

Abstract
Yersinia ruckeri is the causative agent of enteric redmouth disease (ERM) of salmonids. There is little information regarding the proteomics of Y. ruckeri. Herein, we perform whole protein identification and quantification of biotype 1 and biotype 2 strains of Y. ruckeri grown under standard culture conditions using a shotgun proteomic approach. Proteins were extracted, digested and peptides were separated by a nano liquid chromatography system and analyzed with a high-resolution hybrid triple quadrupole time of flight mass spectrometer coupled via a nano ESI interface. SWATH-MS technology and sophisticated statistical analyses were used to identify proteome differences among virulent and avirulent strains. GO annotation, subcellular localization, virulence proteins and antibiotic resistance ontology were predicted using bioinformatic tools. A total of 1395 proteins were identified in the whole cell of Y. ruckeri. These included proteases, chaperones, cell division proteins, outer membrane proteins, lipoproteins, receptors, ion binding proteins, transporters and catalytic proteins. In virulent strains, a total of 16 proteins were upregulated including anti-sigma regulatory factor, arginine deiminase, phosphate-binding protein PstS and superoxide dismutase Cu–Zu. Additionally, several virulence proteins were predicted such as Clp and Lon pro-teases, TolB, PPIases, PstS, PhoP and LuxR family transcriptional regulators. These putative virulence proteins might be used for development of novel targets for treatment of ERM in fish. Our study represents one of the first global proteomic reference profiles of Y. ruckeri and this data can be accessed via ProteomeXchange with identifier PXD005439. These proteomic profiles elucidate proteomic mechanisms, pathogenicity, host-interactions, antibiotic resistance ontology and localization of Y. ruckeri proteins.
Electronic supplementary material
The online version of this article (doi:10.1186/s13567-017-0460-3) contains supplementary material, which is available to authorized users.
Introduction
Enteric redmouth disease (ERM) is one of the most important bacterial diseases of salmonids and causes significant economic losses in the aquaculture industry worldwide. ERM can affect fish from all age classes and appears as a more chronic condition in older and larger fish. The disease is caused by Yersinia ruckeri, a Gram-negative rod-shaped enterobacterium [1, 2]. Y. ruckeri enters the fish via the secondary gill lamellae and from there spreads to the blood and internal organs [3]. Clinical signs of the disease include exophthalmia, darkening of the skin in addition to subcutaneous hemorrhages in and around the mouth and throat. The spleen is often enlarged and can be almost black in color and the lower intestine can become reddened and filled with an opaque, yellowish fluid [1, 2]. Focal areas of necrosis can be present in the organs (spleen, kidney and liver). Degenerated renal tubules, glomerular nephritis and a marked increase in melano-macrophages may be observed in the kidney of infected fish [1, 2, 4]. Several virulence factors of Y. ruckeri have been identified such as extra-cellular products and Yrp1. Extra-cellular products have been shown to reproduce the clinical signs of the disease [5]. The 47 kDa metalloprotease Yrp1 is necessary for virulence and degrades fibronectin, actin and myosin of the fish [6].
Strains of Y. ruckeri have been categorized into two biotypes: biotype 1 strains are motile and lipase positive, while biotype 2 strains are negative for these phenotypes [2, 7]. Previously, the majority of epizootic outbreaks in salmonids were caused by biotype 1 strains which could be easily controlled by vaccination with a bacterin vaccine [5]. Nevertheless, biotype 2 strains have recently emerged and have been responsible for outbreaks in both naive and vaccinated fish, thereby suggesting that biotype 2 strains may be less sensitive to the traditional ERM vaccine which is made from a biotype 1 strain [8, 9]. This relationship between vaccine failure and emergence of biotype 2 has led to the hypothesis that the loss of the flagellum is essential for resistance to immersion vaccination [9, 10]. However, bivalent or biotype 2 vaccines provide good protection against the biotype 2 strains [2, 11].
Whole genome sequences of Y. ruckeri strains have been annotated and can now be used for comparative genomic analysis of strains and other research purposes [12]. Global proteomic identification and comparative analysis of Y. ruckeri strains are required to create a proteomic map, understanding proteomic biology, proteomic changes and proteomic differences between strains. Little is known about the proteomics of Y. ruckeri. Outer membrane protein and whole cell protein patterns of Y. ruckeri isolates were described using SDS-PAGE and 2D-PAGE [11, 13, 14]. Reference proteome maps of many bacteria including Y. pestis have been created, and this work is leading to an understanding of the virulence mechanisms and the regulatory networks used by pathogenic bacteria [15]. However, for fish pathogens, in-depth proteomic analysis is not yet well established.
In our previous study, we compared two culture conditions of Y. ruckeri strains and focused only on proteins expressed in response to iron-limited culture conditions [16]. In this study, we identified, quantified and analyzed the global proteomic profiles of Y. ruckeri strains grown under standard culture conditions using a shotgun proteomic approach. Furthermore, we predicted virulence proteins and antibiotic resistance ontology in the proteome of Y. ruckeri.
Materials and methods
Bacterial strains
Two biotype 1 (SP-05 and CSF007-82) and two biotype 2 (7959-11 and YRNC-10) Y. ruckeri strains were used in the present study. These four strains were isolated from rainbow trout (Oncorhynchus mykiss) and all are serotype 01. Strains SP-05 and 7959-11 originated from Austria and the other two strains, CSF007-82 and YRNC-10, originated from the USA. Virulence for rainbow trout was determined previously using an experimental challenge model. Strains CSF007-82, 7959-11 and YRNC-10 were virulent [17, 18] and strain SP-05 was not virulent (Authors unpublished data). The antimicrobial susceptibility of strains was tested using routine clinical laboratory susceptibility methods employing antimicrobial discs [(enrofloxacin (5 µg), florfenicol (30 µg), tetracycline (30 µg), amoxicillin (10 µg), oxolinic acid (2 µg), trimethoprim–sulfamethoxazole (25 µg), flumequine (30 µg) and doxycycline (30 µg)].
Culture conditions
The culture conditions and growth yield of Y. ruckeri strains have been previously described [16]. Briefly, a single colony of each strain was used to inoculate duplicate 5 mL tryptic soy broth cultures. Duplicate starter cultures of each strain (OD600 0.10) were then used to inoculate 25 mL tryptic soy broth cultures and grown overnight at 22 °C until the late log phase. The yield of CSF007-82, 7959-11 and YRNC-10 strains (OD600 1.62) were similar to each other but the yield of SP-05 strain was slightly lower (OD600 1.32) compared to the other three strains [16]. Cells were harvested and washed three times with sterile phosphate buffered saline containing bacterial protease inhibitor cocktail.
Protein extraction and digestion
The protein extraction procedures used have been previously described [16]. Briefly, bacterial cells were resuspended in denaturing lysis buffer (7 M urea, 2 M thiourea, 4% 3-[(3-cholamidopropyl)dimethyl-ammonio]-1-propane sulfonate and 1% dithiothreitol) containing bacterial protease inhibitor cocktail. Cells were then sonicated on ice and cellular debris removed by centrifugation. Protein digestion was performed using the standard two-step in-solution digestion protocol for Trypsin/LysC mix according to the user manual (Promega) and digested samples were acidified.
