

Abstract
There is a long standing perception that AhR ligands are automatically disqualified from pharmaceutical development due to their induction of Cyp1a1 as well as their potential for causing “dioxin-like” toxicities. However, recent discoveries of new AhR ligands with potential therapeutic applications have been reported, inviting reconsideration of this policy. One area of exploration is focused on the activation of AhR to promote the generation of regulatory T cells, which control the intensity and duration of immune responses. Rapidly metabolized AhR ligands (RMAhRLs), which do not bioaccumulate in the same manner as 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) have been discovered that induce Tregs and display impressive therapeutic efficacy in a broad range of preclinical models of immune-mediated diseases. Given the promise of these RMAhRLs, is the bias against AhR activators still valid? Can RMAhRLs be given chronically to maintain therapeutic levels of AhR activation without producing the same toxicity profile as dioxin-like compounds? Based on our review of the data, there is little evidence to support the indiscriminate exclusion of AhR activators/Cyp1a1 inducers from early drug developmental pipelines. We also found no evidence that short-term treatment with RMAhRLs produce “dioxin-like toxicity” and, in fact, were well tolerated. However, safety testing of individual RMAhRLs under therapeutic conditions, as performed with all promising new drugs, will be needed to reveal whether or not chronic activation of AhR leads to unacceptable adverse outcomes.
Keywords: aryl hydrocarbon receptor, AhR ligands, immune-mediated diseases, Tregs, immunotherapy
Graphical Abstract
1. Introduction and Background
1.1 Immune-mediated diseases and treatment options
Immune-mediated diseases (IMD) encompass a broad range of chronic and debilitating inflammatory conditions that affect upwards of 75 million people in the United States alone. The underlying common etiology of IMD is an aberrant or inappropriate activation of the immune system which can attack any organ system in the body, causing significant morbidity and mortality. Some of the most prevalent IMD include the autoimmune diseases, type 1 diabetes (T1D) and multiple sclerosis, graft-versus-host disease following bone marrow transplantation, as well as allergic hypersensitivity responses. Life-long therapy is often required to combat the underlying immune activation. Current treatments include non-specific immunosuppressive drugs (e.g., corticosteroids, methotrexate) and newer, targeted therapies that neutralize critical immune mediators (e.g., antibodies to tumor necrosis factor-α, interleukin-6). However, adverse effects of immunosuppressive treatments and the high cost of monoclonal antibodies support the need for the development of more affordable, effective and well-tolerated drugs.
Induction of immune suppression by regulatory T cells (Tregs) is a promising new approach for the treatment of immune-mediated diseases [1]. Tregs comprise a heterogeneous subset of CD4+ T cells, which act through a variety of mechanisms to control the generation and/or function of activated immune cells. The loss of Tregs or impairment in their function is associated with the development and progression of IMD; conversely, the restoration of immune tolerance by Treg expansion has been shown to effectively control immunopathology. The efficacy of treating autoimmune diseases by the infusion of in vitro-generated Tregs or ex vivo-expanded autologous Tregs is currently being tested in clinical trials. However, these approaches are constrained by technical challenges related to personalized Treg generation and the associated high costs. To circumvent the challenges associated with Treg transfusion, direct expansion or activation of Tregs in vivo by targeting specific pathways that regulate Treg development (e.g., low dose IL-2, rapamycin) represent alternative approaches to treatment of IMD.
1.2 AhR-mediated induction of Tregs by TCDD
The aryl hydrocarbon receptor (AhR), is a ligand-activated transcription factor that regulates many aspects of immunological function, but is most recognized for its role in suppression of adaptive immune responses. Based primarily on studies with TCDD, the prototypic AhR ligand, immune suppression results from activation of AhR in dendritic cells (DCs) [2,3] and T cells [4,5] during early stages of the response to antigen. AhR activation directly alters gene expression and subsequent T cell differentiation leading to the AhR-dependent generation of Tregs [6,7]. AhR activation in DCs induces a tolerogenic phenotype that promotes the differentiation of Foxp3+ Tregs [8,9]. In addition, AhR activation in CD4+ T cells induces CD25+CTLA4+ IL-10 producing Tr1-like Tregs [4,6,9]. These AhR-dependent Tregs suppress the generation of T helper cell-dependent (Th1, Th2, Th17) immune responses. The induction of AhRTregs by TCDD has been shown to be effective in preventing the development of several different IMD, including murine models of T1D [10], multiple sclerosis [8], autoimmune uveitis [11], inflammatory bowel disease [12–14], as well as several models of transplant tolerance [5,15] and allergic diseases [16–18]. Given the striking efficacy of TCDD to suppress IMD, AhR has become a promising target for therapeutic development. Unfortunately, TCDD itself is not pharmacologically suitable for human use given its long half-life and associated toxicities.
