

Abstract
The neurotransmitter dopamine (DA) regulates multiple behaviors across phylogeny, with disrupted DA signaling in humans associated with addiction, attention-deficit/ hyperactivity disorder, schizophrenia, and Parkinson's disease. The DA transporter (DAT) imposes spatial and temporal limits on DA action, and provides for presynaptic DA recycling to replenish neurotransmitter pools. Molecular mechanisms that regulate DAT expression, trafficking, and function, particularly in vivo, remain poorly understood, though recent studies have implicated rho-linked pathways in psychostimulant action. To identify genes that dictate the ability of DAT to sustain normal levels of DA clearance, we pursued a forward genetic screen in Caenorhabditis elegans based on the phenotype swimming-induced paralysis (Swip), a paralytic behavior observed in hermaphrodite worms with loss-of-function dat-1 mutations. Here, we report the identity of swip-13, which encodes a highly conserved ortholog of the human atypical MAP kinase ERK8. We present evidence that SWIP-13 acts presynaptically to insure adequate levels of surface DAT expression and DA clearance. Moreover, we provide in vitro and in vivo evidence supporting a conserved pathway involving SWIP-13/ERK8 activation of Rho GTPases that dictates DAT surface expression and function.
SIGNIFICANCE STATEMENT Signaling by the neurotransmitter dopamine (DA) is tightly regulated by the DA transporter (DAT), insuring efficient DA clearance after release. Molecular networks that regulate DAT are poorly understood, particularly in vivo. Using a forward genetic screen in the nematode Caenorhabditis elegans, we implicate the atypical mitogen activated protein kinase, SWIP-13, in DAT regulation. Moreover, we provide in vitro and in vivo evidence that SWIP-13, as well as its human counterpart ERK8, regulate DAT surface availability via the activation of Rho proteins. Our findings implicate a novel pathway that regulates DA synaptic availability and that may contribute to risk for disorders linked to perturbed DA signaling. Targeting this pathway may be of value in the development of therapeutics in such disorders.
Keywords: C. elegans, dopamine, genetics, kinase, neurotransmitters, transporters
Introduction
The modulation of behavior by the neurotransmitter dopamine (DA) is evident in animals that range greatly in complexity, from the soil-dwelling nematode Caenorhabditis elegans (McDonald et al., 2006) to humans (Roe, 1997). In humans, DA is a critical neuromodulator that regulates circuits supporting reward, attention, and movement, with perturbed DA signaling associated with disorders that include addiction, attention-deficit/hyperactivity disorder (ADHD), schizophrenia, and Parkinson's disease (Viggiano et al., 2004; Segura-Aguilar et al., 2014; Howes et al., 2015; Nutt et al., 2015). Among the proteins required to sustain and regulate DA signaling, the presynaptic DA transporter (DAT) has received considerable attention, because of its powerful control of extracellular DA availability and its interactions with psychotropic drugs, including cocaine and amphetamine (Zhu and Reith, 2008; Schmitt and Reith, 2010). DAT proteins exhibit a 12 transmembrane domain structure with cytoplasmic N- and C-termini (Kristensen et al., 2011; Penmatsa et al., 2013). Mutations in human DAT (SLC6A3) that alter transporter trafficking and/or function have been identified in subjects with ADHD (Mazei-Robison et al., 2005, 2008; Kurian et al., 2009; Sakrikar et al., 2012; Mergy et al., 2014), bipolar disorder (Grünhage et al., 2000), autism (Hamilton et al., 2013), and juvenile- and adult-onset parkinsonism/dystonia (Kurian et al., 2009; Hansen et al., 2014), underscoring the essential contributions made by DAT to normal DA signaling.
DAT proteins are increasingly understood to be subject to post-translational regulatory mechanisms that impact surface trafficking, membrane microdomain localization, and functional activity states (Torres et al., 2003; Melikian, 2004; Foster et al., 2006; Bermingham and Blakely, 2016), though to date the bulk of evidence supporting these mechanisms derives from in vitro studies. The model organism C. elegans is a particularly attractive model for the elucidation of in vivo contributors to DAT regulation, and DA signaling more generally, because of its simple nervous system, genetic tractability, and the high conservation of DA signaling molecules (Ellis and Horvitz, 1986; Chalfie and Jorgensen, 1998; McDonald et al., 2006). In the adult hermaphrodite, DA is synthesized by eight neurons (4 cephalic sensilla [CEP], 2 anterior deirid [ADE] and 2 posterior deirid [PDE] neurons; Sulston et al., 1975; McDonald et al., 2006) that make well documented contributions to movement, egg-laying, and behavioral plasticity (Schafer and Kenyon, 1995; Weinshenker et al., 1995; Sawin et al., 2000; Chase et al., 2004). These neurons express an Slc6a3 family member (DAT-1) that transports DA and DA-like neurotoxins (e.g., 6-OHDA; Nass et al., 2000) in a Na+ and Cl−-dependent manner, with transport blocked by psychostimulants as well as other agents in vitro and in vivo (Jayanthi et al., 1998; Bermingham et al., 2016).
Worms deficient in DAT-1 expression demonstrate a particularly striking motor phenotype in water termed swimming-induced paralysis (Swip; McDonald et al., 2007; Hardaway et al., 2012). Swip in dat-1 mutants arises due to increased levels of extrasynaptic DA that can act on inhibitory, D2-type DA receptors present on motor neurons (Allen et al., 2011), reducing neuronal activity and/or neurotransmitter secretion and leading to paralysis. The Swip phenotype allows for both a functional evaluation of structural determinants of DAT-1 localization and activity (McDonald et al., 2007; Hardaway et al., 2012; Robinson et al., 2017) as well as for unbiased, forward genetic screens to identify DAT-1 regulators or other determinants of DA neuron excitability, secretion and signaling (Hardaway et al., 2012, 2015). Here we report the molecular lesion present in a worm line (vt32) previously isolated in an ethyl methanesulfonate (EMS)-based mutagenesis screen designed to recover DA-dependent Swip mutants (Hardaway et al., 2012). As we show, the gene, designated swip-13, is the C. elegans ortholog of rat and mouse ERK7/human ERK8 (Mapk15). Using genetic, imaging, and behavioral approaches, we demonstrate that SWIP-13 acts in C. elegans DA neurons to support synaptic membrane DAT-1 availability and DA-dependent swimming behavior. Moreover, we provide in vitro and in vivo evidence that SWIP-13/ERK8 activates Rho signaling to produce physiological levels of surface DAT protein.
Materials and Methods
Materials
All buffers, salts, and biochemical reagents, unless otherwise noted, were obtained from Sigma-Aldrich and were of the highest purity possible. All solutions were prepared in MilliQ water (Millipore) unless obtained as liquid preparations.
C. elegans strains and husbandry
Strains were grown on bacterial lawns of OP50 and maintained at 12–20°C as previously described (Brenner, 1974). N2 Bristol served as our wild-type strain. The strain VC2695 contains the allele gk1234, which possesses a large deletion of the swip-13 gene (C05D10.2), was obtained from the Caenorhabditis elegans Genetics Center (CGC; University of Minnesota, Minneapolis, MN). The strain LX703 that contains a deletion in dop-3 (dop-3(vs106)) was also obtained from the CGC, and the cat-2(tm2261) strain was obtained from Shohei Mitani at the National Bioresource Project at the Tokyo Women's Medical University. The strain IR724 (N2; uvEX724 [Pasic-1:SNB-1::SEpHluorin, pRF4]) that was used for FRAP experiments was obtained from Nektarios Tavernarakis (University of Crete, Heraklion, Crete, Greece). The following strains were generated by genetic cross: BY989: cat-2(tm2261); swip-13(gk1234), BY990: swip-13(gk1234); dop-3(vs106), BY991: swip-13(gk1234); vtIs7 (Pdat-1::GFP), BY992: swip-13(gk1234); vtIs18 (Pdat-1::GFP::dat-1), BY993: swip-13(vt32); vtIs7 (Pdat-1::GFP), BY1117: swip-13(gk1234); uvEx724 (Pasic-1::SNB-1::SEpHluorin), BY1063: swip-13(gk1234) dat-1(ok157). The following strains were generated by microinjection: BY972-974: swip-13(gk1234); vtEx139-141 (C05D10.2 genomic fragment), BY975-977: swip-13(gk1234); vtEx142-144 (Pdat-1::swip-13), BY1062: N2; vtEx191 (Pswip-13::GFP), BY1096: N2; vtEx200 (Pdat-1::GFP::swip-13 Pdat-1::mCherry::RAB-3), BY1064-B1066: swip-13(gk1234); vtEx193-195 (Pdat-1::GFP::swip-13), BY1142-1144: swip-13(gk1234); vtEx239-241 (Pdat-1::GFP::swip-13(K42R)), BY1221-1223: swip-13(gk1234); vtEx277-279 (Pdat-1::rho-1 cDNA).
mouse ERK7/human that we previously demonstrated to be functional (Nass et al., 2002). As shown in Figure 6E, overexpres Fleming PA, Fleming KA, Whitaker
Figure 6.
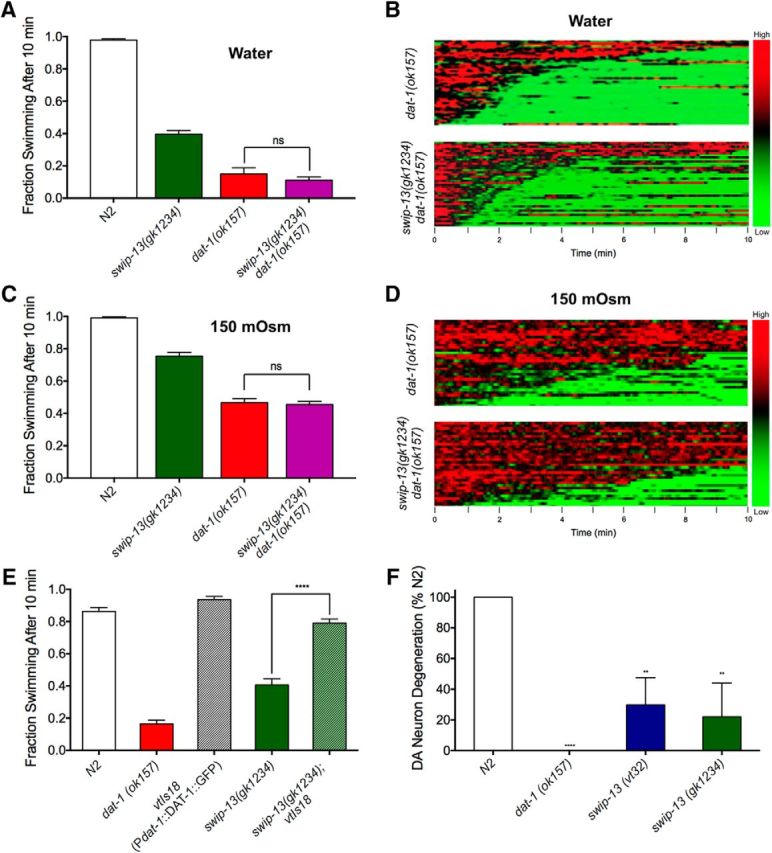
SWIP-13 regulates DAT-1 to control DA signaling. A, In water, dat-1(ok157) and swip-13(gk1234) dat-1(ok157) mutants display similar Swip behavior, with no significant difference between genotypes (t(92) = 1.11, p = 0.27, Bonferroni's post-tests). B, To increase the range in which we might see additivity between swip-13(gk1234) and dat-1(ok157), we performed Swip assays in 150 mOsm sucrose-supplemented water to suppress Swip. In this context, there is still no significant difference between dat-1(ok157) and swip-13(gk1234) dat-1(ok157) animals (t(92) = 0.44, p = 0.66, Bonferroni's post-tests). Data were analyzed using a one-way ANOVA with Bonferroni's post-tests comparing dat-1(ok157) to swip-13(gk1234) dat-1(ok157) in both A and B. C, D, Heat map analysis shows no enhancement of Swip in swip-13(gk1234) dat-1(ok157) animals compared with dat-1(ok157) mutants in either water (C) or 150 mOsm solution (D). E, A functional DAT-1::GFP-expressing transgene vtIs18 (Pdat-1::dat-1::gfp) rescues the paralysis of swip-13(gk1234) mutant animals. This is demonstrated by the significant difference between swip-13(gk1234) and swip-13(gk1234); vtIs18 animals (t(155) = 14.52, p = 0.27, Bonferroni's post-tests). Data were analyzed using a one-way ANOVA with Bonferroni's post-tests comparing swip-13(gk1234) to swip-13(gk1234); vtIs18. Significance was set at p < 0.05. ****p < 0.0001. F, Treatment of N2 animals with GFP-labeled DA neurons with the DA-neuron selective, DAT-1-dependent neurotoxin 6-OHDA leads to robust degeneration of DA neurons, as measured by loss of GFP-labeled CEP dendrites. dat-1(ok157) mutants are insensitive to this neurotoxin, shown as an absence of DA neuron degeneration. Both swip-13(vt32) and swip-13(gk1234) have reduced sensitivity to 6-OHDA compared with N2 animals [swip-13(v32): t(8) = 3.51, p < 0.01, Bonferroni's post-tests; swip-13(gk1234): t(8) = 3.9, p < 0.01, Bonferroni's post-tests]. Data were analyzed using a one-way ANOVA with Bonferroni's post-tests comparing all genotypes to N2. Significance was set at p < 0.05. **p < 0.01 ****p < 0.0001.