Nano LC–MS/MS analysis
Tryptic peptides were separated by a nano liquid chromatography system (Dionex Ultimate 3000 RSLC) and analyzed with a high-resolution hybrid triple quadrupole time of flight mass spectrometer (TripleTOF 5600+, Sciex) coupled via a nano-ESI interface. Preconcentration and desalting of samples were accomplished with a 5 mm Acclaim PepMap µ-Precolumn (Dionex). Details of the LC–MS/MS procedure were described previously [16]. Briefly, 370 ng of digested protein were used per injection and peptide separation was performed on a 25 cm Acclaim PepMap C18 column with a flow rate of 300 nL/min. The gradient started with 4% mobile phase B (80% acetonitrile with 0.1% formic acid) and increased to 35% B over 120 min. MS1 survey scans were collected in the range of 400–1500 mass-to-charge ratio (m/z). The 25 most intense precursors with charge state 2–4, which exceeded 100 counts per second, were selected for fragmentation for 250 ms. MS2 product ion scans were collected in the range of 100–1800 m/z for 110 ms. Precursor ions were dynamically excluded from reselection for 12 s.
For quantitative measurements, data independent sequential window acquisition of all theoretical spectra (SWATH) technology based on MS2 quantification was used [19, 20]. Peptides from biological and technical replicates were fragmented in 35 fixed fragmentation windows of 20 Dalton (Da) in the range of 400–1100 Da with an accumulation time of 50 ms in TOF MS mode and 80 ms in product ion mode. The nano-HPLC system was operated by Chromeleon 6.8 (Dionex) and the MS by Analyst Software 1.6 (Sciex).
Data analysis
Database searches of raw files of data dependent acquisition were carried out with Protein Pilot Software version 5.0 (Sciex). UniProt database (Released 10_2016) was restricted to Y. ruckeri. Mass tolerance in MS mode was set with 0.05 and 0.1 Da in MS/MS mode for the rapid recalibration search as well as 0.0011 Da in MS and 0.01 Da in MS/MS mode for the final search. The following sample parameters were applied: trypsin digestion, cysteine alkylation set to iodoacetamide and the search effort set was to rapid identification. False discovery rate analysis was performed using the integrated tools in ProteinPilot. The global false discovery rate (FDR) was set to < 1% on the protein level, peptide level as well as spectra level. Information dependent data acquisition identification results were used to create the SWATH ion library with the MS/MS (ALL) with SWATH Acquisition MicroApp 2.0 in PeakView 2.2 (both Sciex). Peptides were chosen based on a FDR rate < 1%, excluding shared and modified peptides. Up to six peptides per protein and up to 6 transitions per peptide were used. MarkerView 1.2.1 (Sciex) was used for calculation of peak areas of SWATH samples after retention time alignment and normalization using total area sums. The resulting protein lists were then used for visualization of data after principal component analysis (PCA) in form of loading plots and score plots to get a first impression of the overall data structure and to assess variability between technical and biological replicates.
Differentially expressed proteins were determined by statistical analysis in R programming language [21]. Raw peak areas after normalization to total area sums were log2-transformed to approach a normal distribution. On a logarithmic scale, technical replicates were aggregated by arithmetic mean before application of statistical tests. This procedure is equivalent to the application of a hierarchical model in the subsequent ANOVA, as the same number of technical replicates was measured per biological replicate. Differential expression of proteins in each strain was assessed using one-way ANOVA for each protein. To adjust for multiple testing, the method of Benjamini and Hochberg [22] was used to control the FDR. Differences were considered significant if adjusted p-values were smaller than the significance level of α = 0.001. For those proteins, Tukey’s honest significant difference method was applied as post hoc test to assess the significance of the pairwise comparisons. Protein expression was considered differential if the adjusted p-value was below α and the absolute fold change was at least three (fold change < −3 or > +3).
GO annotation and prediction of virulent proteins
Venn diagrams were used to show the differences between protein lists originating from different strains [23]. Gene ontology annotation of all identified proteins was classified using the software tool for researching annotations of proteins [24]. Subcellular localization of proteins was predicted by PSORTb version 3.0 [25]. Virulence proteins were predicted by a method based on bi-layer cascade Support Vector Machine using VirulentPred [26].
Antibiotic resistance ontology and their validation
Antibiotic resistance ontology was identified using a comprehensive antibiotic resistance database [27]. The antibiotic resistance phenotypes predicted by in silico analysis were validated using the disc diffusion technique and minimal inhibitory concentration (MIC) determination [28, 29]. The antimicrobial commercial Oxoid discs (µg disc/mL, Thermo Scientific): gentamicin (10 µg), polymyxin B (300 UI), erythromycin (15 µg), rifampin (5 µg), novobiocin (5 µg) and mupirocin (5 µg) were applied to inoculated Mueller–Hinton agar (Thermo Scientific) in triplicate. In parallel, MIC ranges for the same antibiotics were determined using microtiter plates and solutions of antibiotics prepared from powders of known potencies (Sigma-Aldrich). All plates were incubated for 48 h at 22 °C. The diameter of the inhibition halo of antimicrobial discs and lowest concentration of antibiotic that inhibited visible growth of bacteria were defined and categorized as susceptible or resistant (Additional file 1) as previously using standard methods [28, 29].
Results
Protein identification
A total of 1395 proteins in the whole cell of Y. ruckeri were identified (Additional file 2). The number of proteins identified in each strain was 1193 for SP-05, 1263 for CSF007-82, 1244 for 7959-11 and 1208 for YRNC-10. The list of identified proteins in each strain is given in Additional file 3. Forty-six proteins in SP-05, 43 proteins in CSF007-82, 31 proteins in 7959-11 and 13 proteins in YRNC-10 were uniquely identified (Figure 1). PCA score plots of all strains suggested that strain SP-05 differs from the other three strains (CSF007-82, 7959-11 and YRNC-10) but the latter three strains showed minor proteomic differences (Additional file 4). The list of uniquely identified proteins in each strain is given in Additional file 5.
Figure 1.
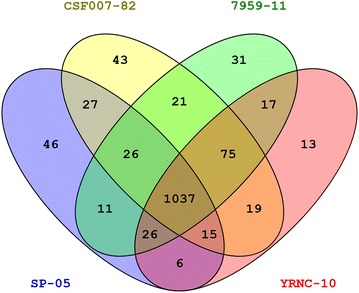
Venn diagram showing the number of proteins identified in Yersinia ruckeri strains. The number of unique or shared proteins in each strain is indicated in each set or subset.
Protein quantification
Sophisticated statistical evaluation revealed a total number of 36 differentially expressed proteins within the four analyzed Y. ruckeri strains. Of these, 16 were upregulated (SP-05 strain versus the other strains) (Table 1) and 20 were downregulated (Additional file 6). As can be seen in Table 1, upregulated proteins were related to iron ion homeostasis, regulation of transcription, transporter activity and metabolic processes. Similarly, downregulated proteins were related to flagellar motility, phosphotransferase system, glycolysis and metabolic processes. We observed upregulation of two proteins: phosphoenolpyruvate (> 25.1-fold) and asparagines synthase (4.3-fold) in biotype 2 strains [biotype 1 strain (CSF007-82) versus biotype 2 strains (7959-11 and YRNC-10)] but saw no significant expression differences between biotype 2 strains (7959-11 versus YRNC-10).
Table 1.
