp B. C.; Ferry J. G.; Silverman D. N. Bicarbonate as a Proton Donor in Catalysis by Zn(II)- and CO(II)- Containing Carbonic Anhydrases. J. Am. Chem. Soc. 2001, 123, 5861–5866. 10.1021/ja010301o. [DOI] [PubMed] [Google Scholar]
- Trummal A.; Lipping L.; Kaljurand I.; Koppel I. A.; Leito I. Acidity of Strong Acids in Water and Dimethyl Sulfoxide. J. Phys. Chem. A 2016, 120, 3663–3669. 10.1021/acs.jpca.6b02253. [DOI] [PubMed] [Google Scholar]
- Bard A. J.; Faulkner L. R.. Electrochemical Methods Fundamentals and Applications, 2nd ed.; John Wiley and Sons: New York, 2001; p 339. [Google Scholar]
- Hori Y.; Murata A.; Takahashi R.; Suzuki S. Electroreduction of CO to CH4 and C2H4 at a Copper Electrode in Aqueous Solutions at Ambient Temperature and Pressure. J. Am. Chem. Soc. 1987, 109, 5022–5023. 10.1021/ja00250a044. [DOI] [Google Scholar]
- Hori Y.; Koga O.; Yamazaki H.; Matsuo T. Infrared Spectroscopy of Adsorbed CO and Intermediate Species in Electrochemical Reduction of CO2 to Hydrocarbons on a Cu Electrode. Electrochim. Acta 1995, 40, 2617–2622. 10.1016/0013-4686(95)00239-B. [DOI] [Google Scholar]
- Hori Y.; Murata A.; Takahashi R. Formation of Hydrocarbons in the Electrochemical Reduction of Carbon Dioxide at a Copper Electrode in Aqueous Solution. J. Chem. Soc., Faraday Trans. 1 1989, 85, 2309–2326. 10.1039/f19898502309. [DOI] [Google Scholar]
- Wuttig A.; Liu C.; Peng Q.; Yaguchi M.; Hendon C. H.; Motobayashi K.; Ye S.; Osawa M.; Surendranath Y. Tracking a Common Surface-Bound Intermediate during CO2-to-Fuels Catalysis. ACS Cent. Sci. 2016, 2, 522–528. 10.1021/acscentsci.6b00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.-J.; Sethuraman V.; Michalsky R.; Peterson A. A. Competition between CO2 Reduction and H2 Evolution on Transition-Metal Electrocatalysts. ACS Catal. 2014, 4, 3742–3748. 10.1021/cs5012298. [DOI] [Google Scholar]
- Martinez-Hincapie R.; Berna A.; Rodes A.; Climent V.; Feliu J. M. Surface Acid-Base Properties of Anion-Adsorbed Species at Pt(111) Electrode Surfaces in Contact with CO2-Containing Perchloric Acid Solutions. J. Phys. Chem. C 2016, 120, 16191–16199. 10.1021/acs.jpcc.6b00589. [DOI] [Google Scholar]
- Zeebe R. E. On the Molecular Diffusion Coefficients of Dissolved CO2, HCO3–, and CO32- and their Dependence on Isotopic Mass. Geochim. Cosmochim. Acta 2011, 75, 2483–2498. 10.1016/j.gca.2011.02.010. [DOI] [Google Scholar]
- Housecroft C. E.; Sharpe A. G.. Inorganic Chemistry, 2nd ed.; Ashford Colour Press Ltd.: Essex, U.K., 2005; p 368. [Google Scholar]
- Dodds W. S.; Stutzman L. F.; Sollami B. J. Carbon Dioxide Solubility in Water. Chem. Eng. Data Ser. 1956, 1, 92–95. 10.1021/i460001a018. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.