1.3 Rapidly metabolized AhR ligands (RMAhRLs) mimic TCDD for the treatment of
IMD In addition to TCDD, AhR can be activated by many aromatic compounds (e.g., indoles, imidazoles, polyphenols) with different pharmacokinetic properties. Several RMAhRLs have been discovered that mimic the Treg induction and corresponding therapeutic effects of TCDD in different IMD models (Table I). However, to achieve sufficient AhR activation to induce the therapeutic response, these RMAhRL need to be administered at higher doses (mg/kg versus μg/kg) and more frequently than TCDD. When AhR is activated by RMAhRLs to the same extent as TCDD, differential gene expression is highly concordant with that of TCDD [19,20] suggesting that these compounds alter similar downstream signaling pathways following AhR activation.
Table I.
List of diseases treated with RMAhRL in preclinical mouse models
| Disease | Ligand | Dose | Route | Frequency | Foxp3+ Tregs | Foxp3− Tregs | Tolerogenic DCs | Reference | |
|---|---|---|---|---|---|---|---|---|---|
| Allograft | Parent-into-F1 GVHD | Cl-BBQ | 10mg/kg | i.p. | daily | + | [20] | ||
| Islet Transplant | VAG539 | 30mg/kg | oral | daily | + | + | [9] | ||
|
| |||||||||
| Autoimmune | Type 1 Diabetes | Cl-BBQ | 60mg/kg | oral | 3x/week | + | [35] | ||
| EAE | I3C | 40mg/kg | i.p. | daily | + | [47] | |||
| DIM | 40mg/kg | i.p. | daily | + | [47] | ||||
| FICZ | 10mg/kg* | i.p. | 1x | [48] | |||||
| Laquinimod | 5mg/kg | oral | daily | [49] | |||||
| Laquinimod | 25mg/kg | oral | daily | + | [50] | ||||
| ITE | 10mg/kg* | i.p. or oral | daily | + | + | [51] | |||
| EAU | ITE | 10mg/kg* | i.p. | daily | + | [52] | |||
| Psoriasis | FICZ | 100ug/kg | i.p. | daily | [53] | ||||
| Colitis | ITE | 10mg/kg* | i.p. | daily | + | + | [54] | ||
| DIM | 50mg/kg | i.p. | daily | + | [55] | ||||
| βNF | 50mg/kg* | oral | daily | [56] | |||||
|
| |||||||||
| Allergy | Asthma Atopic | FICZ | 30μg/kg | i.p. | 1x | [57] | |||
| Dermatitis | P. cocos | 25mg/kg | oral | daily | + | [58] | |||
| Food Allergy | P. cocos | 25mg/kg | oral | daily | + | [58] | |||
estimated based on 20g mouse
Abbreviations used: GVH, graft-versus-host disease; EAE, experimental autoimmune encephalomyelitis; EAU, experimental autoimmune uveitis; Cl-BBQ, chloro-7H-benzimidazo[2,1-a]benzo[de]Iso-quinolin-7-one; VAG539, [4-(3-chloro-phenyl)-pyrimidin-2-yl]-(4-trifluoromethyl-phenyl)-carbamic acid 2-[(2-hydroxy-ethyl)-methyl-amino]-ethyl ester; I3C, indole-3-carbinol; DIM, 3,3′-diindolylmethane; FICZ, 5,11-Dihydroindolo[3,2-b]carbazole-6-carboxaldehyde; ITE, 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester; βNF, beta-naphthoflavone; P. cocos, Poria cocos bark extract
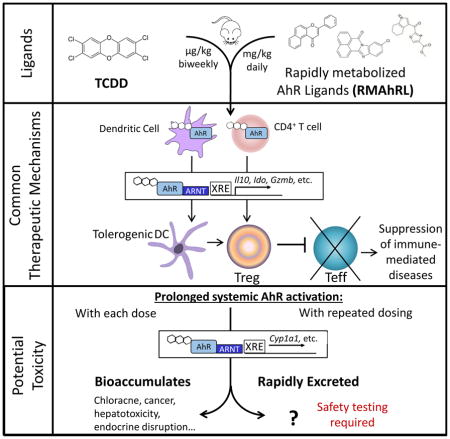
Going forward, the question arises: since RMAhRLs share the same therapeutic mechanism as TCDD, do they also share the same toxicity profile? (Figure 1).