Crosses were performed using integrated fluorescent markers in trans. To verify presence of mutations, single worm PCR was performed using a three primer multiplex strategy. N2 and mutant control reactions were conducted in parallel, along with a synthetic heterozygote reaction containing both N2 and mutant DNA. For generation of the swip-13(gk1234) dat-1(ok157) double-mutant, recombination was required due to the location of both genes on LGIII. After selfing of the double-heterozygote, lines that appeared to be homozygous for either dat-1(ok157) or swip-13(gk1234), and heterozygous for the other mutation were selected. These lines were then selfed and a double-homozygous knock-out line was selected. Platinum PCR Supermix (Life Technologies) was used for all genotyping PCRs and reactions were analyzed via 2% agarose gel electrophoresis.
Creation of plasmids and transgenic animals
Plasmids.
Oligonucleotide primer sequences and PCR conditions used in individual construct creations are available upon request. All high-fidelity PCR amplifications for creation of plasmids were performed with Kapa HiFi Hotstart ReadyMix PCR kit (Kapa Biosystems). The plasmid Pdat-1::swip-13 (pRB1341) was generated by amplification of genomic swip-13 with primers that introduced AscI and KpnI sites in the 5′ and 3′ ends, respectively. This AscI/KpnI (New England BioLabs) digested fragment was then ligated to AscI/KpnI digested plasmid pRB1106 (containing the dat-1 promoter and unc-54 3′UTR) using T4 DNA ligase (New England BioLabs). A similar strategy was used to generate Pdat-1::GFP::swip-13 (pRB1342), except an initial overlap PCR amplification reaction was first used to generate a GFP::swip-13 fragment with AscI/KpnI sites. To generate the K42R kinase dead mutation in the Pdat-1::GFP::swip-13 plasmid (pRB1343), we used a QuikChange II Site-Directed Mutagenesis kit (Agilent) following the manufacturer's protocol. To generate the Pdat-1::rho-1 cDNA (pRB1348) construct, rho-1 cDNA was first amplified from a cDNA library generated from N2 animals using Superscript III First Strand Synthesis (ThermoFisher). Overlap sequence for pRB1106 was engineered into the ends of the rho-1 cDNA PCR fragment, and a PCR fragment of pRB1106 containing the dat-1 promoter and unc-54 3′UTR was generated with overlap sequence for the rho-1 cDNA on both ends. Fusion of these products was performed using NEBuilder HiFi DNA Assembly (New England BioLabs). For mammalian ERK8 expression constructs, we used plasmid pCR3.1 HA-ERK8, generously provided by Dr. Mark Abe (University of Chicago). The R59Q and K42R mutations were engineered using a similar strategy as described above for K24R mutation.
Transgenic worm lines.
Transgenic animals were generated via microinjection following methods previously described (Brenner, 1974). Transgenic F1 progeny were selected based on presence of fluorescent co-injection markers. Lines with F2s demonstrating stable expression of the transgene marker were selected for analysis.
Swip assays
For both manual and automated Swip assays, staged L4 animals were generated by hypochlorite treatment of gravid adults and plating of synchronized L1 animals and growth for various days at 12–20°C. For manual assays, experimenters were blinded to genotype, and ∼10 early L4 animals were picked into 100 μl of water In Pyrex Spot Plates (Catalog #13-748B, Fisher Scientific) and the number of paralyzed animals was quantified after 10 min. For each genotype/treatment, ∼80 animals were assayed per experiment, with at least three experiments performed by one to two experimenters. For reserpine experiments, synchronized L1s were plated on NGM/OP50 plates containing 0.6 mm reserpine or dimethyl sulfoxide vehicle pipetted onto OP50 bacterial lawn and were grown for 2 d at 20°C to reach the early L4 stage before Swip testing. For transgenic rescue experiments, 40–60 transgenic and nontransgenic animals were picked based on co-injections markers, with three independent lines scored per transgene. For automated analyses, animals were picked one at a time into 10 μl of water and 10 min movies of individual worm swimming behavior were captured and analyzed as previously described (Hardaway et al., 2014). Briefly, videos were first analyzed using the in-house tracking software Worm Tracker, which fits a two-segment spine to the worm and calculates the frequency of body bends over time. This frequency information is then processed using SwimR, which calculates various parameters related to movement frequency and paralysis latency and that generates visual representations (heat maps, frequency plots) of thrashing data. In some assays, Swip was suppressed using a medium osmolarity of 150 mOsm to assess allele additivity (Hardaway et al., 2012), achieved through addition to sucrose to MilliQ water. For nisoxetine experiments, nisoxetine hydrochloride was dissolved to a final concentration of 20 μm in MilliQ water or 150mOsm sucrose solution as described above. For fasudil experiments, fasudil hydrochloride (Tocris Bioscience) was dissolved to a final concentration of 500 μm in MilliQ water. Data from Swip assays were graphed and analyzed using Prism 6.0 (GraphPad) software with details of statistical tests provided in the figure legends.
Single-nucleotide polymorphism mapping
Previously (Hardaway et al., 2012), we reported the execution of a nonclonal, F2 screen for EMS-generated mutants that exhibit reserpine-reversible Swip, identifying multiple lines with at least 50% paralysis as a population after 10 min, including line vt32. Mapping of the vt32 mutation locus was performed as described previously (Davis et al., 2005). Briefly, stable outcrossed vt32 animals were crossed to the CB4856 (Hawaiian) strain. For bulk segregant analysis, worm lysates extracted using proteinase K from both Swip-positive and Swip-negative F2 populations were generated and used as the input for genome-wide, 96-well PCR using Platinum PCR Supermix (Life Technologies). Linkage groups identified through bulk segregant analysis were subjected to fine-interval mapping where individual Swip-positive F2s were cloned, and their F3 progeny were tested for a stable Swip phenotype. Experiments were replicated at least twice using separate populations to demonstrate consistent linkage. Mutations were considered homozygous if the F3 population demonstrated Swip comparable to the original vt32 line. Populations were manually scored in at least four to five assays using 40–50 worms. DNA from individual clones extracted using proteinase K was then used as the input for PCR amplification of individual intervals using Platinum PCR Supermix (Life Technologies) to ascertain a Bristol-specific island on the mapped linkage group.
Whole-genome sequencing and sequence analysis
Genomic DNA was isolated as described previously (Sarin et al., 2008). Briefly, BY200 (parental strain) and vt32 worms were harvested from a 10 cm 8p/NA22 plate by rinsing with M9 (in mm: 42 Na2HPO4, 22 KH2PO4, 86 NaCl, 1 MgSO4). After a brief preclearing wash with M9, worms were rocked in M9 for 2–3 h to clear ingested bacteria and then washed with M9 and pelleted for DNA extraction. Before extraction, the worm pellet was incubated at −80°C for 1 h to overnight. Genomic DNA was extracted from the worm pellet using a Qiagen Gentra Puregene kit as described by the manufacturer and a post hoc phenol/chloroform extraction, RNase A digestion, and additional phenol/chloroform extraction. Quality of the gDNA was confirmed on a 2% agarose gel before submitting the samples for Illumina sequencing (Vanderbilt Genome Technology Core). Sequencing libraries were generated from gDNA as described previously (Sarin et al., 2008). Each sample was assigned a unique barcode so that samples could be pooled onto several flow cells of an Illumina Genome Analyzer Iix for sequencing as single-end 76mers. Sequence reads were filtered for quality and offloaded in Fastq format for subsequent analysis. Sequence data were analyzed in MaqGene (Bigelow et al., 2009) and a text file containing mutations against the reference sequence (WS180) was extracted. We then compared this list against sequence of the parental strain (BY200) to identify mutations that reside within the mapped interval on LGIII so as to nominate genes containing the vt32 mutation.
Confocal imaging
Confocal images were acquired on a Zeiss LSM 510 inverted confocal microscope in the imaging laboratory of the Vanderbilt Cell Imaging Shared Resource. Worms for imaging were prepared by placing young adult transgenic animals onto 2% agarose pads after immobilization with 0.05% levamisole in M9 medium. Slides were covered with 1 mm coverslips and sealed with paraffin wax before imaging (Smith et al., 2010). Captured images were analyzed using ImageJ software. Magnification power for captured images is noted in figure legends.
6-OHDA degeneration assay
We assessed sensitivity of DA neurons to 6-hydroxydopamine (6-OHDA) as a proxy for in vivo DAT activity. Assays were performed as previously described (Nass et al., 2002), with minor modifications. BY250 animals that express GFP in DA neurons and processes were assayed in parallel with swip-13 animals crossed to BY250, allowing for complete visualization of the degenerative response of DA neurons to toxin. Synchronized L1 worms were plated on 10 cm 8P plates seeded with NA22 bacteria (8P/NA22) for 1 d at 20°C until they reached the L2-L3 stage. These animals were then washed in M9 three times, and pelleted worms were then treated with 25, 35, or 50 mm 6-OHDA supplemented with ascorbic acid (5, 7, or 10 mm, respectively). Worms were covered and gently agitated for 1 h at room temperature, and then the entire volume of worm solution was plated on 8P/NA22 plates. Plates were placed at 20°C for 3 d, allowing worms to grow to adults, at which time animals were scored for degeneration. Semiquantitative scoring was performed on animals mounted on 2% agarose pads with 2.5 mm levamisole used as a paralytic, at a concentration of 6-OHDA that caused robust, yet incomplete DA neuron degeneration in BY250. CEP neuron degeneration was scored by assigning a number 0–4 for each animal, with 0 representing complete loss of CEP dendrites, and 4 representing intact dendrites for all four neurons. For each genotype, 50 worms were scored in triplicate with experimenter blinded to genotype and with experiments performed on 3 separate days.
FRAP assay
Fluorescence recovery after photobleaching (FRAP) assays (Voglis and Tavernarakis, 2008; Hardaway et al., 2015) were used to assess the impact of swip-13 mutation on DA vesicle fusion rate. Young adult animals were staged by picking L4 animals and growing at 20°C overnight. The day of the experiment, animals were mounted on 2% agarose pads and immobilized using 0.05% levamisole. FRAP experiments were performed using a Zeiss LSM 510 inverted confocal microscope. PDE synapses were chosen for these analyses due to the more two dimensional layout of their processes relative to ADE and CEP synapses and a lack of overlap of individual PDE processes. DA synapses were identified by SNB-1::SEpHluorin fluorescence and were bleached at 488 nm, 15 mW for 5–10 s to an intensity 20–30% that of the original fluorescence value. Fluorescence recovery was then monitored every 10 s for 2 min and analyzed using Zeiss LSM 510 software. Percentage recovery was calculated as the fluorescence at each time point divided by the initial fluorescence value after bleaching. We analyzed 20–25 PDE synapses per genotype, with an average of 3–5 synapses per animal.
In vitro expression of ERK8/DAT and regulators
Human SH-SY5Y cells (ATCC, CRL2266, RRID:CVCL0019) were grown in 1:1 F12/DMEM solution supplemented with 10% fetal bovine serum (FBS; Hyclone), 100 IU penicillin and 100 μg/ml streptomycin (Sigma-Aldrich). Cells were plated in either 24-well (single-point DA uptake experiments), 96-well (DA uptake saturation analyses), or 10 cm culture plates (biochemical experiments) at a density of 9 × 104 cells per cm2. For in vitro kinase assays and Rho activation assays, HEK-293T (ATCC, CRL3216, RRID:CVCL0063) cells were grown in DMEM supplemented with 10% FBS with pen/strep and were plated at 2 × 105 cells per well in 6-well dishes. For DA uptake and biotinylation experiments, plates were first coated with 50 μg/ml poly-d-lysine to enhance cell adherence. In all experiments, cells were grown at 37°C for 1 d before transfection with TransIT LT1 transfection reagent (Mirus) in Opti-MEM (ThermoFisher) medium at a ratio of 1:3 DNA/Transit reagent, with the amount of DNA scaled to the size of the plate according to the manufacturer's instructions. The following ratios of DNA were used for each experiment: DA uptakes and biotinylations: 1:4 DAT/pcDNA3.1, 1:4 DAT/HA-ERK8; Rho activation assays: 100% pcDNA3.1 or HA-ERK8; GFP-C3 DA uptakes: 1:4 DAT/pcDNA3.1, 1:1:3 DAT/pcDNA3.1/HA-ERK8, 1:1:3; DAT/GFP-C3/pcDNA3.1, 1:1:3 DAT/GFP-C3/HA-ERK8. Cells were then grown for an additional 2 d before DA uptake or biochemical experiments.