Fold changes of differentially expressed proteins of Yersinia ruckeri strains compared to each other
| UniProt accession number | Protein | Function | SP-05 vs CSF007-82 | SP-05 vs 7959-11 | SP-05 vs YRNC-10 | CSF007-82 vs 7959-11 | CSF007-82 vs YRNC-10 | 7959-11 vs YRNC-10 |
|---|---|---|---|---|---|---|---|---|
| A0A085U6V7_YERRU | Bacterioferritin | Ferric iron binding | 6.8* | 5.7* | 6.5* | −1.2 | −1.0 | 1.1 |
| A0A085U4B6_YERRU | DNA protection during starvation protein | Iron ion homeostasis | 3.2* | 3.8* | 1.9 | 1.2 | −1.7 | −2.1 |
| A0A085U5L5_YERRU | Anti-sigma factor antagonist | Regulation of transcription | 3.6* | 3.7* | 3.8* | 1.0 | 1.1 | 1.0 |
| A0A085U5L7_YERRU | Anti-sigma regulatory factor | Serine/threonine kinase activity | 3.9* | 3.9* | 4.3* | −1.0 | 1.1 | 1.1 |
| A0A085UBQ1_YERRU | Arginine deiminase | Arginine catabolic process | 5.7* | 5.2* | 5.8* | −1.1 | 1.0 | 1.1 |
| A0A085U605_YERRU | Amino acid transporter | Transporter activity | 4.3* | 4.2* | 4.0* | −1.0 | −1.1 | −1.0 |
| A0A085U8U0_YERRU | Phosphate-binding protein PstS | Phosphate ion transmembrane transport | 3.0 | 3.1* | 3.4* | 1.0 | 1.1 | 1.1 |
| A0A0A5FQB4_YERRU | Superoxide dismutase Cu–Zn | Superoxide dismutase activity | 2.8 | 3.2* | 3.4* | 1.2 | 1.2 | 1.1 |
| A0A0A5FMC5_YERRU | Arginine decarboxylase, catabolic | Amino acid metabolic process | 7.9* | 6.5* | 6.2* | −1.2 | −1.3 | −1.0 |
| A0A085UBP8_YERRU | Glutamate decarboxylase | Glutamate metabolic process | 10.4* | 8.5* | 6.8* | −1.2 | −1.5 | −1.3 |
| A0A0A8VE52_YERRU | Glutaminase | Glutamine metabolic process | 6.7* | 6.5* | 8.2* | −1.0 | 1.2 | 1.3 |
| A0A085U745_YERRU | Glucose-1-phosphate adenylyltransferase | Glycogen biosynthetic process | 5.3* | 5.0* | 3.8* | −1.1 | −1.4 | −1.3 |
| A0A085UBM7_YERRU | 3-Oxoacyl-ACP reductase | Oxidoreductase | 6.2* | 6.1* | 6.8* | −1.0 | 1.1 | 1.1 |
| A0A085U7G0_YERRU | Uncharacterized protein | Unknown | 9.1* | 7.9* | 9.5* | −1.2 | 1.0 | 1.2 |
| A0A085UBQ0_YERRU | Uncharacterized protein | Unknown | 5.0* | 4.9* | 4.3* | −1.0 | −1.2 | −1.2 |
| A0A085U732_YERRU | Putative exported protein | Unknown | 2.9 | 2.9 | 3.4* | 1.0 | 1.2 | 1.2 |
ANOVA was performed for UniProt database searches.
* Denotes statistically significant difference according to Tukey’s honest significant difference post hoc test with FDR-adjusted p < 0.001 and fold change < −3 or > +3.
GO annotation and subcellular localization of proteins
The identified proteins were associated with cellular process, metabolic process, regulation, localization and response to stimulus (Figure 2A). Proteins were localized in the cytoplasm, plasma membrane, ribosome, macromolecular complex, nucleus, chromosome and others (Figure 2B). Proteins involved in catalytic activity and binding were the most abundant among those identified proteins, 51 and 39%, respectively (Figure 2C). The identified proteins were predicted in the cytoplasmic space (67%), unknown (16%), cytoplasmic membrane (8%), periplasmic space (6%), outer membrane (2%) and extracellular space (1%) (Figure 3). The unknown group included proteins with multiple subcellular and unknown localizations.
Figure 2.
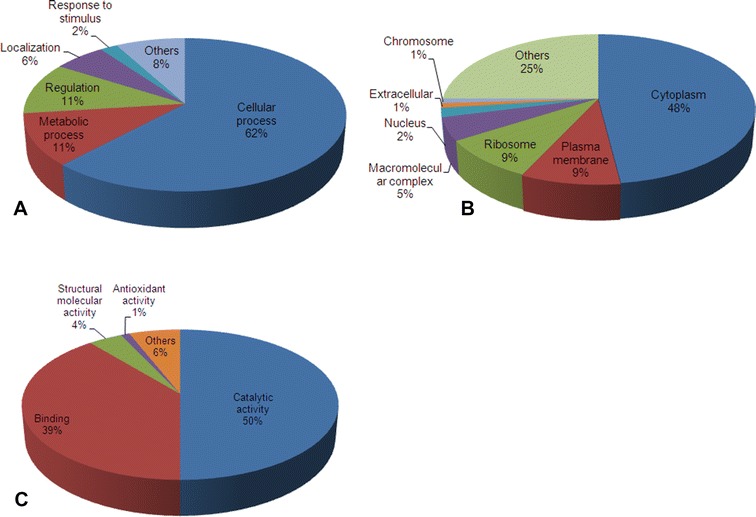
Proteomic profiles of identified proteins of Yersinia ruckeri. Proteins were classified by gene ontology terms for biological processes, cellular components and molecular functions using software tool for researching annotations of proteins (A) biological process, (B) cellular component, and (C) molecular function.
Figure 3.
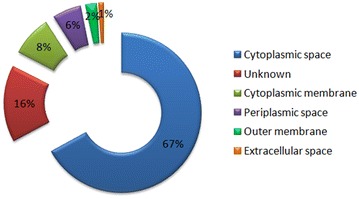
Subcellular locations of Yersinia ruckeri proteins. Cellular location of proteins was predicted by PSORTb version 3.0 and particular location of proteins was shown in percentage. Unknown location includes proteins with multiple localization sites or unknown location.
Virulence proteins and antibiotic resistance ontology
Several predicted virulence proteins were identified: HtrA protease, protein TolB, peptidyl-prolyl cis–trans isomerase, UvrY response regulator, chaperone protein fimC, lipoprotein NlpD, putative exported protein, MltA-interacting protein, superoxide dismutase Cu–Zn, PhoP, LuxR and AsnC family transcriptional regulators (Table 2 and Additional file 7).
Table 2.
Lists of important virulence proteins of Yersinia ruckeri
| Protein | Function | Cascade of SVMs and PSI-BLAST, score |
|---|---|---|
| Gene expression modulator/haemolysin expression modulating protein | Haemolysin expression | 1.0520 |
| HtrA protease | Serine-type endopeptidase activity | 0.5097 |
| Outer membrane stress sensor protease DegQ serine protease | Serine-type endopeptidase activity | 1.0012 |
| Anti-sigma regulatory factor | Protein serine/threonine kinase activity | 0.9363 |
| Beta-barrel assembly-enhancing protease | Chaperone and a metalloprotease | 1.0339 |
| BarA-associated response regulator UvrY | Regulation of transcription | 0.9837 |
| Peptidyl-prolyl cis–trans isomerase | Protein folding | 0.9128 |
| PhoP family transcriptional regulator | Regulation of transcription | 1.0898 |
| LuxR family transcriptional regulator | Regulation of transcription | 1.0031 |
| AsnC family transcriptional regulator | Regulation of transcription | 0.7053 |
| RNA-binding protein Hfq | Regulation of transcription | 1.0260 |
| Anti-sigma factor antagonist | Regulation of transcription | 0.9804 |
| Attachment invasion locus protein | Invasion | 1.0123 |
| Invasin | Cell adhesion | 1.0130 |
| Superoxide dismutase Cu–Zn | Superoxide dismutase activity | 1.0003 |
| Molybdenum ABC transporter periplasmic molybdenum-binding protein ModA | Transporter activity | 0.8399 |
| DcrB protein | Required for phage C1 adsorption | 1.0009 |
| Methyl-accepting chemotaxis protein I | Chemotaxis | 0.9279 |
| Methyl-accepting chemotaxis protein III | Chemotaxis | 0.9922 |
Proteins were predicted by a method based on bi-layer cascade support vector machine using VirulentPred.