Figure 1.

TCDD and RMAhRL share the same therapeutic mechanism to suppress immune-mediated diseases. Further testing is needed to determine if RMAhRL will exhibit dioxin-like toxicities following prolonged systemic AhR activation, which may be required for therapeutic efficacy.
2. TCDD drives the unique safety issues associated with AhR ligands
2.1. What is “dioxin-like” toxicity?
“Dioxin” is a commonly used term for 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) which gained fame as the toxic component of Agent Orange used in the Vietnam War. TCDD represents the most toxic member of a large class of environmental contaminants that include 75 polychlorinated dibenzo-p-dioxins (PCDD), as well as structurally similar polychlorinated dibenzofurans and polychlorinated biphenyls. Structure-activity relationships (SAR) for individual congeners within these classes of compounds are defined by the position of the chlorine molecules and predict the ability of the congener to bind and activate the AhR, to induce Cyp1a1, and to resist oxidative metabolism and excretion resulting in bioaccumulation [21]. The same SARs predict an in vivo pattern of toxicity that has come to be known as “dioxin-like” toxicity.
The characteristics of dioxin-like toxicity range from a lethal wasting syndrome, hepatotoxicity and thymic involution at the high end of the dose response curve to alterations in immune and endocrine system homeostasis at low doses. Carcinogenicity has also been associated with low but chronic exposure to dioxin-like compounds [22]. Although TCDD is classified as a human carcinogen by WHO and USEPA, the epidemiological evidence causally linking TCDD exposure to increased cancer risk in humans is weak and inconsistent between exposure cohorts [23]. Furthermore, most epidemiologic studies on the health effects of TCDD are complicated by exposure to low levels of dioxin within a complex mixture of other potentially carcinogenic chemicals. However, even in the Seveso cohort exposed to high levels of TCDD based on chloracne development (n=183), no cases of cancer were identified twenty years later [24]. Mechanistically, TCDD is not genotoxic nor mutagenic. However, carcinogenicity testing of TCDD in rodents has shown an overall increased incidence of certain cancers (e.g., liver, lung, skin), as well as decreased incidence of others. A reduced incidence of tumors in the pituitary, mammary gland, uterus and pancreas is likely related to the altered endocrine status of TCDD-treated animals. The potential for TCDD to cause developmental toxicity has been recognized for many years and is considered to be the most sensitive endpoint of TCDD exposure in laboratory animals [25]. In mice, the specific teratogenic response is hydronephrosis and cleft palate. The likelihood of developmental toxicity in humans from exposure to dioxin-like compounds is less clear, although changes in sex ratio, and other birth defects have been associated with populations exposed to dioxin-like compounds.