DA uptake assays
DA uptake assays were performed in triplicate or quadruplicate as previously described (Apparsundaram et al., 1998) using [3H]DA (PerkinElmer; NET673001MC). Briefly, cells were washed with Krebs Ringers-HEPES (KRH) buffer (in mm: 130 NaCl, 1.3 KCl, 2.2 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 10 HEPES, pH 7.4) three times before incubation in KRH containing 10 μm desipramine (to block background endogenous norepinephrine transport activity), 100 μm pargyline, 100 μm tropolone, and 100 μm L-ascorbic acid with or without 10 μm cocaine at 37°C for 15 min. A 5× concentrated solution of 5% [3H]DA/95% unlabeled DA was then added to each well for to achieve a 1× concentration of DA, and cells were again incubated at 37°C for 15 min. For DA uptake saturation analysis, final total concentrations of DA used were 500 nm, 1 μm, 2 μm, 3 μm, and 6 μm. For single-point DA uptake assays, 6 μm DA was used. Specific DA uptake was defined in parallel incubations using 10 μm cocaine. Following assay, cells were washed three times with ice-cold KRH and then incubated with Microscint 20 scintillation fluid (PerkinElmer) for least 1 h before quantitation using a TopCount Microplate Scintillation Counter (Packard). BCA protein assays (ThermoFisher) were conducted following the manufacturer's recommendations using cells from parallel transfections in each condition to normalize uptake to total protein.
Cell surface biotinylation assay
Transfected cells in 10 cm dishes were washed twice in ice-cold PBS supplemented with 0.1 mm CaCl2 and 1 mm MgSO4. Cells were then incubated in Sulfo-NHS-Biotin (ThermoFisher) at 1 mg/ml for 30 min at 4°C with gentle agitation. Excess biotinylation reagent was quenched with 0.1 m glycine in PBS and then cells were lysed by incubation in ice-cold hypotonic lysis buffer (10 mm Tris-HCl, pH7.5) containing Sigma-Aldrich protease inhibitor cocktail. Lysed cells and cell fragments were collected and centrifuged for 1 min at 13,000 × g using a tabletop microcentrifuge to collect membrane pellets. Pellets were then solubilized in ice-cold lysis buffer (20 mm Tris-HCl, pH 8, 137 mm NaCl, 2 mm EDTA, 1% Triton X-100), and protein lysates quantified by BCA assay (ThermoFisher) to determine protein concentration. Equal amounts of protein were either dissolved directly in Laemmli sample buffer for SDS-PAGE to determine total protein levels, or were incubated with prewashed Streptavidin-conjugated beads (ThermoFisher) for cell surface protein isolation, with incubation at room temp for 1 h. Beads were then washed three times with ice-cold lysis buffer, and samples were eluted in 1× Laemmli sample buffer before analysis via SDS-PAGE and Western blotting. A rat anti-hDAT antibody (Millipore; MAB 369; RRID: AB_2190413; 1:1000) was used to visualize DAT and mouse anti-transferrin receptor (TfR; ThermoFisher; H68.4, 13-6890; RRID: AB_2533030; 1:1000) antibody was used to detect transferrin receptor as a loading control. HRP-conjugated goat anti-rat (Santa Cruz Biotechnology; sc-2006; RRID: AB_1125219; 1:10,000) and goat anti-mouse secondary antibodies (Jackson ImmunoResearch; 115-035-003; RRID: AB_10015289; 1:10,000) were used. Total DAT was normalized to total TfR, and surface DAT was normalized to surface TfR.
In vitro kinase assay
Lysates from transfected HEK 293T cells were incubated with Anti-HA Affinity Matrix (Roche) at 4°C for 1 h with constant agitation. Beads were then pelleted and washed five times with lysis buffer for 5 min each, again with constant agitation. Beads were then washed once in kinase assay buffer [50 mm Tris, 0.1 mm EGTA, 10 mm MgAc2, 0.1% (v/v) β-mercaptoethanol, 1 mm sodium othrovanadate] for 5 min, and then beads were pelleted and resuspended in kinase assay buffer plus 5 μg of myelin basic protein (MBP; Active Motif) and 50 μm [32P]-γATP (2500 cpm/pmol; PerkinElmer). Reactions were conducted at 30°C for 15 min and then terminated by the addition of SDS sample buffer. After pelleting to remove beads, proteins were resolved by SDS-PAGE and gels were stained with Coomassie blue (Bio-Rad). Gels were dried by vacuum and incorporation of [32P] was assessed by autoradiography on x-ray film (Phenix Research Products) with multiple exposures taken to insure quantitation in the linear range of the film.
Rho activation assay
For Rho activation assays, a RhoA pull-down activation assay kit (Cytoskeleton) was used. Transfected HEK-293T cells were washed in ice-cold TBS (50 mm Tris-HCl, pH 7.5, 150 mm NaCl) and lysed in supplied lysis buffer with protease inhibitor cocktail. Lysates were clarified by centrifugation and immediately snap frozen in liquid nitrogen and stored at −80 to prevent loss of active Rho due to rho-GAP activity. Protein concentrations were determined using BCA protein assay (ThermoFisher), frozen lysates were rapidly thawed, and 800 μg of protein was added to 30 μl of GST-rho-binding domain (RBD) beads and incubated with mixing at 4°C for 1 h. Beads were then washed once with supplied wash buffer and 2× Laemmli buffer was added to elute protein from the beads. Eluates, along with pure RhoA protein and 30 μg of total lysates from each sample were resolved on a 12% SDS-PAGE gel and then transferred to Bio-Rad Immun-Blot PVDF with 0.2 μm pore size for better retention of small proteins. After transfer, membrane was dried for 30 min, and then re-wet with methanol followed by TBST. Membrane was blocked in 5% dry milk in TBS + 0.1% Tween 20 (TBST) for 1 h, and then incubated in anti-RhoA antibody (Cytoskeleton, ARH04; 1:500) in TBST overnight at 4°C. Membrane was then washed once in TBST, and incubated with HRP-conjugated goat anti-mouse antibody (Jackson ImmunoResearch, 115-035-003; RRID: AB_10015289; 1:10,000) in TBST for 1 h at room temp followed by five 10 min washes in TBST. Membranes were imaged using GE ImageQuant LAS4000 (GE Healthcare Life Sciences). Membranes were stripped and reprobed with monoclonal mouse anti-HA antibody (Vanderbilt Antibody and Protein Resource, 12CA5; RRID: AB_2532070; 1:1000; goat anti-mouse, 1:10,000), followed by HRP-conjugated anti-β-actin antibody (Sigma-Aldrich; A3854; RRID: AB_262011; 1:20,000). Total RhoA was normalized to β-actin, and active RhoA was normalized to this value.
Experimental design and statistical analysis
Data were analyzed and graphed using either SwimR software (Hardaway et al., 2014), or using Prism 6.0 (GraphPad). Protein sequence alignments were generated in the MegAlign module of Lasergene for the Mac (DNAStar). For all manual Swip assays, each replicate represents the fraction of animals swimming in one well of ∼10 animals. For more specifics on the number of animals tested in each assay, see “Swip Assays” section. Either a two-tailed unpaired Student's t test or a one-way ANOVA with Bonferroni's post-test was used to compare groups (figure legends, test used, groups compared, and test details noted). In the 6-OHDA experiments, each replicate represents the average degeneration score for each line on each of three separate scoring days, and a one-way ANOVA with Bonferroni's post-tests was used to compare each group to N2. For analysis of both DAT and Rho Western blots, three separate experiments were performed, and band intensities were normalized to control and a two-tailed unpaired Student's t test was used to compare values from ERK8-transfection to control. For DA uptake experiments comparing ERK8 mutants, as well as for GFP-tagged exotoxin C3 (GFP-C3) experiments, three experimental replicates were performed on 3 separate days. For ERK8 mutant uptake experiments, a one-way ANOVA was used with a Bonferroni's post-tests to compare all values to the control (DAT transfection alone). For GFP-C3 experiments, a one-way ANOVA was used with Bonferroni's post-tests to compare DAT transfection alone to both DAT/ERK8 transfection and DAT/GFP-C3 transfection, as well as DAT/GFP-C3 transfection to DAT/ERK8/GFP-C3 triple transfection. See figure legends for more specifics on statistical methods and results for each experiment.
Results
Loss-of-function mutations in the nematode MAP kinase C05D10.2 gene perturb DA-dependent swimming behavior
Previously (Hardaway et al., 2012), we reported our results with an EMS-based mutagenesis screen designed to isolate mutant lines that transmit DA-dependent Swip, as assessed through rescue with the vesicular monoamine transporter inhibitor reserpine. Lines were recovered that demonstrated Swip in at least 50% of the population following a 10 min incubation in water. Three additional evaluations were used before selecting lines for mutation mapping and sequence analysis. First, lines were not advanced if they appeared compromised in crawling or response to touch that might indicate major changes in motor neuron architecture. Second, as we mutagenized N2 hermaphrodite worms carrying an integrated, dat-1 promoter-driven green fluorescent protein (GFP) transgene (BY200; Nass et al., 2002), we examined lines for gross alterations in DA neuron structure or processes, moving forward lines with qualitatively intact morphology. Last, because dat-1 loss-of-function alleles generate highly penetrant Swip, we sequenced the dat-1 gene. In this effort, we documented recovery of two lines, vt21 and vt22, that bear loss-of-function dat-1 alleles (Hardaway et al., 2012), validating the capacity of the screen to identify genes supporting DAT function and DA signaling. Recently, we reported the gene mutated in lines vt29 and vt33 (Hardaway et al., 2015), designated swip-10, as an ortholog of the mammalian, metallo β-lactamase domain-containing protein MBLAC1, demonstrating that swip-10 loss in glia elevates DA neuron excitability and DA vesicular release via increased glutamatergic signaling onto DA neurons. Here, we report our efforts to identify and characterize mutant line vt32.
As demonstrated in Figure 1A, vt32 displays reserpine-sensitive Swip, consistent with a hyperdopaminergic state and the pharmacological rescue demonstrated in the initial screen (Hardaway et al., 2012). Notably, the Swip penetrance of vt32 Swip is less than that of dat-1(ok157) animals, which can be further appreciated by visualizing swimming behavior of a population of individual animals in a thrashing behavior heatmap generated by SwimR software (see Materials and Methods; Fig. 1B). In this illustration, movement frequency is color-coded (red, active; green, paralysis) and depicted as a function of time horizontally. The behavior of individual animals is stacked vertically, with animals paralyzing first located on the bottom of the plot. This depiction also allows visualization of a property of Swip not seen with dat-1 animals, specifically a greater tendency to revert to normal swimming frequency during the analysis period (see also Fig. 2D).
Figure 1.

vt32 mutation in the gene C05D10.2 causes reserpine-sensitive Swip. A, dat-1(ok157) animals show robust Swip in water after 10 min, and vt32 animals show a more intermediate Swip phenotype. Reserpine pretreatment rescues Swip behavior in both dat-1(ok157) animals (t(190) = 16.98, p > 0.0001, Bonferroni's post-tests) and vt32 animals (t(190) = 8.29, p < 0.0001, Bonferroni's post-tests). Significance was calculated using a one-way ANOVA with Bonferroni's post-tests comparing each genotype without reserpine to the same genotype with reserpine. Significance was set at p < 0.05. *p < 0.01 ****p < 0.0001. B, Heat maps showing the swimming behavior of N2, dat-1(ok157), and vt32 animals. Each horizontal line represents the frequency of swimming over time for a single worm, going from 0 to 10 min from left to right, with red representing high-frequency values and green representing low-frequency values. N2 animals show very little paralysis, as demonstrated by the minimal green colored lines, and dat-1(ok157) animals show robust paralysis as demonstrated by the high prevalence of green lines. vt32 Animals have more intermediate paralysis. C, C05D10.2 is located on chromosome III at −1.42 cm. The vt32 mutation was located in exon 3, and results in an Arg to Gln substitution. gk1234 is a large deletion allele of C05D10.2 that deletes all of exons 5–7, and part of exon 8. D, A genomic PCR fragment containing the C05D10.2 genomic locus, including 1 kb upstream and downstream to include putative promoter and 3′ UTR sequences, was transgenically expressed in vt32 mutant animals. Comparison of transgenic and nontransgenic animals revealed a significant suppression of Swip with expression of WT C05D10.2 (t(58) = 7.83, p < 0.0001, unpaired t test). Bars represent the average of three transgenic lines with at least 100 animals per line. Significance was calculated using a two-tailed Student's t test with significance set at p < 0.05. ****p < 0.0001.