We also predicted antibiotic resistance ontology in 12 antibiotic classes (Table 3) in the proteome of Y. ruckeri, which contains 14 proteins such as bacterial regulatory protein (cyclic AMP receptor protein), membrane fusion protein of the resistance-nodulation-division (RND) family multidrug efflux pump, bifunctional polymyxin resistance protein ArnA and RND efflux system inner membrane transporter CmeB.
Table 3.
Details of antibiotic resistance ontology of Yersinia ruckeri
| Protein | Antibiotic resistance ontology | Bit score |
|---|---|---|
| Bacterial regulatory, crp family protein (cyclic AMP receptor protein) | Fluoroquinolone (enrofloxacin), beta-lactam (amoxicillin), Macrolide (erythromycin) | 431.409 |
| Cys regulon transcriptional activator CysB | Aminocoumarin (novobiocin) | 620.928 |
| Copper-sensing two-component system response regulator CpxR | Aminoglycoside (gentamicin), aminocoumarin (novobiocin) | 389.808 |
| Alanine tRNA ligase | Aminocoumarin | 1487.63 |
| Transcription repair-coupling factor | Fluoroquinolone | 1938.7 |
| Dihydropteroate synthase | Sulfonamide | 440.654 |
| Membrane fusion protein of RND family multidrug efflux pump | Fluoroquinolone, beta-lactam, Macrolide, Rifampin, Chloramphenicol, Tetracycline, Aminocoumarin | 548.125 |
| Beta-lactamase | beta-lactam (amoxicillin) | 608.601 |
| Outer membrane channel protein | Fluoroquinolone, beta-lactam, Macrolide, Rifampin, Chloramphenicol, Tetracycline, Aminocoumarin | 689.878 |
| Elongation factor Tu | Elfamycin | 583.178 |
| Bifunctional polymyxin resistance protein ArnA | Polymyxin B | 951.814 |
| DNA gyrase subunit A | Fluoroquinolone | 751.895 |
| RND efflux system inner membrane transporter CmeB | Fluoroquinolone, tetracycline | 751.125 |
| Isoleucine-tRNA ligase | Mupirocin | 219.55 |
Antibiotic resistance ontology was predicted in the proteome of Y. ruckeri using a comprehensive antibiotic resistance database.
Antimicrobial susceptibility test
Strains of Y. ruckeri were susceptible to enrofloxacin, florfenicol, tetracycline, amoxicillin, oxolinic acid, trimethoprim–sulfamethoxazole, flumequine and doxycycline (data not shown). Additional file 1 shows the diameter of the inhibition zone and MIC of antimicrobial agents used for validation of antibiotic resistance ontology. Novobiocin and mupirocin discs displayed no inhibition zone against Y. ruckeri strains, while strains showed intermediate susceptibility to gentamicin and polymyxin B. Y. ruckeri strains were resistant to erythromycin (MIC = 1024 μg/mL), rifampin (MIC = 32 μg/mL), novobiocin (MIC = 16–32 μg/mL) and mupirocin (MIC = 32–64 μg/mL). Three antibiotic resistance ontologies: novobiocin, mupirocin and erythromycin were fully consistent with the proteomic data such as cys regulon transcriptional activator CysB, alanine tRNA ligae and isoleucine-tRNA ligase.
Discussion
Here we identify global proteomic reference profiles of Y. ruckeri strains (PXD005439) grown under standard culture conditions. These global proteomic profiles help us to understand the physiology, protein biology, virulence factors, host-interactions, localization and antibiotic resistance of Y. ruckeri. The total number of proteins identified was 1395 in Y. ruckeri (Additional file 2). These included proteases, chaperones, cell division proteins, outer membrane proteins, chromosome partitioning proteins and transporters. Proteins have been classified into different functional categories such as biological process and molecular function (Figure 2) and this information will be useful for further studies in the direction of extracellular (flagellin and flagellar hook-associated protein), interaction with cells (invasin and manganese ABC transporter, periplasmic-binding protein SitA), antioxidant (thioredoxin reductase and glutathione amide-dependent peroxidase) and molecular transducer (methyl-accepting chemotaxis protein) activity. The identification of predicted virulence proteins (Table 2; Additional file 7) and antibiotic resistance ontology (Table 3) contributes to our understanding of this pathogen and will aid in the rational design of novel treatment strategies for ERM disease.
Biotype 2 strains showed minor proteomic differences among each other (Additional file 4). However, the Austrian biotype 1 strain (SP-05) showed major proteomic differences when compared to the USA biotype 1 strain (CSF007-82) and biotype 2 strains (7959-11 and YRNC-10). These major differences may be due to the slightly lower yield and growth rate of SP-05 strain compared to the other three strains (CSF007-82, 7959-11 and YRNC-10) or its avirulent nature toward the fish.
Sixteen upregulated proteins were identified in virulent Y. ruckeri strains using a sophisticated statistical analysis (avirulent SP-05 strain versus virulent strains). We found strong upregulation of bacterioferritin (5.7- to 6.8-fold) and DNA protection during starvation protein (3.2- to 3.8-fold) in Y. ruckeri strains. However, iron dependent proteins (bacterioferritin and iron-sulfur cluster assembly scaffold protein IscU) were downregulated (−3-fold) in Y. ruckeri strains in response to iron-limited culture conditions [16]. The phosphate-binding protein PstS is a high affinity phosphate binding protein of the Pst transport system and has been shown to be involved in pathogenesis, invasion and biofilm formation of many bacteria [30]. Superoxide dismutase Cu–Zn is an important for oxidative stress and has been shown to contribute to the pathogenicity of many bacteria [31]. Arginine deiminase protects bacterial cells against the damaging effects of acidic environments and enhances the ability of cells to survive in acidic extracellular conditions [32]. We observed strong upregulation of phosphate-binding protein PstS (> 3-fold), superoxide dismutase Cu–Zn (3.3- to 3.4-fold) and arginine deiminase (5.2- to 5.8-fold) in Y. ruckeri strains. Based on the results of the present study, it appears that upregulated proteins (avirulent strain versus virulent strains) such as PstS, SOD-Cu–Zn and arginine deiminase may be involved in the establishment of disease inside the host and the survival of Y. ruckeri during the infection process.