2.2 Cancer risk due to CYP1 induction
The expression of the cytochrome P450 enzymes, CYP1A1, CYP1A2 and CYP1B1, are directly regulated by AhR and catalyze the bioactivation of polycyclic aromatic hydrocarbons (PAHs) and other aromatic compounds (e.g., estrogens). PAHs are ubiquitous compounds found in cigarette smoke, charred and smoked food, and diesel exhaust particles and their oxygenation by CYP1 enzymes results in the formation of epoxides and other reactive species that form DNA adducts. The potential increased risk of cancer from the formation of these DNA adducts forms the basis of the long-standing paradigm that coexposure to PAHs and Cyp1a1 inducers (e.g., TCDD) increases carcinogenesis [26,27]. At the same time, induction of CYP is also critical for the detoxification and metabolic clearance of PAHs, which reduces the probability of bioactivation and DNA adduct formation. For example, carcinogenicity and DNA adduct formation following benzo-a-pyrene exposure were greatly increased in the absence of CYP1A1 [28]. Likewise, topical application of coal tar, a complex mixture of PAHs, used for the treatment of atopic dermatitis and psoriasis is not associated with an increased long-term risk of skin cancer or other malignancies despite the systemic induction of Cyp1a1 [29]. These results suggest that AhR activation in conjunction with PAH exposure may be less hazardous than originally thought. Nonetheless, minimizing exposure to PAHs (e.g. quitting smoking, limiting intake of charcoal-grilled food, etc.) would reduce any potential risk associated with RMAhRL therapy.
2.3. What is known about RMAhRL Toxicity?
The association of “dioxin-like toxicity” as well as carcinogen bioactivation with AhR activation cast a pall over the development of AhR ligands as potential therapeutic agents for many years. However, as the unique mechanisms of action of TCDD on the immune system were discovered, perceptions of AhR as a therapeutic target began to change. With TCDD as the prototype for AhR-Treg induction, several potential therapeutic RMAhRLs were identified. Like TCDD, these RMAhRLs are lipophilic and bind the AhR with high affinity, but unlike TCDD are rapidly metabolized, resulting in short half-lives in the body, with presumably no bioaccumulation. Some RMAhRLs are endogenous compounds or are commonly found in the diet (e.g., ITE, FICZ, I3C), with the implicit perception that they are therefore non-toxic and safe to use. Publications often mention the “non-toxic” status of different RMAhRLs, however, apart from 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE) and 11-chloro-7H-benzimidazo[ 2,1-a]benzo[de]Iso-quinolin-7-one (Cl-BBQ), very little chronic toxicity data have been published.
ITE was first identified from porcine lung tissue following a search for endogenous AhR ligands [30,31]. Subsequently toxicity studies have been performed in rodents and in cell culture. In developmental toxicity studies, ITE (5.6 mg/kg) was administered to dams on gestation days 10–12, or TCDD (24 μg/kg on gestation day 10) as a positive control. TCDD, but not ITE, caused cleft palate and hydronephrosis in the fetuses [31]. Similarly, pregnant rats were treated with TCDD (1.6 or 8 μg/kg) or ITE (1.6 or 8 mg/kg) on gestation day 15. Intrauterine fetal death and placental vascular remodeling only occurred at the 8 μg/kg dose of TCDD [32]. Thymic atrophy, another endpoint of TCDD toxicity, did not occur following treatment of mice on postnatal day 35 with ITE (5.6 mg/kg), although, there was decreased thymic cellularity [31]. In other studies, ITE was assessed for chloracnegenic potential using a human epidermal equivalent model. TCDD but not ITE decreased epidermal cell layer thickness and compaction of the stratum corneum, consistent with a chloracne-like phenotype [33]. Thus, dioxin-like toxicities were not observed with ITE treatment, however, these studies did not utilize chronic activation of AhR as occurs with TCDD.
Toxicity following chronic exposure to a relatively high dose of the RMAhRL, beta-naphthoflavone (BNF, 150mg/kg followed by 500ppm in the diet for 10 weeks) was evaluated in mice in relation to iron loading [34]. In the absence of iron, no adverse effects were observed in BNF treated mice.
Another RMAhRL with some chronic toxicity data is Cl-BBQ, which was discovered through a small molecule screen designed to identify AhR ligands that acted similarly to TCDD in regard to Treg induction, but displayed more favorable pharmacokinetic properties [20]. In a study to assess the therapeutic potential of Cl-BBQ in preventing type 1 diabetes, mice were treated for 13 weeks by gavage with Cl-BBQ (60mg/kg, 3x/week), or TCDD (30μg/kg, every other week) as a positive control [35]. Standard clinical chemistry analysis and liver histopathology following chronic treatment did not reveal any abnormalities in renal or hepatic function. In the same set of studies, TCDD treated mice also had normal serum chemistry and pathology results (unpublished data), suggesting these are not sensitive endpoints for assessing dioxin-like toxicities of RMAhRLs.