Figure 2.

gk1234 Mutation fails to complement vt32 mutation, generates DA-dependent Swip. A, Like swip-13(vt32) animals, swip-13(gk1234) show significant Swip, and this behavior is also rescued by reserpine pretreatment (t(190) = 13.3, p > 0.0001, Bonferroni's post-tests). Data were analyzed using a one-way ANOVA with Bonferroni's post-tests comparing each genotype without reserpine to the same genotype with reserpine. Significance was set at p < 0.05. *p < 0.01, ****p < 0.0001. B, Heterozygous vt32/+ and gk1234/+ animals swim normally, but double-heterozygous gk1234/vt32 animals demonstrate significant paralysis. Data were analyzed using a one-way ANOVA with Bonferroni's post-tests comparing gk1234/vt32 double-heterozygotes to both vt32/+ (t(69) = 8.55, p < 0.0001, Bonferroni's post-tests) and gk1234/+ (t(69) = 9.76, p < 0.0001, Bonferroni's post-tests) single-heterozygotes. Significance was set at p < 0.05. ****p < 0.0001. C, Both dop-3(vs106) and cat-2(tm2261) mutations significantly suppress Swip in swip-13(gk1234) animals. Data were analyzed using a one-way ANOVA with Bonferroni's post-tests comparing swip-13(gk1234) to both swip-13(gk1234);dop-3(vs106) (t(197) = 18.77, p < 0.0001, Bonferroni's post-tests) and cat-2(tm2261);swip-13(gk1234) (t(197) = 24.76, p < 0.0001, Bonferroni's post-tests). Significance was set at p < 0.05. ****p < 0.0001. (D) Heat map analysis again shows a near complete rescue of Swip in swip-13(gk1234);dop-3(vs106) and cat-2(tm2261);swip-13(gk1234) animals.
SNP-based mapping of vt32 DNA elements that cosegregate with Swip behavior revealed the likely mutation locus to be on LGIII, which also harbors the dat-1 gene (Fig. 1C). As noted above, sequencing efforts revealed no dat-1 mutations, and thus we moved to whole-genome sequencing to identify mutations within the mapped region, relative to the parental strain (BY200). Among the genes with coding polymorphisms in this region is the gene C05D10.2 (WBGene00015478). Using the online tool WormViz (Spencer et al., 2011), which reports mRNA expression levels in a variety of C. elegans cell types across development, we observed expression of C05D10.2 mRNA levels to be enriched in late embryonic and L3/L4 DA neurons compared with other cell types (data not shown). As illustrated in Figure 1C, the C05D10.2 gene is located on LGIII at −1.42cM, with the longest open reading frame (ORF) predicted to derive from 10 exons, and with possibilities for alternatively spliced products derived from translation of 3 or 8 exons (Wormbase:Version WS256). Our sequence analysis revealed a nonconservative, missense mutation in exon 3 that produces the substitution Arg59Gln. We found that transgenic overexpression of the wild-type C05D10.2 gene in a vt32 background resulted in significant suppression of Swip (Fig. 1D), bolstering our suspicions that C05D10.2 might be the site of the molecular lesion producing Swip in vt32 animals.
As an independent test of the hypothesis that the vt32 mutation perturbs C05D10.2 function to generate Swip, we tested the deletion mutation and predicted null allele C05D10.2(gk1234) for reserpine-sensitive Swip and complementation with vt32. As shown in Figure 2A, gk1234 animals demonstrated significant Swip, similar to that observed with vt32. Moreover, as observed for vt32 and dat-1(ok157), growth on reserpine plates led to an essentially complete reversal of Swip. In complementation tests, gk1234/+ and vt32/+ animals demonstrated relatively normal swimming behavior (Fig. 2B). However, the compound heterozygote demonstrated significant Swip, comparable to that observed in homozygous gk1234 or vt32 mutants (Figs. 1A, 2A). Based on the ability of a wild-type copy of C05D10.2 to rescue Swip in vt32, of a second, deletion allele (gk1234) of C05D10.2 to generate reserpine-sensitive Swip, and of gk1234's failure to complement vt32 in Swip assays, we conclude that C05D10.2 is the gene harboring the Swip-inducing mutation in vt32 and that the Arg59Gln mutation is a loss-of-function allele. Based on the order of identification of mutant lines in our initial screen (Hardaway et al., 2012), we hereafter designate the C05D10.2 gene as swip-13 and its encoded protein as SWIP-13.
We performed additional genetic tests to evaluate the idea that swip-13 loss-of-function alleles result in hyperdopaminergic signaling. To rule out the possibility that reserpine-dependent suppression of swip-13 mutant Swip (Fig. 2) arises from inhibition of the VMAT2 transporter CAT-1 in serotonergic neurons (Duerr et al., 1999), we determined that a loss-of-function mutation in the gene encoding the rate-limiting step in DA synthesis, cat-2 [tyrosine hydroxylase also suppresses the Swip behavior of swip-13(gk1234); Fig. 2C,D]. This finding replicates our earlier observation that cat-2 mutation also precludes Swip in dat-1(ok157) animals (McDonald et al., 2007). Swip behavior in the dat-1 mutants can also be reversed by a loss-of-function mutation (vs106) of the D2-type DA receptor DOP-3 (McDonald et al., 2007). Consistent with these studies, and supportive of swip-13 animals as generating excess DA signaling, we observed significant suppression of Swip in swip-13(gk1234);dop-3(vs106) double-mutant animals (Figs. 2C,D).
The predicted, 10 exon ORF of the swip-13 gene (validated by RT-PCR, data not shown) encodes a 470 aa member of the atypical MAPK gene family Mapk15 that is highly conserved with rat (ERK7) and human (ERK8) orthologs (Abe et al., 1999, 2002). Sequence alignment (Fig. 3) demonstrates ∼40% overall amino acid identity of SWIP-13 with ERK7 and ERK8, with identity rising to ∼60% within the kinase domain (blue outline; Strambi et al., 2013). Notably, the substitution comprising the vt32 allele impacts an Arg residue in the predicted N-terminal, nucleotide-binding lobe of the kinase domain that is conserved between worm, mouse, and man. All three proteins also encode a TEY sequence (Fig. 3) that is specific to ERK-family MAP kinases and is proposed to be a site of phosphorylation-dependent activation (Anderson et al., 1990; Abe et al., 1999, 2002).
Figure 3.

C05D10.2/swip-13 encodes an ortholog of mammalian ERK7/8. Sequence alignment of C05D10.2 with rat ERK7 and human ERK8 shows a high level of sequence identity, including the site of the vt32 mutation (red highlight), as well as the TEY phosphoactivation motif (yellow highlight) and the conserved Lys residue that can be mutated to Arg to abolish kinase activity (green highlight).Conservation between these proteins is highest in the putative kinase domain (Abe et al., 2002; blue box), and the majority of divergence appears to be in the long C-terminal tail.
C05D10.2/SWIP-13 functions in DA neurons to regulate DA neuron signaling and promote swimming behavior
Reported contributions of ERK7/8 to cell physiology include roles in macroautophagy, oocyte meiotic maturation, ciliogenesis, and the regulation of hormone receptors (Saelzler et al., 2006; Rossi et al., 2011; Colecchia et al., 2012; Yang et al., 2013; Miyatake et al., 2015). Despite evidence of its expression in the brain, as demonstrated by microarray analysis (Hawrylycz et al., 2012), no reports to date have implicated ERK7/8 in regulation of nervous system function. To evaluate the pattern of expression of swip-13, we generated a construct with 1kb of swip-13 sequences upstream of the ATG start codon fused to GFP as a transcriptional reporter (Pswip-13::GFP). Confocal images of worms expressing the transgene revealed GFP expression in a number of head neurons (Fig. 4A) and in a pair of neurons resembling PDE (Fig. 4B), as well as expression by a small number tail neurons and by developing oocytes (Fig. 4E). Dual labeling studies with animals expressing a dopaminergic mCherry reporter (Pdat-1::mCherry) revealed colabeling in all DA neurons (Fig. 4C–E). Evidence of swip-13 expression in DA neurons suggests a cell-autonomous action of the swip-13 mutation in generating Swip. To pursue this hypothesis, we generated a construct providing for DA-neuron-specific expression of swip-13 genomic sequences (Pdat-1::swip-13) and injected this construct into swip-13(gk1234) animals. Consistent with our hypothesis, we observed a significant rescue of the Swip phenotype in these transgenic animals (Fig. 4F). Given that Swip is a hyperdopaminergic phenotype, our findings argue that SWIP-13 functions in a DA neuron pathway that acts normally to constrain DA signaling.
Figure 4.

swip-13 Acts in DA neurons to regulate DA signaling. A, A fusion construct using 1 kb upstream of the swip-13 ATG start site fused to GFP drives GFP expression in swip-13-expressing cells. GFP (green) is observed in a number of neurons in the (A) head, (B) body, and (E) tail of the animal. Coexpression of Pdat-1::mCherry (red) to label DA neurons shows complete colocalization of swip-13-driven GFP and DA neurons (arrows). This is shown by yellow overlap in the CEP and ADE neurons in the head (C) and in the PDE neurons in the body (D). All images were acquired with a 63× objective and represent compressed z-stacks. F, A transgene driving genomic swip-13 by the dat-1 promoter (Pdat-1::swip-13) was made to restrict swip-13 expression to DA neurons. swip-13(gk1234) animals expressing this transgene showed a significant suppression of Swip compared with nontransgenic swip-13(gk1234) animals (t(73) = 8.34, p < 0.0001, unpaired t test). Bars represent the average of three transgenic lines with at least 100 animals per line. Significance was calculated using a two-tailed Student's t test with significance set at p < 0.05. ****p < 0.0001.
To explore where SWIP-13 might be acting within DA neurons to alter DA signaling, we generated a translational fusion construct with GFP cloned in frame at the N-terminus of the swip-13 genomic sequence placing these sequences downstream of the dat-1 promoter to restrict expression to DA neurons (Pdat-1::GFP::swip-13). Confocal images of animals expressing this transgene demonstrated a distribution of the fusion protein throughout the cell, including the cell bodies and processes (Fig. 5A,C,E). Colabeling for DA synapses using Pdat-1::mCherry::RAB-3 demonstrated robust colocalization with GFP::SWIP-13 in CEP (Fig. 5B), ADE (data not shown), and PDE neurons (Fig. 5D), suggesting that a fraction of SWIP-13 is localized to DA synapses. Additional sites of expression include the cell body and the ciliated endings (inset in Fig. 5E) in all three types of DA neurons. Importantly, the GFP::SWIP-13 fusion protein rescued Swip when expressed in swip-13(gk1234) mutants, indicating that the sites of expression noted above derive from the localization of a functional protein (Fig. 5F). Using this construct, we engineered a mutation to mimic mutations known to disable the activities of essentially all protein kinases, including ERK7 and ERK8 (K42R; Abe et al., 1999, 2002). Mutation of the equivalent, highly conserved Lys residue to Arg or Ala prevents the binding and/or hydrolysis of ATP. Notably, the K42R mutated protein was unable to rescue the Swip phenotype (Fig. 5F), although the pattern of protein expression remained qualitatively equivalent (data not shown). These findings indicate that SWIP-13 kinase activity is required for the regulatory actions of SWIP-13 on DA signaling in vivo.
Figure 5.

Subcellular expression of SWIP-13. A, GFP::SWIP-13 fusion protein is seen in CEP and ADE cell bodies and processes. C, In PDE, GFP::SWIP-13 is again seen in the cell body and processes, including the cilia (#). B, D, Colabeling with synaptic marker mCherry::RAB-3 shows colocalization with GFP::SWIP-13 at putative synapses (*) in CEP, ADE (B), and PDE (D). E, In CEP dendrites, GFP::SWIP-13 is seen along the processes, and enriches at the ciliated endings (#). All images were acquired with a 63× objective and represent compressed z-stacks. F, DA neuron-specific expression of GFP::swip-13 in swip-13(gk1234) mutants results in significant Swip suppression (t(58) = 20.31, p < 0.0001, unpaired t test). A kinase-dead GFP::swip-13 fusion, made by introduction a K42R mutation, does not significantly suppress swip-13(gk1234) Swip (t(58) = 1.58, p = 0.12, unpaired t test). Bars represent the average of three transgenic lines with at least 100 animals per line. Significance was calculated using a two-tailed Student's t test for each transgene (transgenic vs nontransgenic) with significance set at p < 0.05. ****p < 0.0001.