Several proteases such as HtrA, Lon, carboxy-terminal, signal peptidase I, La Type II, HslUV, pyrrolidone–carboxylate, FtsH, protease III, Clp, protease 4, putative protease, peptidase B, T and M37 were identified. These proteases were serine, threonine, cysteine, metalloproteinase and ATP-dependent type proteases, and belonged to the C15, M16, M17, M20B, M23, S16, S26, S41A, S49, U32, AAA ATPase and Clp families including PDZ domains. Proteases play critical roles in the invasion of host tissues, contribute to virulence and damage host tissue during infection [33]. The Yrp1 protease of Y. ruckeri has been implicated in the hydrolysis of different matrix and muscle proteins of fish and vaccination with Yrp1 elicits a strong protection against the development of enteric redmouth disease [6]. Additionally, the Clp and Lon pro-teases have been shown to have a role in the regulation of the type III secretion systems (T3SS) in various bacterial pathogens. The T3SS forms a needle-like structure in several Gram negative bacteria that allows direct transfer of bacterial virulence factors into the cytoplasm of host cells. The T3SS has been linked to flagellum biosynthesis [34]. We also identified flagellar biosynthesis proteins (FliC, FliG, FliH and FliN), flagellar hook proteins (FlgD, FlgE and FlgK), flagellar brake protein YcgR, flagellar motor protein MotB and pilus assembly protein PilW in Y. ruckeri. FliC and FliH flagellar proteins have been linked with pathogenesis in the fish pathogen, Edwardsiella tarda [35]. Additionally, the Y. ruckeri flagellin protein has been shown to elicit a robust innate immune response and protect fish against biotype 1 and biotype 2 Y. ruckeri strains [36]. More research on the role of proteases and T3SS in Y. ruckeri virulence is needed to more fully understand the pathogenicity of Y. ruckeri.
We also identified other important virulence proteins such as the UvrY response regulator, peptidyl-prolyl cis–trans isomerase (PPIases), TolB, PhoP and LuxR family transcriptional regulators. UvrY is a response regulator of the BarA-UvrY two-component system and has been shown to be involved in the pathogenesis of Y. ruckeri, probably through its regulation of both the invasion of epithelial cells and protection against oxidative stress induced by immune cells [37]. PPIases are FKBP domain-containing ubiquitous folding proteins and have been reported as virulence factors in several bacterial pathogens [38]. Upregulation of FKBP-type peptidyl-prolyl cis–trans isomerases has been observed in iron-starved biotype 2 Y. ruckeri strains [16], which may be involved in virulence of Y. ruckeri. PhoP is part of a two component system and is important for bacterial survival and replication in macrophages [39]. TolB is the periplasmic component of the Tol–Pal system and is important for antibiotic resistance and pathogenicity in Gram negative pathogens and has been suggested as a suitable candidate for the development of novel drugs against Pseudomonas aeruginosa [40]. The LuxR transcriptional regulator is a key player in quorum sensing and affects survival, virulence, antibiotic biosynthesis and biofilm formation of bacteria [41].
A number of chaperone proteins (CbpA, ClpB, DnaK, DnaJ, fimC, HscA, HscB, HtpG, skp, SurA, ProQ), an acid stress chaperone HdeB, universal stress protein E, cold shock (CspC and CspE) and a phage shock protein were identified in Y. ruckeri. Bacterial pathogens produce a number of chaperone proteins for survival during changing environments and stress conditions [42]. Some chaperone proteins have also been implicated in bacterial virulence [43]. DnaK chaperone protein plays a role in protein folding and interacts with ClpB in reactivating proteins which have become aggregated after heat shock [44]. The DnaK/DnaJ chaperone machinery and ClpB have been shown to be involved in the invasion of epithelial cells and survival within macrophages of the host, leading to systemic infection of Salmonella enterica and Francisella tularensis in mice [43, 45]. Upregulation of ClpB, HtpG and universal stress protein A have been observed in Flavobacterium psychrophilum during in vivo growth in fish and were suggested to play an important role in the pathogenesis of F. psychrophilum [46]. Based on these data, we suggest that some chaperone proteins may be important for in vivo survival and pathogenesis of Y. ruckeri.
A number of cell division proteins (BolA, DedD, DamX, FtsA, FtsE, FtsH, FtsP, FtsZ, ZapA, ZapB and ZapD), chromosome partitioning proteins (ParA, ParB, MukB and MukE) and biosynthesis proteins (iscR, MraZ, basR/pmrA, IF-1, IF-3, S2-S21, L1-L6, RsmA-RsmC and RsmG-RsmI) were identified. The FtsZ and ParA proteins have been identified as potential drug targets against clinically important bacterial pathogens [47]. Protein synthesis (transcriptional and translational) proteins have been targeted for inhibition of bacterial pathogens [48]. However, cell division and chromosome partitioning proteins may act as new drug targets for Y. ruckeri. Additionally, we predicted 12 antibiotic resistance classes (Table 3) in the Y. ruckeri proteome, particularly for cys regulon transcriptional activator CysB, bifunctional polymyxin resistance protein ArnA, copper-sensing two-component system response regulator CpxR and isoleucine-tRNA ligase. We observed intermediate susceptibility of aminoglycoside (gentamicin, MIC = 4–8 μg/mL) and polymyxin B (MIC = 4 μg/mL) antibiotics against Y. ruckeri strains. Similar results were previously reported in French Y. ruckeri isolates with aminoglycoside (gentamicin) [49] and greatest variation (MIC = 2–512 μg/mL) in antibiotic sensitivity of polymyxin B was reported among Y. ruckeri strains [50]. These higher MIC values suggest that Y. ruckeri strains may harbor acquired or intrinsic resistance mechanisms to aminoglycosides and polymyxin B. Additionally, our Y. ruckeri strains were highly resistant to erythromycin (MIC = 1024 μg/mL) and rifampin (MIC = 32 μg/mL), consistent with observations by Calvez et al. [49] and Stock et al. [51], who found Y. ruckeri strains to be resistant to erythromycin (MIC = 32–64 μg/mL) and rifampin (MIC = 8–16 μg/mL). Erythromycin and novobiocin discs did not show inhibition zone against the Chinese Y. ruckeri strain H01 [52]. Similarly, we did not observe any inhibition zone of novobiocin and mupirocin discs against the Y. ruckeri strains examined. Inherent resistance to erythromycin and rifampin has been described for the other Yersinia species (Y. enterocolitica, Y. mollaretii and Y. aldovae) [51]. Our results support these findings and suggest that Y. ruckeri strains might also be resistant to novobiocin and mupirocin. Moreover, two efflux pumps of the RND family were identified. This family is widespread among Gram negative bacteria and, in Enterobacteriaceae such as E. coli, contributes to the intrinsic resistance against several antibiotics, including macrolide and novobiocin [53]. This is consistent with our present results that found Y. ruckeri to be resistant to both antibiotics. Finally, it is important to note that the antimicrobial agents used to validate the results of our antibiotic resistance ontology are generally not approved for use in aquaculture. Y. ruckeri strains are susceptible to commonly applied antimicrobial agents such as florfenicol and oxytetracycline to treat fish diseases [49].
The outer membrane proteins (OmpA, OmpC, OmpF and OmpW), outer membrane assembly factors (BamA, BamB, BamC, BamD and BamE), outer membrane lipoproteins (Blc, pcp, RcsF, LolB, LolD, Omp16, RcsF and YfeY), lipoproteins (NlpD, NlpE and NlpI) and lipopolysaccharide biosynthesis proteins (LptA, LptD and LptE) were identified. These proteins play an important role in pathogen-host interactions and pathogenicity [54]. Additionally, OMPs help in resisting host defense mechanisms and have been shown to confer protection in fish [54, 55]. The outer membrane assembly factor YeaT and OmpC have been shown to induce a strong immune response and protect Labeo rohita and Japanese flounder against Edwardsiella tarda infection [56, 57].