Taken together, while these results do not raise red flags, they do highlight the limited amount of data available that can be used to predict the likelihood of RMAhRLs having dioxin-like toxicities following chronic treatment. Adverse outcomes associated with TCDD such as endocrine disruption and cancer, remain to be assessed with RMAhRLs using appropriate long-term therapeutic dosing regimens. Furthermore, species-specific differences in AhR and CYP expression need to be taken into account when designing safety studies.
3. Evidence that AhR ligands are not inherently risky
Despite hesitations regarding the therapeutic use of AhR ligands, many currently marketed FDA-approved drugs activate the AhR. The therapeutic mechanism of action of these compounds, however, is not necessarily through AhR activation, and it wasn’t until after FDA approval, and corresponding safety testing, did their status as AhR ligands become apparent. In 2007, Hu et al., screened 596 pharmaceutically active drugs for their ability to induce Cyp1a1 [36]. Of the 239 compounds that led to Cyp1a1 induction, 158 were drugs approved by the FDA, and six of these were shown to be true AhR ligands. These findings alone call into question the concern over the safety of compounds based solely on their induction of Cyp1a1. The six AhR ligands identified in the screen were flutamide (an androgen receptor antagonist used to treat prostate cancer), leflunomide (a pyrimidine synthesis inhibitor used for the treatment of rheumatoid arthritis), nimodipine (a calcium channel blocker to treat subarachnoid hemorrhage), omeprazole (a proton pump inhibitor commonly used to treat gastroesophageal reflux disease), and sulindac (a non-steroidal anti-inflammatory drug). These drugs were shown to activate the AhR based on a battery of in vitro and in vivo testing including ability to induce downstream AhR genes (Cyp1a1, Cyp1a2, Ugt1a1 and Nqo1), the ability to transform AhR into a DNA-binding complex, induce DRE-driven reporter gene expression, and bind to rat AhR. Later, raloxifene, a selective estrogen receptor modulator used to prevent osteoporosis, was added to the list of FDA approved AhR ligands [37].
Each of these drugs has diverse therapeutic indications, different therapeutic mechanisms of action, and different binding affinities for AhR. Although AhR binding affinities are generally low in comparison to TCDD, high therapeutic doses and chronic treatment with these drugs are likely to significantly activate AhR. Yet their long-term use is not associated with dioxin-like toxicities.
Additionally, and in contrast to concerns about carcinogenicity, several of these drugs have been shown to suppress the growth, invasion, and/or metastases of experimental cancers in an AhR dependent manner [37–41]. Taken together, these findings provide further evidence that TCDD should not set the precedent for the selective discrimination against the development of RMAhRLs.
4. Final Remarks
Toxicity is often just high-dose pharmacology, the difference being whether or not the outcome is desirable or undesirable. The goal with RMAhRLs is to take advantage of the desirable immunosuppression (immunotoxicity with TCDD) caused by AhR activation, while limiting the toxicities associated with bioaccumulation. There is, however, outstanding concern regarding the expression of AhR in many cell types and tissues [42]. Targeted delivery of RMAhRLs, through nanoparticle or liposome formulations, may further improve the safety profile of therapeutic AhR activation by directing the compounds to CD4+ T cells and/or DCs, bypassing off target effects in other tissue types [43,44].
In this review, we did not go into depth regarding increased cancer and infection risk stemming from immunosuppression, as this is not a unique concern with AhR ligands but rather with immunosuppressive drugs in general. However, there are several unique characteristics of AhR-induced immune suppression that may improve the safety profile in comparison to global immunosuppressants. First, memory responses are less sensitive to immune suppression by AhR activation than primary immune responses [45, 46], suggesting RMAhRL therapy will not interfere with established immunity. Second, the AhR needs to be activated in order to suppress the generation of pathogenic immune responses [10]. Depending on the IMD and the frequency of autoreactive T cell generation, intermittent dosing may be sufficient for effective disease control. Thus, because of the short half-life of RMAhRLs, treatment could be temporarily withdrawn during new infections or if vaccinations are needed to allow for the generation of protective immune responses.