SWIP-13 regulates DAT-1 to control DA signaling in vivo
After demonstrating the DA dependence of swip-13 mutants, establishing DA neurons as the relevant site of expression for SWIP-13 in suppressing Swip, and documenting localization of a GFP::SWIP-13 fusion protein to DA synaptic terminals, we reasoned that the kinase might normally support the synaptic activity of DAT-1. To test this model, we crossed the swip-13(gk1234) deletion allele onto a dat-1(ok157) background and assessed the Swip phenotype of double-mutants. In standard water-based Swip assays, we observed no significant enhancement of Swip in swip-13(gk1234);dat-1(ok157) animals when compared with single-mutant dat-1(ok157) animals (Fig. 6A). Because of potential floor effects with the highly penetrant Swip of dat-1(ok157) animals in water, we elevated medium osmolarity to 150 mOsm using sucrose to diminish Swip activity (Hardaway et al., 2012) and thereby enhance the potential dynamic range for observation of Swip additivity in double-mutants. Under these conditions, we again observed a lack of additivity in swip-13(gk1234);dat-1(ok157) double-mutants (Fig. 6C). This result can be seen at a population level in a comparison of heat maps between dat-1(ok157) single- and swip-13(gk1234);dat-1(ok157) double-mutants, where the paralysis of swip-13(gk1234);dat-1(ok157) and dat-1(ok157) were comparable regardless of osmotic strength of the medium (Fig. 6B,D). A similar result was also observed using the DAT-1 inhibitor nisoxetine (NIS) to acutely induce Swip. We have previously shown that acute treatment of worms with NIS induces Swip in a dat-1-dependent manner (Bermingham et al., 2016). Importantly, we observed no difference in the fraction of animals swimming between wild-type (WT) animals treated with NIS versus swip-13(gk1234) animals treated with NIS in both water [WT: 0.11 ± 0.02; swip-13(gk1234): 0.08 ± 0.01, n = 24 p = 0.14, Student's two-tailed t test] and osmosuppressed conditions [WT: 0.34 ± 0.02; swip-13(gk1234): 0.33 ± 0.02, n = 28, p = 0.70, Student's two-tailed t test]. The inability of swip-13 mutation to demonstrate additivity in Swip assays with pharmacological blockade of dat-1, as with genetic experiments, supports contribution of SWIP-13 to a DAT-1 associated pathway.
Further evidence that SWIP-13 acts in the same pathway as DAT-1 arises from experiments where we produced overexpression of DAT-1 in swip-13(gk1234) animals, using an integrated GFP-tagged DAT-1 fusion that we previously demonstrated to be functional (Nass et al., 2002). As shown in Figure 6E, overexpression of DAT-1 restored thrashing behavior to near wild-type levels (Fig. 6E). As more direct proof that SWIP-13 regulates DAT-1 availability or function, we examined the sensitivity of swip-13(gk1234) animals to 6-OHDA, a DAT-1 substrate that induces DA neural degeneration in a DAT-1-dependent manner (Nass et al., 2002). To monitor cell death, we crossed both the swip-13(vt32) and the swip-13(gk1234) alleles onto an N2 background expressing an integrated transgene that expressed GFP in DA neurons (vtIs7), scoring the resulting animals for DA neuron degeneration, as described in Materials and Methods. As documented in Figure 6F, WT animals displayed a virtually complete loss of DA neurons after 6-OHDA treatment, whereas dat-1(ok157) animals are highly resistant. Both swip-13(vt32) and swip-13(gk1234) alleles show reduced sensitivity to 6-OHDA (Fig. 6F). These results support a model where SWIP-13 functions upstream of DAT-1 to sustain DA transport capacity.
Although reduced 6-OHDA sensitivity supports an impact of SWIP-13 on DAT-1 surface expression or function, it is possible that swip-13 mutants could also drive paralysis by enhancing DA release. To test this idea, we used a FRAP assay that makes use of a pH-sensitive SNB-1::SepHluorin transgene expressed in DA neurons (Voglis and Tavernarakis, 2008; Hardaway et al., 2015). This technique takes advantage of the fact that the fluorophore is localized intracellularly to the synaptic vesicle lumen, where the pH is low and the fluorophore is quenched. When the DA vesicle fuses with the plasma membrane (Fig. 7A), fluorescence increases as a result of exposure of the fluorophore to a more neutral environment. After photobleaching DA synapses to diminish actively fluorescing, surface-exposed reporter, the rate of recovery of fluorescence from SNB-1::SepHluorin molecules resident in intracellular vesicles can be used as a proxy for the rate of vesicle fusion. Using this approach, we previously showed that swip-10 mutants induced an enhanced fusion recovery rate, consistent with changes in neuronal excitability (Hardaway et al., 2015). In contrast, we observed no difference in fluorescence recovery rates between WT and swip-13(gk1234) animals (Fig. 7B), suggesting that elevated DA release is unlikely to drive Swip, favoring alterations in mechanisms that impact DA clearance.
Figure 7.

FRAP measurement of vesicle fusion. A, A transgene driving expression of a SNB-1::SEpHluorin fusion protein in DA neurons (Pasic-1::SNB-1::SEpHluorin) was used to measure SV fusion rate. This fluorophore is quenched by the low pH of the SV lumen, and fluoresces upon vesicle fusion due to the higher pH of the extracellular space. Bleaching of this fluorescence and analysis of the recovery of the fluorescent signal allows for a measurement of the rate of SV fusion. B, Comparison of this recovery between N2 and swip-13(gk1234) animals expressing this transgene revealed no significant difference between genotypes. Recovery plots were fit by nonlinear regression methods to a one-phase exponential model, and data were analyzed using repeated-measures ANOVA, with significance value set at p < 0.05. No difference was seen in the K (rate constant) between genotypes (0.035 ± 0.005 s−1 for N2 vs 0.034 ± 0.04 s−1 for swip-13(gk1234), p = 0.93, Student's two-tailed t test).
ERK8-dependent kinase activity elevates DAT activity/surface expression
Given the high conservation between SWIP-13 and its human ortholog ERK8, we wished to determine whether SWIP-13 regulation of DAT-1 is conserved. To test this idea, we coexpressed human DAT and ERK8 cDNAs in the human catecholaminergic neuroblastoma cell line SH-SY5Y. As shown in Figure 8A, ERK8 induced a significant increase in DA transport activity, manifested as a 97% increase in the VMAX of DA uptake compared with cells expressing DAT alone. We also observed a small, but significant increase in DA KM (DAT: 0.67 ± 0.11; DAT/ERK8: 1.16 ± 0.10, two-tailed Student's t test, p = 0.017). A DA transport VMAX increase could be established through the activation of inactive, surface DAT proteins or by an increase in DAT surface expression. The latter possibility was examined using cell-surface biotinylation assays. Here we observed that ERK8 generated a >2-fold increase in surface-labeled DAT protein compared with cells transfected with DAT alone (Fig. 8B,C). Interestingly, total DAT protein levels were increased to a similar degree, suggesting that ERK8 may influence DAT mRNA stability, protein translation and/or stability, the latter perhaps via its effects on DAT trafficking. Transcriptional regulation of the DAT gene is unlikely as the DAT expressed by our transfected cDNA construct is driven by the CMV promoter, and we found that ERK8 had no effect on the levels of GFP protein driven by the CMV promoter in the same cells (data not shown).
Figure 8.

Human ERK8 regulates DAT activity and protein expression. A, Transfection of human DAT into SH-SY5Y (black line) cells results in saturable, cocaine-sensitive [3H]DA uptake. Cotransfection of ERK8 with DAT (red line) leads to an increase in this [3H]DA uptake, as shown by a ∼98% increase in the Vmax of DA uptake. Data are the average of four experiments, and specific DA uptake was calculated by subtracting DA uptake in the presence of cocaine. In each experiment, DA uptake was normalized to protein levels for each transfection condition, and then each data point was normalized to the VMAX of the DAT transfection alone control from that day, calculated using the Michaelis–Menton equation. Curves were generated using a Michaelis–Menton nonlinear fit in Prism. B, Lysates from SH-SY5Y cells treated with the surface biotinylating agent Sulfo-NHS-biotin were subjected to pull-down using Streptavidin-conjugated beads and eluates from these beads are labeled as “Surface”. Total protein before pulldown was run in parallel (“Total”). Mock transfected cells show no anti-DAT staining at the expected molecular weight of DAT of ∼80 kDa, and DAT/ERK8 transfected cells show a robust increase in labeling compared with DAT transfection alone. A similar increase is seen in surface DAT. Anti-TfR was used as a loading control. C, Quantification of B, total DAT was normalized to TfR and these values were normalized to control DAT transfection alone to generate a percentage control value. For surface levels, surface DAT was normalized to surface TfR, and again values were normalized to control surface levels to generate a percentage control value. ERK8 transfection significantly elevated both total (t(4) = 2.94, p = 0.0425, unpaired t test) and surface DAT (t(4) = 10.02, p = 0.0006, unpaired t test). Data are the average of three experiments and were analyzed using two-tailed Student's t tests for each group of data (total and surface), and a significance threshold was set at p < 0.05. ***p < 0.001. D, Transfection of kinase-dead ERK8(K42R) and vt32-analogous ERK8(R59Q) mutants do not increase DA uptake in SH-SY5Y cells cotransfected with DAT [ERK8(K42R): t(8) = 0.23, p > 0.05, Bonferroni's post-tests; ERK8(R59Q): t(8) = 0.45, p > 0.05, Bonferroni's post-tests]. As previously shown, wild-type ERK8 significantly increases DAT activity (t(8) = 3.40, p < 0.05, Bonferroni's post-tests). Data are the average of three experiments, and were analyzed using a one-way ANOVA with Bonferroni's post-tests comparing each condition to vector control. Significance was set at p < 0.05. *p < 0.01. E, In vitro kinase assay of HA-ERK8, isolated with HA beads, using purified MBP as the substrate. Wild-type HA-ERK8, but neither the kinase-dead HA-ERK8(K42R) nor the HA-ERK8(R59Q) mutant, significantly phosphorylates MBP above background levels. Importantly, all conditions had similar levels of total MBP protein as visualized by Coomassie stain, and ERK8 mutants were expressed at similar levels to WT ERK8, as measured by Western blot analysis of cell lysates using an anti-HA antibody.
To test whether the ERK8-induced increase in whole-cell DAT activity, like Swip, is dependent on ERK8 kinase activity, we quantified DA uptake with cells transfected with the kinase-inactivating mutation K42R (Fig. 8E, kinase activity assays). As shown in Figure 8D, this mutation eliminated the stimulatory effect on ERK8 cotransfection on DAT activity. Additionally, we introduced the R59Q mutation in ERK8 to reproduce the molecular lesion that we identified in swip-13(vt32). Similar to the lack of DAT regulation imparted by the K42R mutation, we found no significant elevation of DAT activity with cotransfection of the ERK8 R59Q mutation. (Fig. 8D). Finally, we sought confirmatory evidence that the inability of ERK K24R and R59Q mutants to increase DAT activity is determined by a loss of ERK8 kinase activity. In these experiments, we compared the ability of WT and mutant ERK8 proteins to phosphorylate the artificial substrate MBP using an in vitro kinase assay (Abe et al., 2002). Whereas wild-type ERK8 reliably phosphorylated MBP, neither the kinase-dead ERK8(K42R) nor ERK8 bearing the swip-13(vt32) mutation [ERK8(R59Q)] increased MBP phosphorylation above background levels (Fig. 8E).
SWIP-13/ERK7/8 regulates Rho GTPases to control DA signaling and DAT activity
As noted earlier GFP::SWIP-13 was expressed at synapses and in anterior ciliated endings of DA neuron dendrites (Fig. 5). ERK7 has been implicated in a Wnt signaling pathway regulating ciliogenesis via its interactions with Dishevelled (Miyatake et al., 2015), Another protein that binds to Dishevelled and regulates ciliogenesis is the small GTPase RhoA, and loss of RhoA produces similar alterations as Dishevelled mutations to cilia structure (Pan et al., 2007). Moreover, the Amara laboratory recently demonstrated that RhoA regulates DAT endocytosis in response to AMPH treatment (Wheeler et al., 2015), nominating this pathway as a possible contributor to the SWIP-13/ERK7/8 regulation of DAT proteins. To pursue this idea, we first sought to determine whether ERK8 might influence RhoA activity. Due to low efficiency of our transfections with SH-SY5Y cells, we pursued this effort using transfected HEK-293T cells. We performed pull-down assays on extracts of mock- or ERK8-transfected HEK-293T cells using beads conjugated to a RBD that only binds active Rho proteins, blotting bead eluates for RhoA. In these assays, we detected significantly increased levels of active RhoA following ERK8 transfection (Fig. 9A,B). To assess whether RhoA activation might underlie the ERK8 regulation of DAT, we transfected DAT/ERK8 coexpressing SH-SY5Y cells with GFP-C3, which is an inhibitor of Rho proteins. We observed an inability of ERK8 to enhance DA transport activity with GFP-C3 transfection (Fig. 9C). Importantly, we found no significant effect of GFP-C3 transfection on the DAT activity observed in the absence of ERK8 cotransfection, suggesting that endogenous Rho activity in SH-SY5Y cells does not contribute significantly to basal DA uptake induced by DAT transfection. Additionally, this finding indicates that GFP-C3 transfection did not significantly alter cell viability, as cells coexpressing GFP-C3 and DAT were able to transport DA at levels comparable to cells expressing DAT alone.