In conclusion, our study provides the first global proteomic profiles of Y. ruckeri and this work will provide a better understanding of the physiology, proteomic biology, proteomic changes, virulence mechanisms and localization of Y. ruckeri proteins. The most commonly expressed proteins such as SOD-Cu–Zn and PstS might be useful to develop a single vaccination protocol or single drug therapy for both biotype 1 and biotype 2 strains. Additionally, proteins associated with virulence and antigenicity such as Clp and Lon pro-teases, TolB, PPIases, PhoP and LuxR family transcriptional regulators may be used for the construction of novel vaccines for yersiniosis in fish. The comprehensive data set generated in this study will serve as a reference proteome for future studies such as protein–protein interaction and network analysis.
Data deposition
Shotgun proteomics data have been deposited in the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository [58] with the dataset identifier PXD005439.
Additional files
Additional file 1. Antimicrobial susceptibility of Yersinia ruckeri strains. Antibiotic susceptibility was determined using the disc diffusion technique on Mueller–Hinton agar and minimal inhibitory concentration was determined with the same antibiotics using micro dilution on microtiter plates. The diameter of the inhibition halo and lowest concentration of antibiotic that inhibited visible growth of bacteria was defined after incubation 48 h at 22 °C. Novobiocin and mupirocin discs displaced no inhibition zone against Y. ruckeri strains. Note: I = intermediate and R = resistant.
Additional file 2. Details of total identified proteins of Yersinia ruckeri. Number of proteins was identified at false discovery rate 1% with more than one peptide.
Additional file 3. Details of identified proteins of Yersinia ruckeri strains. Number of proteins was identified at false discovery rate 1% with more than one peptide.
Additional file 4. Principal component analysis of Yersinia ruckeri strains. The score plots show that strain SP-05 differs from the three strains (CSF007-82, 7959-11 and YRNC-10) but the latter three strains showed minor proteomic differences.
Additional file 5. Lists of uniquely identified proteins in each strain of Yersinia ruckeri. Forty-six proteins in SP-05, 43 proteins in CSF007-82, 31 proteins in 7959-11 and 13 proteins in YRNC-10 were uniquely identified.
Additional file 6. Fold changes of differentially down regulated proteins of Yersinia ruckeri strains compared to each other. ANOVA was performed for UniProt database searches. * Denotes statistically significant difference according to Tukey’s honest significant difference post hoc test with false discovery rate-adjusted p-value < 0.001 and fold change < −3 or > +3.
Additional file 7. Lists of virulence proteins of Yersinia ruckeri. Proteins were predicted by a method based on bi-layer cascade support vector machine using VirulentPred.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
GK and MEM conceived and designed the experiment. GK performed the experiment. KH, GK and ERF performed the LC–MS/MS. GK analyzed the data and drafted the manuscript. TJW, ERF and MEM revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This study was funded by the Austrian Science Fund (FWF) Project No. P 27489-B22 to Gokhlesh Kumar. We are thankful to Mag. Katharina Nöbauer and Dipl.-Biol. Sarah Schlosser for their technical assistance.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- ERM
enteric redmouth disease
- LC–MS
liquid chromatography–mass spectrometry
- TOF
triple quadrupole time of flight
- FDR
false discovery rate
- SWATH
sequential window acquisition of all theoretical spectra
- IDA
information dependent data acquisition
- PCA
principal component analysis
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s13567-017-0460-3) contains supplementary material, which is available to authorized users.
Contributor Information
Gokhlesh Kumar, Phone: +43 1250774730, Email: Gokhlesh.Kumar@vetmeduni.ac.at.
Karin Hummel, Email: Karin.Hummel@vetmeduni.ac.at.
Timothy J. Welch, Email: Tim.Welch@ARS.USDA.GOV
Ebrahim Razzazi-Fazeli, Email: Ebrahim.Razzazi@vetmeduni.ac.at.
Mansour El-Matbouli, Email: Mansour.El-Matbouli@vetmeduni.ac.at.
References
- 1.Horne MT, Barnes AC. Enteric redmouth disease (Yersinia ruckeri) In: Woo PTK, Bruno DW, editors. Fish diseases and disorders. Viral, bacterial and fungal infections. Wallingford: CABI Publishing; 1999. pp. 445–477. [Google Scholar]
- 2.Kumar G, Menanteau-Ledouble S, Saleh M, El-Matbouli M. Yersinia ruckeri, the causative agent of enteric redmouth disease in fish. Vet Res. 2015;46:103. doi: 10.1186/s13567-015-0238-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ohtani M, Villumsen KR, Strøm H, Raida MK. 3D Visualization of the initial Yersinia ruckeri infection route in rainbow trout (Oncorhynchus mykiss) by optical projection tomography. PLoS One. 2014;9:e89672. doi: 10.1371/journal.pone.0089672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tobback E, Decostere A, Hermans K, Ryckaert J, Duchateau L, Haesebrouck F, Chiers K. Route of entry and tissue distribution of Yersinia ruckeri in experimentally infected rainbow trout Oncorhynchus mykiss. Dis Aquat Org. 2009;84:219–228. doi: 10.3354/dao02057. [DOI] [PubMed] [Google Scholar]
- 5.Romalde JL, Toranzo AE. Pathological activities of Yersinia ruckeri, the enteric redmouth (ERM) bacterium. FEMS Microbiol Lett. 1993;112:291–300. doi: 10.1111/j.1574-6968.1993.tb06465.x. [DOI] [PubMed] [Google Scholar]
- 6.Fernández L, Lopez JR, Secades P, Menendez A, Marquez I, Guijarro JA. In vitro and in vivo studies of the Yrp1 protease from Yersinia ruckeri and its role in protective immunity against enteric red mouth disease of salmonids. Appl Environ Microbiol. 2003;69:7328–7335. doi: 10.1128/AEM.69.12.7328-7335.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davies RL. Outer membrane protein profiles of Yersinia ruckeri. Vet Microbiol. 1991;26:125–140. doi: 10.1016/0378-1135(91)90049-L. [DOI] [PubMed] [Google Scholar]
- 8.Austin DA, Robertson PAW, Austin B. Recovery of a new biogroup of Yersinia ruckeri from diseased rainbow trout (Oncorhynchus mykiss, Walbaum) Syst Appl Microbiol. 2003;26:127–131. doi: 10.1078/072320203322337416. [DOI] [PubMed] [Google Scholar]
- 9.Welch TJ, Verner-Jeffreys DW, Dalsgaard I, Wiklund T, Evenhuis JP, Cabrera JA, Hinshaw JM, Drennan JD, LaPatra SE. Independent emergence of Yersinia ruckeri biotype 2 in the United States and Europe. Appl Environ Microbiol. 2011;77:3493–3499. doi: 10.1128/AEM.02997-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fouz B, Zarza C, Amaro C. First description of non-motile Yersinia ruckeri serovar I strains causing disease in rainbow trout, Oncorhynchus mykiss (Walbaum), cultured in Spain. J Fish Dis. 2006;29:339–346. doi: 10.1111/j.1365-2761.2006.00723.x. [DOI] [PubMed] [Google Scholar]