Taken together, while there may be some remaining concerns about risks associated with RMAhRL therapy, safety testing, as performed with all promising new drugs, is the only way to address these concerns.
Highlights.
AhR ligands promote Treg differentiation and suppress immune-mediated diseases in mice
TCDD toxicity drives concerns regarding the safety of AhR ligands for therapy
Dioxin-like toxicity has not been associated with rapidly metabolized AhR ligands
Safety testing is needed to determine toxicity associated with chronic AhR activation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
* of special interest
- 1*.Singer BD, King LS, D’Alessio FR. Regulatory T cells as immunotherapy. Front Immunol. 2014;5:46. doi: 10.3389/fimmu.2014.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vorderstrasse BA, Kerkvliet NI. 2,3,7,8-Tetrachlorodibenzo-p-dioxin affects the number and function of murine splenic dendritic cells and their expression of accessory molecules. Toxicol Appl Pharmacol. 2001;171:117–125. doi: 10.1006/taap.2000.9119. [DOI] [PubMed] [Google Scholar]
- 3.Bankoti J, Rase B, Simones T, Shepherd DM. Functional and phenotypic effects of AhR activation in inflammatory dendritic cells. Toxicol Appl Pharmacol. 2010;246:18–28. doi: 10.1016/j.taap.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4*.Funatake CJ, Marshall NB, Steppan LB, Mourich DV, Kerkvliet NI. Cutting edge: activation of the aryl hydrocarbon receptor by 2,3,7,8-tetrachlorodibenzo-p-dioxin generates a population of CD4+ CD25+ cells with characteristics of regulatory T cells. J Immunol. 2005;175:4184–4188. doi: 10.4049/jimmunol.175.7.4184. [DOI] [PubMed] [Google Scholar]
- 5.Kerkvliet NI, Shepherd DM, Baecher-Steppan L. T lymphocytes are direct, aryl hydrocarbon receptor (AhR)-dependent targets of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD): AhR expression in both CD4+ and CD8+ T cells is necessary for full suppression of a cytotoxic T lymphocyte response by TCDD. Toxicol Appl Pharmacol. 2002;185:146–152. doi: 10.1006/taap.2002.9537. [DOI] [PubMed] [Google Scholar]
- 6.Marshall NB, Vorachek WR, Steppan LB, Mourich DV, Kerkvliet NI. Functional characterization and gene expression analysis of CD4+ CD25+ regulatory T cells generated in mice treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin. J Immunol. 2008;181:2382–2391. doi: 10.4049/jimmunol.181.4.2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rohlman D, Pham D, Yu Z, Steppan LB, Kerkvliet NI. Aryl Hydrocarbon Receptor-Mediated Perturbations in Gene Expression during Early Stages of CD4(+) T-cell Differentiation. Front Immunol. 2012;3:223. doi: 10.3389/fimmu.2012.00223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8*.Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453:65–71. doi: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- 9*.Hauben E, Gregori S, Draghici E, Migliavacca B, Olivieri S, Woisetschlager M, Roncarolo MG. Activation of the aryl hydrocarbon receptor promotes allograft-specific tolerance through direct and dendritic cell-mediated effects on regulatory T cells. Blood. 2008;112:1214–1222. doi: 10.1182/blood-2007-08-109843. [DOI] [PubMed] [Google Scholar]
- 10.Kerkvliet NI, Steppan LB, Vorachek W, Oda S, Farrer D, Wong CP, Pham D, Mourich DV. Activation of aryl hydrocarbon receptor by TCDD prevents diabetes in NOD mice and increases Foxp3+ T cells in pancreatic lymph nodes. Immunotherapy. 2009;1:539–547. doi: 10.2217/imt.09.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang L, Ma J, Takeuchi M, Usui Y, Hattori T, Okunuki Y, Yamakawa N, Kezuka T, Kuroda M, Goto H. Suppression of experimental autoimmune uveoretinitis by inducing differentiation of regulatory T cells via activation of aryl hydrocarbon receptor. Invest Ophthalmol Vis Sci. 2010;51:2109–2117. doi: 10.1167/iovs.09-3993. [DOI] [PubMed] [Google Scholar]