Figure 9.

ERK8 activates Rho to regulate DAT. A, Cells transfected with HA-ERK8 plasmid show an increase in active RhoA protein compared with mock transfected cells. Active RhoA was measured by pulldown using GST-RBD beads that bind active Rho. B, Quantification of three experiments. Active RhoA was increased in HA-ERK8 transfected cells (t(4) = 3.05, p < 0.05, unpaired t test), with no significant change in total RhoA protein levels (t(4) = 0.77, p = 0.49, unpaired t test). Significance was calculated using a two-tailed Student's t test comparing empty vector and ERK8 transfection for both total and active RhoA with significance set at p < 0.05. *p < 0.05. C, As shown previously, ERK8 overexpression significantly increases DAT activity compared with vector-transfected cells (t(8) = 4.47, p < 0.01, Bonferroni's post-tests). Transfection of GFP-C3 prevents any increase in DAT activity with ERK8 overexpression (DAT/ERK8/GFP-C3 vs DAT/GFP-C3: t(8) = 0.43, p > 0.05, Bonferroni's post-tests). Data were analyzed using a one-way ANOVA with Bonferroni's post-tests comparing DAT/ERK8 to DAT, and DAT/ERK8/GFP-C3 to DAT/GFP-C3. Significance was set at p < 0.05. **p < 0.01. D, Transgenic expression of Pdat-1::rho-1 cDNA in swip-13(gk1234) animals drives overexpression of WT rho-1 in DA neurons, and significantly rescues Swip compared with nontransgenic swip-13(gk1234) animals (t(59) = 15.85, p < 0.0001, unpaired t test). Bars represent the average of three transgenic lines with at least 100 animals per line. Significance was calculated using a two-tailed Student's t test with significance set at p < 0.05. ****p < 0.0001. E, Treatment of N2 animals with the ROCK inhibitor fasudil HCl induces significant Swip that is significantly reduced in cat-2(e1112) and cat-2(tm2261) mutants. Significance was calculated using a one-way ANOVA with Bonferroni's post-tests comparing N2 in water to N2 in fasudil (t(156) = 16.58, p < 0.0001, Bonferroni's post-tests), N2 in fasudil to cat-2(e1112) in fasudil (t(156) = 6.41, p <0.0001, Bonferroni's post-tests), and N2 in fasudil to cat-2(tm2261) in fasudil (t(156) = 6.19, p <0.0001, Bonferroni's post-tests). Significance was set at p < 0.05. ****p < 0.0001.
Finally, we sought to determine whether the an ERK8/Rho/DAT regulatory interaction is functional in vivo. To pursue this objective, we overexpressed a cDNA encoding the C. elegans RhoA ortholog RHO-1 in the DA neurons of swip-13(gk1234) animals, followed by Swip assays. As shown in Figure 9D, we observed a significant suppression of Swip with rho-1 overexpression, consistent with SWIP-13 acting upstream of RHO-1 to regulate DAT-1. As a second, independent test of this this idea, and to avoid potential indirect effects associated with chronic RHO-1 overexpression, we treated WT worms acutely with an inhibitor (fasudil, 0.5 mm), of rho-associated coiled-coil containing kinase (ROCK), a downstream effector of Rho that has been implicated in the DAT trafficking effects of amphetamine (Wheeler et al., 2015). As shown in Figure 9E, fasudil induced Swip in WT animals that could be suppressed by two loss-of-function alleles of the tyrosine hydroxylase ortholog cat-2. Together, our studies support a model in which SWIP-13/ERK8 acts in DA neurons to sustain normal levels of DAT trafficking via activation of RHO-1/Rho thereby sustaining normal levels of DA clearance.
Discussion
We first described Swip as a feature of loss-of-function dat-1 mutants and have used it for in vivo DAT-1 structure–function studies (McDonald et al., 2007; Robinson et al., 2017), to isolate novel dat-1 alleles (Hardaway et al., 2012), and to identify genes whose mutation phenocopies a dat-1 mutation (Hardaway et al., 2015). Our forward genetic Swip screen was designed to identify genes with contributions to DA signaling, and although it was based on the properties of dat-1 mutants, such that positive DAT-1 regulators could be identified, the Swip phenotype has sufficient breadth to allow identification of genes that function at multiple levels within DA neurons (e.g., DA neuron excitability, DA synthesis, DA secretion, DA metabolism), as well as in non-DA cells whose function normally regulates DA neuron signaling. For example, we recently reported identification of the gene swip-10, as a glial expressed gene where loss-of-function mutations enhance DA neuron excitability and DA secretion, leading to extrasynaptic DA overflow and DOP-3-dependent paralysis (Hardaway et al., 2015). Here, we identify the previously unstudied gene C05D10.2 as the site of the mutation in the vt32 line (Hardaway et al., 2012), now designated as swip-13. At a population level, swip-13 mutant animals have a milder paralysis phenotype compared with dat-1 (or swip-10) mutants, suggesting a less robust and/or less well sustained hyperdopaminergic state that arises in the context of a loss of functional SWIP-13 protein.
Through a combination of genetic, biochemical, imaging and behavior experiments, we present evidence that swip-13 mutation produces Swip by reducing dat-1 availability and/or function. Specifically, we show that (1) genomic swip-13 expression in DA neurons rescues swip-13 paralysis, (2) swip-13;dat-1 double-mutants do not show additive Swip compared with single-mutants, (3) dat-1 overexpression rescues swip-13 mutant Swip, (4) GFP::SWIP-13 protein localizes to DA synapses, (5) DAT activity-dependent 6-OHDA toxicity is significantly reduced in swip-13 animals (notably not seen with swip-10 animals; Hardaway et al., 2015), and (6) swip-13 animals lack the enhanced rate of DA vesicular release observed with swip-10 mutants (Hardaway et al., 2015). Although we show that all DA neurons express swip-13, consistent with prior microarray studies (Spencer et al., 2011), swip-13 expression is not exclusive to these cells and thus DA neuron-specific rescue of Swip provides key support for this model. Additionally, we identified sites in DA neurons other than synapses where SWIP-13 protein localizes (e.g., terminal cilia), suggesting potential contributions to DA neuron structure/function/excitability that may be independent of, or could synergize with the DAT regulatory model we present. In this regard, our FRAP experiments support changes in DA clearance versus release as the basis for the hyperdopaminergia that drives swip-13 Swip. However, we recognize that these data are gathered from immobilized animals versus the thrashing-induced context of water immersion. We cannot as yet rule out that SWIP-13 makes a contribution to context-dependent DA neuron excitability.
The swip-13 gene has not been previously characterized in nematodes, but is highly related to the human atypical MAP kinases ERK7/8. Other MAP kinases, specifically ERK1/2 have been implicated in the regulation of DA signaling, including regulation of DAT activity and trafficking (Morón et al., 2003; Bolan et al., 2007; Subramanian and Morozov, 2011; Yu et al., 2011; Kivell et al., 2014). Although a more complete elaboration of mechanisms downstream of ERK1/2 that support DAT regulation awaits, there is some evidence that ERK1/2 phosphorylates a site (Thr53 in mouse/rat/human DAT) on the DAT N terminus to elevate DAT surface expression and function (Gorentla et al., 2009; Foster et al., 2012). Presynaptic D2-type DA autoreceptors are thought to activate ERK1/2 to enhance DAT surface expression (Morón et al., 2003; Bolan et al., 2007; Kivell et al., 2014). A presynaptically expressed D2-type DA receptor (DOP-2) also regulates DA signaling in worms (Suo et al., 2003; Bermingham et al., 2016). Possibly, SWIP-13 may play an analogous role to mammalian ERK1/2 in autoreceptor control of DAT-1, though our studies support a pathway dependent on Rho activation versus direct DAT phosphorylation. The combination of in vitro and in vivo approaches we have implemented should be useful in further dissecting and integrating the elements of DAT regulation supported by typical and atypical MAPKs.
Not only are SWIP-13/ERK7/8 highly related, but the site of the vt32 mutation, R59, is conserved from worms to humans, suggesting an important contribution to kinase structure/function, possibly in substrate recognition and/or kinase activation (Kannan and Neuwald, 2004). Importantly, we observed no reduction of HA-ERK8 protein levels arising from either the R59Q mutation or the kinase-dead K42R mutation. Most likely, SWIP-13 R59Q lacks kinase activity, supported by the loss of kinase activity seen with our in vitro phosphorylation studies using ERK8 R59Q as well as the fact that a deletion mutation of swip-13 phenocopies the allele we recovered.
In the human catecholaminergic neuroblastoma cell line SH-SY5Y, coexpression of ERK8 and human DAT led to a doubling of DA transport capacity (VMAX) compared with cells transfected with DAT alone. Biotinylation studies reveal this effect to be due to an increase in DAT surface expression. We also observed an effect of ERK8 on total DAT protein levels. Further studies are needed to determine whether ERK8 increases DAT mRNA stability, transporter protein translation, or reduces DAT degradation. Interestingly, both surface expression and degradation of mammalian DAT appear to be regulated by the MAP kinase phosphatase MKP3 (Mortensen et al., 2008), with evidence suggesting that the phosphatase modulates the activity of a non-classical MAP kinase that could include ERK7/8. With respect to changes in DAT protein levels induced by ERK8 expression, multiple reports suggest that DAT can be routed to different subcellular compartments upon endocytosis, with differential targeting contributing to whether the transporter is degraded or recycled (Sorkina et al., 2006; Gabriel et al., 2013; Hong and Amara, 2013; Vuorenpää et al., 2016). It is therefore possible that ERK8 induces a shift in DAT endocytic fate that both elevates DAT surface trafficking and routes the transporter away from compartments that support degradation. Clarification of this issue may well have disease significance. We have reported that the ADHD-associated DAT mutation R615C displays altered trafficking such that PKC-dependent endocytosis is precluded in favor of a rapid, constitutively recycling pathway. Interestingly, MKP3 also opposes PKC-dependent endocytosis (Mortensen et al., 2008), further stimulating our interest in exploring the convergence of ERK8/MKP3/PKC-linked pathways in future studies.
With respect to a potential target of SWIP-13/ERK8 in regulating DAT availability, we provide evidence for a critical role of the Rho class of small GTPase proteins. Our initial orientation toward an examination of Rho proteins in SWIP-13/ERK7/8 action arose from finding GFP::SWIP-13 localized to the cilia-containing endings of DA neuron (CEP) dendrites, combined with evidence that ERK7 both regulates ciliogenesis and interacts with the RhoA-interacting protein Dishevelled (Park et al., 2008; Miyatake et al., 2015). Moreover, loss of RhoA produces similar alterations as Dishevelled mutations to cilia structure (Pan et al., 2007). That SWIP-13/ERK8 effects on DAT depend on RhoA activation is supported by (1) the ability of ERK8 overexpression to increase active RhoA in HEK-293T cells; (2) the ability of C3 exotoxin, which ADP ribosylates and inactivates Rho family GTPases (Aktories and Just, 2005), to eliminate ERK8-driven elevation of DAT activity; (3) the ability of the ROCK inhibitor fasudil to produce a phenotype (Swip) seen in dat-1 mutants; and (4) the ability of overexpression of rho-1 to reduce Swip in swip-13 mutants. Recently, Wheeler et al. (2015) reported that amphetamine reduces DAT surface expression via Rho activation. Similarly, Rho activation has been suggested to support the ability of the Rho family guanine nucleotide exchange factor Vav2 to drive DAT endocytosis following activation of the GDNF receptor Ret (Zhu et al., 2015). In contrast, both our in vitro and in vivo studies support a model whereby SWIP-13/ERK8 enhances DAT surface expression via Rho family member activation. These contrasting effects may derive from distinct modes of Rho activation (SWIP-13/ERK8 vs Ret/amphetamine), engagement of different and potentially antagonistic Rho family isoforms (Tashiro et al., 2000; Duan et al., 2010; Shoval and Kalcheim, 2012), and/or different subcellular and membrane compartments supporting DAT trafficking. Indeed, the Rho family GTPase Cdc42 was recently shown to act as a brake on DAT endocytosis, supporting roles for Rho GTPases in both promoting and antagonizing DAT surface expression (Wu et al., 2015). Altogether, our studies bring to light a contribution of a novel pathway regulating DAT and extracellular DA availability, and they suggest that the further study of SWIP-13/ERK8 mechanisms can provide a more complete picture of how the presynaptic terminal controls DA signaling and possibly clues to disorders associated with DA perturbations, as well as opportunities for improved therapeutics.