- 11.Tinsley JW, Lyndon AR, Austin B. Antigenic and cross-protection studies of biotype 1 and biotype 2 isolates of Yersinia ruckeri in rainbow trout, Oncorhynchus mykiss (Walbaum) J Appl Microbiol. 2011;111:8–16. doi: 10.1111/j.1365-2672.2011.05020.x. [DOI] [PubMed] [Google Scholar]
- 12.Nelson MC, LaPatra SE, Welch TJ, Graf J. Complete genome sequence of Yersinia ruckeri strain CSF007-82, etiologic agent of red mouth disease in salmonid fish. Genome Announc. 2015;3:e01491–e01494. doi: 10.1128/genomeA.01491-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coquet L, Cosette P, Dé E, Galas L, Vaudry H, Rihouey C, Lerouge P, Junter GA, Jouenne T. Immobilization induces alterations in the outer membrane protein pattern of Yersinia ruckeri. J Proteome Res. 2005;4:1988–1998. doi: 10.1021/pr050165c. [DOI] [PubMed] [Google Scholar]
- 14.Bystritskaya E, Stenkova A, Chistuylin D, Chernysheva N, Khomenko V, Anastyuk S, et al. Adaptive responses of outer membrane porin balance of Yersinia ruckeri under different incubation temperature, osmolarity, and oxygen availability. Microbiologyopen. 2016;5:597–603. doi: 10.1002/mbo3.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou L, Ying W, Han Y, Chen M, Yan Y, Li L, Zhu Z, Zheng Z, Jia W, Yang R, Qian X. A proteome reference map and virulence factors analysis of Yersinia pestis 91001. J Proteom. 2012;75:894–907. doi: 10.1016/j.jprot.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 16.Kumar G, Hummel K, Ahrens M, Menanteau-Ledouble S, Welch TJ, Eisenacher M, Razzazi-Fazeli E, El-Matbouli M. Shotgun proteomic analysis of Yersinia ruckeri strains under normal and iron-limited conditions. Vet Res. 2016;47:100. doi: 10.1186/s13567-016-0384-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Evenhuis JP, Lapatra SE, Verner-Jeffreys DW, Dalsgaard I, Welch TJ. Identification of flagellar motility genes in Yersinia ruckeri by transposon mutagenesis. Appl Environ Microbiol. 2009;75:6630–6633. doi: 10.1128/AEM.01415-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Welch TJ, Wiens GD. Construction of a virulent, green fluorescent protein-tagged Yersinia ruckeri and detection in trout tissues after intraperitoneal and immersion challenge. Dis Aquat Organ. 2005;67:267–272. doi: 10.3354/dao067267. [DOI] [PubMed] [Google Scholar]
- 19.Domon B, Aebersold R. Mass spectrometry and protein analysis. Science. 2006;312:212–217. doi: 10.1126/science.1124619. [DOI] [PubMed] [Google Scholar]
- 20.McQueen P, Spicer V, Schellenberg J, Krokhin O, Sparling R, Levin D, Wilkins JA. Whole cell, label free protein quantitation with data independent acquisition: quantitation at the MS2 level. Proteomics. 2015;15:16–24. doi: 10.1002/pmic.201400188. [DOI] [PubMed] [Google Scholar]
- 21.R Core Team (2015) R: a language and environment for statistical computing. R foundation for statistical computing, Vienna. R version 3.2.2. https://www.R-project.org/. Accessed 8 Mar 2016
- 22.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- 23.Oliveros JC (2007) VENNY. An interactive tool for comparing lists with Venn Diagrams. http://bioinfogp.cnb.csic.es/tools/venny/index.html. Accessed 28 Nov 2016
- 24.Bhatia VN, Perlman DH, Costello CE, McComb ME. Software tool for researching annotations of proteins: open-source protein annotation software with data visualization. Anal Chem. 2009;81:9819–9823. doi: 10.1021/ac901335x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, Dao P, Sahinalp SC, Ester M, Foster LJ, Brinkman FS. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics. 2010;26:1608–1615. doi: 10.1093/bioinformatics/btq249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garg A, Gupta D. VirulentPred: a SVM based prediction method for virulent proteins in bacterial pathogens. BMC Bioinform. 2008;9:62. doi: 10.1186/1471-2105-9-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McArthur AG, Waglechner N, Nizam F, Yan A, Azad MA, Baylay AJ, et al. The comprehensive antibiotic resistance database. Antimicrob Agents Chemother. 2013;57:3348–3357. doi: 10.1128/AAC.00419-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.CLSI (2006) Methods for antimicrobial disk susceptibility testing of bacteria isolated from aquatic animals; approved guideline. CLSI document VET03-A. Clinical and Laboratory Standards Institute, Wayne, PA, USA. https://clsi.org/standards/products/packages/vet03pk/. Accessed 23 Mar 2017
- 29.CLSI (2006) Methods for broth dilution susceptibility testing of bacteria isolated from aquatic animals; approved guideline. CLSI document VET04-A. Clinical and Laboratory Standards Institute, Wayne, PA, USA. https://clsi.org/standards/products/veterinary-medicine/documents/vet04/. Accessed 23 Mar 2017
- 30.Jacobsen SM, Lane MC, Harro JM, Shirtliff M, Mobley HL. The high-affinity phosphate transporter Pst is a virulence factor for Proteus mirabilis during complicated urinary tract infection. FEMS Immunol Med Microbiol. 2008;52:180–193. doi: 10.1111/j.1574-695X.2007.00358.x. [DOI] [PubMed] [Google Scholar]
- 31.Fang FC, DeGroote MA, Foster JW, Bäumler AJ, Ochsner U, Testerman T, Bearson S, Giárd JC, Xu Y, Campbell G, Laessig T. Virulent Salmonella typhimurium has two periplasmic Cu, Zn-superoxide dismutases. Proc Natl Acad Sci U S A. 1999;96:7502–7507. doi: 10.1073/pnas.96.13.7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu B, Yang X, Zhang P, Ma Z, Lin H, Fan H. The arginine deiminase system facilitates environmental adaptability of Streptococcus equi ssp. zooepidemicus through pH adjustment. Res Microbiol. 2016;167:403–412. doi: 10.1016/j.resmic.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 33.Frees D, Brøndsted L, Ingmer H. Bacterial proteases and virulence. Subcell Biochem. 2013;66:161–192. doi: 10.1007/978-94-007-5940-4_7. [DOI] [PubMed] [Google Scholar]
- 34.Fernández L, Prieto M, Guijarro JA. The iron- and temperature-regulated haemolysin YhlA is a virulence factor of Yersinia ruckeri. Microbiology. 2007;153:483–489. doi: 10.1099/mic.0.29284-0. [DOI] [PubMed] [Google Scholar]
- 35.Zhou D, Galán J. Salmonella entry into host cells: the work in concert of type III secreted effector proteins. Microbes Infect. 2001;3:1293–1298. doi: 10.1016/S1286-4579(01)01489-7. [DOI] [PubMed] [Google Scholar]
- 36.He Y, Xu T, Fossheim LE, Zhang XH. FliC, a flagellin protein, is essential for the growth and virulence of fish pathogen Edwardsiella tarda. PLoS One. 2012;7:e45070. doi: 10.1371/journal.pone.0045070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dahiya I, Stevenson RMW. The UvrY response regulator of the BarA-UvrY two-component system contributes to Yersinia ruckeri infection of rainbow trout (Oncorhynchus mykiss) Arch Microbiol. 2010;192:541–547. doi: 10.1007/s00203-010-0582-8. [DOI] [PubMed] [Google Scholar]