- 12.Takamura T, Harama D, Matsuoka S, Shimokawa N, Nakamura Y, Okumura K, Ogawa H, Kitamura M, Nakao A. Activation of the aryl hydrocarbon receptor pathway may ameliorate dextran sodium sulfate-induced colitis in mice. Immunol Cell Biol. 2010;88:685–689. doi: 10.1038/icb.2010.35. [DOI] [PubMed] [Google Scholar]
- 13.Benson JM, Shepherd DM. Aryl hydrocarbon receptor activation by TCDD reduces inflammation associated with Crohn’s disease. Toxicol Sci. 2011;120:68–78. doi: 10.1093/toxsci/kfq360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh NP, Singh UP, Singh B, Price RL, Nagarkatti M, Nagarkatti PS. Activation of aryl hydrocarbon receptor (AhR) leads to reciprocal epigenetic regulation of FoxP3 and IL-17 expression and amelioration of experimental colitis. PLoS One. 2011;6:e23522. doi: 10.1371/journal.pone.0023522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pauly SK, Fechner JH, Zhang X, Torrealba J, Bradfield CA, Mezrich JD. The Aryl Hydrocarbon Receptor Influences Transplant Outcomes in Response to Environmental Signals. Toxicol Environ Chem. 2012;94:1175–1187. doi: 10.1080/02772248.2012.688546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schulz VJ, Smit JJ, Willemsen KJ, Fiechter D, Hassing I, Bleumink R, Boon L, van den Berg M, van Duursen MB, Pieters RH. Activation of the aryl hydrocarbon receptor suppresses sensitization in a mouse peanut allergy model. Toxicol Sci. 2011;123:491–500. doi: 10.1093/toxsci/kfr175. [DOI] [PubMed] [Google Scholar]
- 17.Li XM, Peng J, Gu W, Guo XJ. TCDD-Induced Activation of Aryl Hydrocarbon Receptor Inhibits Th17 Polarization and Regulates Non-Eosinophilic Airway Inflammation in Asthma. PLoS One. 2016;11:e0150551. doi: 10.1371/journal.pone.0150551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luebke RW, Copeland CB, Daniels M, Lambert AL, Gilmour MI. Suppression of allergic immune responses to house dust mite (HDM) in rats exposed to 2,3,7,8-TCDD. Toxicol Sci. 2001;62:71–79. doi: 10.1093/toxsci/62.1.71. [DOI] [PubMed] [Google Scholar]
- 19.Henry EC, Welle SL, Gasiewicz TA. TCDD and a putative endogenous AhR ligand, ITE, elicit the same immediate changes in gene expression in mouse lung fibroblasts. Toxicol Sci. 2010;114:90–100. doi: 10.1093/toxsci/kfp285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20*.Punj S, Kopparapu P, Jang HS, Phillips JL, Pennington J, Rohlman D, O’Donnell E, Iversen PL, Kolluri SK, Kerkvliet NI. Benzimidazoisoquinolines: a new class of rapidly metabolized aryl hydrocarbon receptor (AhR) ligands that induce AhR-dependent Tregs and prevent murine graft-versus-host disease. PLoS One. 2014;9:e88726. doi: 10.1371/journal.pone.0088726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tuomisto J. The toxic equivalency principle and its application in dioxin risk assessment. In: Pohjanvirta R, editor. The AH Receptor in Biology and Toxicology. John Wiley & Sons, Inc; 2012. pp. 317–330. [Google Scholar]
- 22.Knerr S, Schrenk D. Carcinogenicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in experimental models. Mol Nutr Food Res. 2006;50:897–907. doi: 10.1002/mnfr.200600006. [DOI] [PubMed] [Google Scholar]