Footnotes
This work was supported by NIH awards MH090544 (R.D.B.), DA035559 (D.P.B.), MH093102 (J.A.H.) NS078291 (R.J.C.), MH109196 (C.R.M.), and by the Vanderbilt Institute for Clinical and Translational Research Center (D.P.B.), and the Vanderbilt Brain Institute and the Graduate Neuroscience Training Program through NIH award MH064913 to D.P.B. and J.A.H. Experiments were performed through the use of the VUMC Cell Imaging Shared Resource, supported by NIH Grants CA68485, DK20593, DK58404, DK59637, and EY08126. We thank Christina Svitek, Jane Wright, Tracy Moore-Jarrett, and Angela Steele for expert laboratory oversight.
The authors declare no competing financial interests.
References
- Abe MK, Kuo WL, Hershenson MB, Rosner MR (1999) Extracellular signal-regulated kinase 7 (ERK7), a novel ERK with a C-terminal domain that regulates its activity, its cellular localization, and cell growth. Mol Cell Biol 19:1301–1312. 10.1128/MCB.19.2.1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe MK, Saelzler MP, Espinosa R 3rd, Kahle KT, Hershenson MB, Le Beau MM, Rosner MR (2002) ERK8, a new member of the mitogen-activated protein kinase family. J Biol Chem 277:16733–16743. 10.1074/jbc.M112483200 [DOI] [PubMed] [Google Scholar]
- Aktories K, Just I (2005) Clostridial rho-inhibiting protein toxins. Curr Top Microbiol Immunol 291:113–145. 10.1007/3-540-27511-8_7 [DOI] [PubMed] [Google Scholar]
- Allen AT, Maher KN, Wani KA, Betts KE, Chase DL (2011) Coexpressed D1- and D2-like dopamine receptors antagonistically modulate acetylcholine release in Caenorhabditis elegans. Genetics 188:579–590. 10.1534/genetics.111.128512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson NG, Maller JL, Tonks NK, Sturgill TW (1990) Requirement for integration of signals from two distinct phosphorylation pathways for activation of MAP kinase. Nature 343:651–653. 10.1038/343651a0 [DOI] [PubMed] [Google Scholar]
- Apparsundaram S, Galli A, DeFelice LJ, Hartzell HC, Blakely RD (1998) Acute regulation of norepinephrine transport: I. protein kinase C-linked muscarinic receptors influence transport capacity and transporter density in SK-N-SH cells. J Pharmacol Exp Ther 287:733–743. [PubMed] [Google Scholar]
- Bermingham DP, Blakely RD (2016) Kinase-dependent regulation of monoamine neurotransmitter transporters. Pharmacol Rev 68:888–953. 10.1124/pr.115.012260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermingham DP, Hardaway JA, Snarrenberg CL, Robinson SB, Folkes OM, Salimando GJ, Jinnah H, Blakely RD (2016) Acute blockade of the Caenorhabditis elegans dopamine transporter dat-1 by the mammalian norepinephrine transporter inhibitor nisoxetine reveals the influence of genetic modifications of dopamine signaling in vivo. Neurochem Int 98:122–128. 10.1016/j.neuint.2016.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigelow H, Doitsidou M, Sarin S, Hobert O (2009) MAQGene: software to facilitate C. elegans mutant genome sequence analysis. Nat Methods 6:549. 10.1038/nmeth.f.260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolan EA, Kivell B, Jaligam V, Oz M, Jayanthi LD, Han Y, Sen N, Urizar E, Gomes I, Devi LA, Ramamoorthy S, Javitch JA, Zapata A, Shippenberg TS (2007) D2 receptors regulate dopamine transporter function via an extracellular signal-regulated kinases 1 and 2-dependent and phosphoinositide 3 kinase-independent mechanism. Mol Pharmacol 71:1222–1232. 10.1124/mol.106.027763 [DOI] [PubMed] [Google Scholar]
- Brenner S. (1974) The genetics of Caenorhabditis elegans. Genetics 77:71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalfie M, Jorgensen EM (1998) C. elegans neuroscience: genetics to genome. Trends Genet 14:506–512. 10.1016/S0168-9525(98)01623-0 [DOI] [PubMed] [Google Scholar]
- Chase DL, Pepper JS, Koelle MR (2004) Mechanism of extrasynaptic dopamine signaling in Caenorhabditis elegans. Nat Neurosci 7:1096–1103. 10.1038/nn1316 [DOI] [PubMed] [Google Scholar]
- Colecchia D, Strambi A, Sanzone S, Iavarone C, Rossi M, Dall'Armi C, Piccioni F, Verrotti di Pianella A, Chiariello M (2012) MAPK15/ERK8 stimulates autophagy by interacting with LC3 and GABARAP proteins. Autophagy 8:1724–1740. 10.4161/auto.21857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MW, Hammarlund M, Harrach T, Hullett P, Olsen S, Jorgensen EM (2005) Rapid single nucleotide polymorphism mapping in C. elegans. BMC Genomics 6:118. 10.1186/1471-2164-6-118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan L, Chen G, Virmani S, Ying G, Raja SM, Chung BM, Rainey MA, Dimri M, Ortega-Cava CF, Zhao X, Clubb RJ, Tu C, Reddi AL, Naramura M, Band V, Band H (2010) Distinct roles for Rho versus Rac/Cdc42 GTPases downstream of Vav2 in regulating mammary epithelial acinar architecture. J Biol Chem 285:1555–1568. 10.1074/jbc.M109.057976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerr JS, Frisby DL, Gaskin J, Duke A, Asermely K, Huddleston D, Eiden LE, Rand JB (1999) The cat-1 gene of Caenorhabditis elegans encodes a vesicular monoamine transporter required for specific monoamine-dependent behaviors. J Neurosci 19:72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis HM, Horvitz HR (1986) Genetic control of programmed cell death in the nematode C. elegans. Cell 44:817–829. 10.1016/0092-8674(86)90004-8 [DOI] [PubMed] [Google Scholar]
- Foster JD, Cervinski MA, Gorentla BK, Vaughan RA (2006) Regulation of the dopamine transporter by phosphorylation. Handb Exp Pharmacol 175:197–214. 10.1007/3-540-29784-7_10 [DOI] [PubMed] [Google Scholar]
- Foster JD, Yang JW, Moritz AE, Challasivakanaka S, Smith MA, Holy M, Wilebski K, Sitte HH, Vaughan RA (2012) Dopamine transporter phosphorylation site threonine 53 regulates substrate reuptake and amphetamine-stimulated efflux. J Biol Chem 287:29702–29712. 10.1074/jbc.M112.367706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel LR, Wu S, Kearney P, Bellvé KD, Standley C, Fogarty KE, Melikian HE (2013) Dopamine transporter endocytic trafficking in striatal dopaminergic neurons: differential dependence on dynamin and the actin cytoskeleton. J Neurosci 33:17836–17846. 10.1523/JNEUROSCI.3284-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorentla BK, Moritz AE, Foster JD, Vaughan RA (2009) Proline-directed phosphorylation of the dopamine transporter N-terminal domain. Biochemistry 48:1067–1076. 10.1021/bi801696n [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünhage F, Schulze TG, Müller DJ, Lanczik M, Franzek E, Albus M, Borrmann-Hassenbach M, Knapp M, Cichon S, Maier W, Rietschel M, Propping P, Nöthen MM (2000) Systematic screening for DNA sequence variation in the coding region of the human dopamine transporter gene (DAT1). Mol Psychiatry 5:275–282. 10.1038/sj.mp.4000711 [DOI] [PubMed] [Google Scholar]
- Hamilton PJ, Campbell NG, Sharma S, Erreger K, Herborg Hansen F, Saunders C, Belovich AN, Consortium NAAS, Sahai MA, Cook EH, Gether U, McHaourab HS, Matthies HJ, Sutcliffe JS, Galli A (2013) De novo mutation in the dopamine transporter gene associates dopamine dysfunction with autism spectrum disorder. Mol Psychiatry 18:1315–1323. 10.1038/mp.2013.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen FH, Skjorringe T, Yasmeen S, Arends NV, Sahai MA, Erreger K, Andreassen TF, Holy M, Hamilton PJ, Neergheen V, Karlsborg M, Newman AH, Pope S, Heales SJ, Friberg L, Law I, Pinborg LH, Sitte HH, Loland C, Shi L, et al. (2014) Missense dopamine transporter mutations associate with adult parkinsonism and ADHD. J Clin Invest 124:3107–3120. 10.1172/JCI73778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardaway JA, Hardie SL, Whitaker SM, Baas SR, Zhang B, Bermingham DP, Lichtenstein AJ, Blakely RD (2012) Forward genetic analysis to identify determinants of dopamine signaling in Caenorhabditis elegans using swimming-induced paralysis. G3 (Bethesda) 2:961–975. 10.1534/g3.112.003533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardaway JA, Wang J, Fleming PA, Fleming KA, Whitaker SM, Nackenoff A, Snarrenberg CL, Hardie SL, Zhang B, Blakely RD (2014) An open-source analytical platform for analysis of C. elegans swimming-induced paralysis. J Neurosci Methods 232:58–62. 10.1016/j.jneumeth.2014.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardaway JA, Sturgeon SM, Snarrenberg CL, Li Z, Xu XZ, Bermingham DP, Odiase P, Spencer WC, Miller DM 3rd, Carvelli L, Hardie SL, Blakely RD (2015) Glial expression of the Caenorhabditis elegans gene swip-10 supports glutamate-dependent control of extrasynaptic dopamine signaling. J Neurosci 35:9409–9423. 10.1523/JNEUROSCI.0800-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, van de Lagemaat LN, Smith KA, Ebbert A, Riley ZL, Abajian C, Beckmann CF, Bernard A, Bertagnolli D, Boe AF, Cartagena PM, Chakravarty MM, Chapin M, Chong J, Dalley RA, et al. (2012) An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 489:391–399. 10.1038/nature11405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong WC, Amara SG (2013) Differential targeting of the dopamine transporter to recycling or degradative pathways during amphetamine- or PKC-regulated endocytosis in dopamine neurons. FASEB J 27:2995–3007. 10.1096/fj.12-218727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howes O, McCutcheon R, Stone J (2015) Glutamate and dopamine in schizophrenia: an update for the 21st century. J Psychopharmacol 29:97–115. 10.1177/0269881114563634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayanthi LD, Apparsundaram S, Malone MD, Ward E, Miller DM, Eppler M, Blakely RD (1998) The Caenorhabditis elegans gene T23G5.5 encodes an antidepressant- and cocaine-sensitive dopamine transporter. Mol Pharmacol 54:601–609. [PubMed] [Google Scholar]
- Kannan N, Neuwald AF (2004) Evolutionary constraints associated with functional specificity of the CMGC protein kinases MAPK, CDK, GSK, SRPK, DYRK, and CK2alpha. Protein Sci 13:2059–2077. 10.1110/ps.04637904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivell B, Uzelac Z, Sundaramurthy S, Rajamanickam J, Ewald A, Chefer V, Jaligam V, Bolan E, Simonson B, Annamalai B, Mannangatti P, Prisinzano TE, Gomes I, Devi LA, Jayanthi LD, Sitte HH, Ramamoorthy S, Shippenberg TS (2014) Salvinorin A regulates dopamine transporter function via a kappa opioid receptor and ERK1/2-dependent mechanism. Neuropharmacology 86:228–240. 10.1016/j.neuropharm.2014.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen AS, Andersen J, Jørgensen TN, Sørensen L, Eriksen J, Loland CJ, Strømgaard K, Gether U (2011) SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol Rev 63:585–640. 10.1124/pr.108.000869 [DOI] [PubMed] [Google Scholar]
- Kurian MA, Zhen J, Cheng SY, Li Y, Mordekar SR, Jardine P, Morgan NV, Meyer E, Tee L, Pasha S, Wassmer E, Heales SJ, Gissen P, Reith ME, Maher ER (2009) Homozygous loss-of-function mutations in the gene encoding the dopamine transporter are associated with infantile parkinsonism-dystonia. J Clin Invest 119:1595–1603. 10.1172/JCI39060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazei-Robison MS, Couch RS, Shelton RC, Stein MA, Blakely RD (2005) Sequence variation in the human dopamine transporter gene in children with attention deficit hyperactivity disorder. Neuropharmacology 49:724–736. 10.1016/j.neuropharm.2005.08.003 [DOI] [PubMed] [Google Scholar]