- 38.Ünal CM, Steinert M. Microbial Peptidyl-Prolyl cis/trans Isomerases (PPIases): virulence factors and potential alternative drug targets. Microbiol Mol Biol Rev. 2014;78:544–571. doi: 10.1128/MMBR.00015-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Groisman EA, Chiao E, Lipps CJ, Heffron F. Salmonella typhimurium phoP virulence gene is a transcriptional regulator. Proc Natl Acad Sci U S A. 1989;86:7077–7081. doi: 10.1073/pnas.86.18.7077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lo Sciuto A, Fernández-Piñar R, Bertuccini L, Iosi F, Superti F, Imperi F. The periplasmic protein TolB as a potential drug target in Pseudomonas aeruginosa. PLoS One. 2014;9:e103784. doi: 10.1371/journal.pone.0103784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alonso-Hearn M, Eckstein TM, Sommer S, Bermudez LE. A Mycobacterium avium subsp. paratuberculosis LuxR regulates cell envelope and virulence. Innate Immun. 2010;16:235–247. doi: 10.1177/1753425909339811. [DOI] [PubMed] [Google Scholar]
- 42.Neckers L, Tatu U. Molecular chaperones in pathogen virulence: emerging new targets for therapy. Cell Host Microbe. 2008;4:519–527. doi: 10.1016/j.chom.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takaya A, Tomoyasu T, Matsui H, Yamamoto T. The Dnak/Dnaj chaperone machinery of Salmonella enterica serovar typhimurium is essential for invasion of epithelial cells and survival within macrophages, leading to systemic infection. Infect Immun. 2004;72:1364–1373. doi: 10.1128/IAI.72.3.1364-1373.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lund PA. Microbial molecular chaperones. Adv Microb Physiol. 2001;44:93–140. doi: 10.1016/S0065-2911(01)44012-4. [DOI] [PubMed] [Google Scholar]
- 45.Meibom KL, Dubail I, Dupuis M, Barel M, Lenco J, Stulik J, Golovliov I, Sjöstedt A, Charbit A. The heat-shock protein ClpB of Francisella tularensis is involved in stress tolerance and is required for multiplication in target organs of infected mice. Mol Microbiol. 2008;67:1384–1401. doi: 10.1111/j.1365-2958.2008.06139.x. [DOI] [PubMed] [Google Scholar]
- 46.LaFrentz BR, LaPatra SE, Call DR, Wiens GD, Cain KD. Proteomic analysis of Flavobacterium psychrophilum cultured in vivo and in iron-limited media. Dis Aquat Organ. 2009;87:171–182. doi: 10.3354/dao02122. [DOI] [PubMed] [Google Scholar]
- 47.Nisa S, Blokpoel MC, Robertson BD, Tyndall JD, Lun S, Bishai WR, O’Toole R. Targeting the chromosome partitioning protein ParA in tuberculosis drug discovery. J Antimicrob Chemother. 2010;65:2347–2358. doi: 10.1093/jac/dkq311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hong W, Zeng J, Xie J. Antibiotic drugs targeting bacterial RNAs. Acta Pharm Sin B. 2014;4:258–265. doi: 10.1016/j.apsb.2014.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Calvez S, Gantelet H, Blanc G, Douet DG, Daniel P. Yersinia ruckeri biotypes 1 and 2 in France: presence and antibiotic susceptibility. Dis Aquat Organ. 2014;109:117–126. doi: 10.3354/dao02725. [DOI] [PubMed] [Google Scholar]
- 50.De Grandis SA, Stevenson RM. Antimicrobial susceptibility patterns and R plasmid-mediated resistance of the fish pathogen Yersinia ruckeri. Antimicrob Agents Chemother. 1985;27:938–942. doi: 10.1128/AAC.27.6.938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stock I, Henrichfreise B, Wiedemann B. Natural antibiotic susceptibility and biochemical profiles of Yersinia enterocolitica-like strains: Y. bercovieri, Y. mollaretii, Y. aldovae and “Y. ruckeri”. J Med Microbiol. 2002;51:56–69. doi: 10.1099/0022-1317-51-1-56. [DOI] [PubMed] [Google Scholar]
- 52.Shaowu L, Di W, Hongbai L, Tongyan L. Isolation of Yersinia ruckeri strain H01 from farm-raised amur sturgeon Acipenser schrencki in China. J Aquat Anim Health. 2013;25:9–14. doi: 10.1080/08997659.2012.728169. [DOI] [PubMed] [Google Scholar]
- 53.Nikaido H, Takatsuka Y. Mechanisms of RND multidrug efflux pumps. Biochim Biophys Acta. 2009;1794:769–781. doi: 10.1016/j.bbapap.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seltman G, Holst O. The bacterial cell wall. Berlin: Springer; 2002. pp. 9–40. [Google Scholar]
- 55.Kumar G, Rathore G, Sengupta U, Singh V, Kapoor D, Lakra WS. Isolation and characterization of outer membrane proteins of Edwardsiella tarda and its application in immunoassays. Aquaculture. 2007;272:98–104. doi: 10.1016/j.aquaculture.2007.08.054. [DOI] [Google Scholar]
- 56.Kumar G, Rathore G, El-Matbouli M. Outer membrane protein assembly factor YaeT (omp85) and GroEL proteins of Edwardsiella tarda are immunogenic antigens for Labeo rohita (Hamilton) J Fish Dis. 2014;37:1055–1059. doi: 10.1111/jfd.12205. [DOI] [PubMed] [Google Scholar]
- 57.Fuguo L, Xiaoqian T, Xiuzhen S, Jing X, Wenbin Z. Edwardsiella tarda outer membrane protein C: an immunogenic protein induces highly protective effects in Flounder (Paralichthys olivaceus) against Edwardsiellosis. Int J Mol Sci. 2016;17:1117. doi: 10.3390/ijms17071117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vizcaíno JA, Csordas A, del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, Xu QW, Wang R, Hermjakob H. 2016 update of the PRIDE database and related tools. Nucleic Acids Res. 2016;44:D447–D456. doi: 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Antimicrobial susceptibility of Yersinia ruckeri strains. Antibiotic susceptibility was determined using the disc diffusion technique on Mueller–Hinton agar and minimal inhibitory concentration was determined with the same antibiotics using micro dilution on microtiter plates. The diameter of the inhibition halo and lowest concentration of antibiotic that inhibited visible growth of bacteria was defined after incubation 48 h at 22 °C. Novobiocin and mupirocin discs displaced no inhibition zone against Y. ruckeri strains. Note: I = intermediate and R = resistant.
Additional file 2. Details of total identified proteins of Yersinia ruckeri. Number of proteins was identified at false discovery rate 1% with more than one peptide.
Additional file 3. Details of identified proteins of Yersinia ruckeri strains. Number of proteins was identified at false discovery rate 1% with more than one peptide.
Additional file 4. Principal component analysis of Yersinia ruckeri strains. The score plots show that strain SP-05 differs from the three strains (CSF007-82, 7959-11 and YRNC-10) but the latter three strains showed minor proteomic differences.
Additional file 5. Lists of uniquely identified proteins in each strain of Yersinia ruckeri. Forty-six proteins in SP-05, 43 proteins in CSF007-82, 31 proteins in 7959-11 and 13 proteins in YRNC-10 were uniquely identified.
Additional file 6. Fold changes of differentially down regulated proteins of Yersinia ruckeri strains compared to each other. ANOVA was performed for UniProt database searches. * Denotes statistically significant difference according to Tukey’s honest significant difference post hoc test with false discovery rate-adjusted p-value < 0.001 and fold change < −3 or > +3.
Additional file 7. Lists of virulence proteins of Yersinia ruckeri. Proteins were predicted by a method based on bi-layer cascade support vector machine using VirulentPred.