- Mazei-Robison MS, Bowton E, Holy M, Schmudermaier M, Freissmuth M, Sitte HH, Galli A, Blakely RD (2008) Anomalous dopamine release associated with a human dopamine transporter coding variant. J Neurosci 28:7040–7046. 10.1523/JNEUROSCI.0473-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald PW, Jessen T, Field JR, Blakely RD (2006) Dopamine signaling architecture in Caenorhabditis elegans. Cell Mol Neurobiol 26:593–618. 10.1007/s10571-006-9003-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald PW, Hardie SL, Jessen TN, Carvelli L, Matthies DS, Blakely RD (2007) Vigorous motor activity in Caenorhabditis elegans requires efficient clearance of dopamine mediated by synaptic localization of the dopamine transporter dat-1. J Neurosci 27:14216–14227. 10.1523/JNEUROSCI.2992-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikian HE. (2004) Neurotransmitter transporter trafficking: endocytosis, recycling, and regulation. Pharmacol Ther 104:17–27. 10.1016/j.pharmthera.2004.07.006 [DOI] [PubMed] [Google Scholar]
- Mergy MA, Gowrishankar R, Gresch PJ, Gantz SC, Williams J, Davis GL, Wheeler CA, Stanwood GD, Hahn MK, Blakely RD (2014) The rare DAT coding variant Val559 perturbs DA neuron function, changes behavior, and alters in vivo responses to psychostimulants. Proc Natl Acad Sci U S A 111:E4779–4788. 10.1073/pnas.1417294111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyatake K, Kusakabe M, Takahashi C, Nishida E (2015) ERK7 regulates ciliogenesis by phosphorylating the actin regulator CapZIP in cooperation with Dishevelled. Nat Commun 6:6666. 10.1038/ncomms7666 [DOI] [PubMed] [Google Scholar]
- Morón JA, Zakharova I, Ferrer JV, Merrill GA, Hope B, Lafer EM, Lin ZC, Wang JB, Javitch JA, Galli A, Shippenberg TS (2003) Mitogen-activated protein kinase regulates dopamine transporter surface expression and dopamine transport capacity. J Neurosci 23:8480–8488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen OV, Larsen MB, Prasad BM, Amara SG (2008) Genetic complementation screen identifies a mitogen-activated protein kinase phosphatase, MKP3, as a regulator of dopamine transporter trafficking. Mol Biol Cell 19:2818–2829. 10.1091/mbc.E07-09-0980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nass R, Duerr J, Rand JB, Miller DM III, Blakely RD (2000) 6-OHDA sensitivity of dopaminergic neurons in C. elegans: role of the dopamine transporter and cell death pathways. Soc Neurosci Abstr 26:572.577. [Google Scholar]
- Nass R, Hall DH, Miller DM 3rd, Blakely RD (2002) Neurotoxin-induced degeneration of dopamine neurons in Caenorhabditis elegans. Proc Natl Acad Sci U S A 99:3264–3269. 10.1073/pnas.042497999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt DJ, Lingford-Hughes A, Erritzoe D, Stokes PR (2015) The dopamine theory of addiction: 40 years of highs and lows. Nat Rev Neurosci 16:305–312. 10.1038/nrn3939 [DOI] [PubMed] [Google Scholar]
- Pan J, You Y, Huang T, Brody SL (2007) RhoA-mediated apical actin enrichment is required for ciliogenesis and promoted by Foxj1. J Cell Sci 120:1868–1876. 10.1242/jcs.005306 [DOI] [PubMed] [Google Scholar]
- Park TJ, Mitchell BJ, Abitua PB, Kintner C, Wallingford JB (2008) Dishevelled controls apical docking and planar polarization of basal bodies in ciliated epithelial cells. Nat Genet 40:871–879. 10.1038/ng.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penmatsa A, Wang KH, Gouaux E (2013) X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 503:85–90. 10.1038/nature12533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson SB, Hardaway JA, Hardie SL, Wright J, Glynn RM, Bermingham DP, Han Q, Sturgeon SM, Freeman P, Blakely RD (2017) Sequence determinants of the Caenorhabditis elegans dopamine transporter dictating in vivo axonal export and synaptic localization. Mol Cell Neurosci 78:41–51. 10.1016/j.mcn.2016.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe DL. (1997) From DOPA to Parkinson's disease: the early history of dopamine research. J Hist Neurosci 6:291–301. 10.1080/09647049709525715 [DOI] [PubMed] [Google Scholar]
- Rossi M, Colecchia D, Iavarone C, Strambi A, Piccioni F, Verrotti di Pianella A, Chiariello M (2011) Extracellular signal-regulated kinase 8 (ERK8) controls estrogen-related receptor α (ERRα) cellular localization and inhibits its transcriptional activity. J Biol Chem 286:8507–8522. 10.1074/jbc.M110.179523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saelzler MP, Spackman CC, Liu Y, Martinez LC, Harris JP, Abe MK (2006) ERK8 down-regulates transactivation of the glucocorticoid receptor through Hic-5. J Biol Chem 281:16821–16832. 10.1074/jbc.M512418200 [DOI] [PubMed] [Google Scholar]
- Sakrikar D, Mazei-Robison MS, Mergy MA, Richtand NW, Han Q, Hamilton PJ, Bowton E, Galli A, Veenstra-Vanderweele J, Gill M, Blakely RD (2012) Attention deficit/hyperactivity disorder-derived coding variation in the dopamine transporter disrupts microdomain targeting and trafficking regulation. J Neurosci 32:5385–5397. 10.1523/JNEUROSCI.6033-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarin S, Prabhu S, O'Meara MM, Pe'er I, Hobert O (2008) Caenorhabditis elegans mutant allele identification by whole-genome sequencing. Nat Methods 5:865–867. 10.1038/nmeth.1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawin ER, Ranganathan R, Horvitz HR (2000) C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron 26:619–631. 10.1016/S0896-6273(00)81199-X [DOI] [PubMed] [Google Scholar]
- Schafer WR, Kenyon CJ (1995) A calcium-channel homologue required for adaptation to dopamine and serotonin in Caenorhabditis elegans. Nature 375:73–78. 10.1038/375073a0 [DOI] [PubMed] [Google Scholar]
- Schmitt KC, Reith ME (2010) Regulation of the dopamine transporter: aspects relevant to psychostimulant drugs of abuse. Ann N Y Acad Sci 1187:316–340. 10.1111/j.1749-6632.2009.05148.x [DOI] [PubMed] [Google Scholar]
- Segura-Aguilar J, Paris I, Muñoz P, Ferrari E, Zecca L, Zucca FA (2014) Protective and toxic roles of dopamine in Parkinson's disease. J Neurochem 129:898–915. 10.1111/jnc.12686 [DOI] [PubMed] [Google Scholar]
- Shoval I, Kalcheim C (2012) Antagonistic activities of Rho and Rac GTPases underlie the transition from neural crest delamination to migration. Dev Dyn 241:1155–1168. 10.1002/dvdy.23799 [DOI] [PubMed] [Google Scholar]
- Smith CJ, Watson JD, Spencer WC, O'Brien T, Cha B, Albeg A, Treinin M, Miller DM 3rd (2010) Time-lapse imaging and cell-specific expression profiling reveal dynamic branching and molecular determinants of a multi-dendritic nociceptor in C. elegans. Dev Biol 345:18–33. 10.1016/j.ydbio.2010.05.502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkina T, Miranda M, Dionne KR, Hoover BR, Zahniser NR, Sorkin A (2006) RNA interference screen reveals an essential role of Nedd4–2 in dopamine transporter ubiquitination and endocytosis. J Neurosci 26:8195–8205. 10.1523/JNEUROSCI.1301-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer WC, Zeller G, Watson JD, Henz SR, Watkins KL, McWhirter RD, Petersen S, Sreedharan VT, Widmer C, Jo J, Reinke V, Petrella L, Strome S, Von Stetina SE, Katz M, Shaham S, Rätsch G, Miller DM 3rd (2011) A spatial and temporal map of C. elegans gene expression. Genome Res 21:325–341. 10.1101/gr.114595.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strambi A, Mori M, Rossi M, Colecchia D, Manetti F, Carlomagno F, Botta M, Chiariello M (2013) Structure prediction and validation of the ERK8 kinase domain. PLoS One 8:e52011. 10.1371/journal.pone.0052011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian J, Morozov A (2011) Erk1/2 inhibit synaptic vesicle exocytosis through L-type calcium channels. J Neurosci 31:4755–4764. 10.1523/JNEUROSCI.6594-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston J, Dew M, Brenner S (1975) Dopaminergic neurons in the nematode caenorhabditis elegans. J Comp Neurol 163:215–226. 10.1002/cne.901630207 [DOI] [PubMed] [Google Scholar]
- Suo S, Sasagawa N, Ishiura S (2003) Cloning and characterization of a Caenorhabditis elegans D2-like dopamine receptor. J Neurochem 86:869–878. 10.1046/j.1471-4159.2003.01896.x [DOI] [PubMed] [Google Scholar]
- Tashiro A, Minden A, Yuste R (2000) Regulation of dendritic spine morphology by the rho family of small GTPases: antagonistic roles of Rac and Rho. Cereb Cortex 10:927–938. 10.1093/cercor/10.10.927 [DOI] [PubMed] [Google Scholar]
- Torres GE, Gainetdinov RR, Caron MG (2003) Plasma membrane monoamine transporters: structure, regulation and function. Nat Rev Neurosci 4:13–25. 10.1038/nrn1008 [DOI] [PubMed] [Google Scholar]
- Viggiano D, Vallone D, Sadile A (2004) Dysfunctions in dopamine systems and ADHD: evidence from animals and modeling. Neural Plast 11:97–114. 10.1155/NP.2004.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voglis G, Tavernarakis N (2008) A synaptic DEG/ENaC ion channel mediates learning in C. elegans by facilitating dopamine signalling. EMBO J 27:3288–3299. 10.1038/emboj.2008.252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuorenpää A, Jorgensen TN, Newman AH, Madsen KL, Scheinin M, Gether U (2016) Differential internalization rates and postendocytic sorting of the norepinephrine and dopamine transporters are controlled by structural elements in the N termini. J Biol Chem 291:5634–5651. 10.1074/jbc.M115.702050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinshenker D, Garriga G, Thomas JH (1995) Genetic and pharmacological analysis of neurotransmitters controlling egg laying in C. elegans. J Neurosci 15:6975–6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DS, Underhill SM, Stolz DB, Murdoch GH, Thiels E, Romero G, Amara SG (2015) Amphetamine activates Rho GTPase signaling to mediate dopamine transporter internalization and acute behavioral effects of amphetamine. Proc Natl Acad Sci U S A 112:E7138–E7147. 10.1073/pnas.1511670112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Bellve KD, Fogarty KE, Melikian HE (2015) Ack1 is a dopamine transporter endocytic brake that rescues a trafficking-dysregulated ADHD coding variant. Proc Natl Acad Sci U S A 112:15480–15485. 10.1073/pnas.1512957112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SW, Huang H, Gao C, Chen L, Qi ST, Lin F, Wang JX, Hou Y, Xing FQ, Sun QY (2013) The distribution and possible role of ERK8 in mouse oocyte meiotic maturation and early embryo cleavage. Microsc Microanal 19:190–200. 10.1017/S1431927612013918 [DOI] [PubMed] [Google Scholar]
- Yu HS, Kim SH, Park HG, Kim YS, Ahn YM (2011) Intracerebroventricular administration of ouabain, a Na/K-ATPase inhibitor, activates tyrosine hydroxylase through extracellular signal-regulated kinase in rat striatum. Neurochem Int 59:779–786. 10.1016/j.neuint.2011.08.011 [DOI] [PubMed] [Google Scholar]
- Zhu J, Reith ME (2008) Role of the dopamine transporter in the action of psychostimulants, nicotine, and other drugs of abuse. CNS Neurol Disord Drug Targets 7:393–409. 10.2174/187152708786927877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Zhao C, Wu Y, Yang Q, Shao A, Wang T, Wu J, Yin Y, Li Y, Hou J, Zhang X, Zhou G, Gu X, Wang X, Bustelo XR, Zhou J (2015) Identification of a Vav2-dependent mechanism for GDNF/Ret control of mesolimbic DAT trafficking. Nat Neurosci 18:1084–1093. 10.1038/nn.4060 [DOI] [PubMed] [Google Scholar]