- 23.Boffetta P, Mund KA, Adami H-O, Cole P, Mandel JS. TCDD and Cancer: A critical review of epidemiologic studies. Crit Rev Toxicol. 2011;41:622–636. doi: 10.3109/10408444.2011.560141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pesatori AC, Consonni D, Rubagotti M, Grillo P, Bertazzi PA. Cancer incidence in the population exposed to dioxin after the “Seveso accident”: twenty years of follow-up. Environ Health. 2009;8:39. doi: 10.1186/1476-069X-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.EPA US. Reanalysis of key issues related to dioxin toxicity and response to NAS Comments. Edited by. 2012;1:521. [Google Scholar]
- 26*.Nebert DW, Dalton TP, Okey AB, Gonzalez FJ. Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J Biol Chem. 2004;279:23847–23850. doi: 10.1074/jbc.R400004200. [DOI] [PubMed] [Google Scholar]
- 27*.Ma Q, Lu AY. CYP1A induction and human risk assessment: an evolving tale of in vitro and in vivo studies. Drug Metab Dispos. 2007;35:1009–1016. doi: 10.1124/dmd.107.015826. [DOI] [PubMed] [Google Scholar]
- 28.Uno S, Dalton TP, Derkenne S, Curran CP, Miller ML, Shertzer HG, Nebert DW. Oral exposure to benzo[a]pyrene in the mouse: detoxication by inducible cytochrome P450 is more important than metabolic activation. Mol Pharmacol. 2004;65:1225–1237. doi: 10.1124/mol.65.5.1225. [DOI] [PubMed] [Google Scholar]
- 29*.Pittelkow MR, Perry HO, Muller SA, Maughan WZ, O’Brien PC. Skin cancer in patients with psoriasis treated with coal tar. A 25-year follow-up study. Arch Dermatol. 1981;117:465–468. [PubMed] [Google Scholar]
- 30.Song J, Clagett-Dame M, Peterson RE, Hahn ME, Westler WM, Sicinski RR, DeLuca HF. A ligand for the aryl hydrocarbon receptor isolated from lung. Proc Natl Acad Sci USA. 2002;99:14694–14699. doi: 10.1073/pnas.232562899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henry EC, Bemis JC, Henry O, Kende AS, Gasiewicz TA. A potential endogenous ligand for the aryl hydrocarbon receptor has potent agonist activity in vitro and in vivo. Arch Biochem Biophys. 2006;450:67–77. doi: 10.1016/j.abb.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 32.Wu Y, Chen X, Zhou Q, He Q, Kang J, Zheng J, Wang K, Duan T. ITE and TCDD differentially regulate the vascular remodeling of rat placenta via the activation of AhR. PLoS One. 2014;9:e86549. doi: 10.1371/journal.pone.0086549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Forrester AR, Elias MS, Woodward EL, Graham M, Williams FM, Reynolds NJ. Induction of a chloracne phenotype in an epidermal equivalent model by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is dependent on aryl hydrocarbon receptor activation and is not reproduced by aryl hydrocarbon receptor knock down. J Dermatol Sci. 2014;73:10–22. doi: 10.1016/j.jdermsci.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Francis JE, Smith AG. Polycyclic aromatic hydrocarbons cause hepatic porphyria in iron-loaded C57BL/10 mice: Comparison of uroporphyrinogen decarboxylase inhibition with induction of alkoxyphenoxazone dealkylations. Biochem Biophys Res Comm. 1987;146:13–20. doi: 10.1016/0006-291x(87)90683-8. [DOI] [PubMed] [Google Scholar]
- 35*.Ehrlich AK, Pennington JM, Wang X, Rohlman D, Punj S, Lohr CV, Newman MT, Kolluri SK, Kerkvliet NI. Activation of the Aryl Hydrocarbon Receptor by 10-Cl-BBQ Prevents Insulitis and Effector T Cell Development Independently of Foxp3+ Regulatory T Cells in Nonobese Diabetic Mice. J Immunol. 2016;196:264–273. doi: 10.4049/jimmunol.1501789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36*.Hu W, Sorrentino C, Denison MS, Kolaja K, Fielden MR. Induction of cyp1a1 is a nonspecific biomarker of aryl hydrocarbon receptor activation: results of large scale screening of pharmaceuticals and toxicants in vivo and in vitro. Mol Pharmacol. 2007;71:1475–1486. doi: 10.1124/mol.106.032748. [DOI] [PubMed] [Google Scholar]
- 37.O’Donnell EF, Koch DC, Bisson WH, Jang HS, Kolluri SK. The aryl hydrocarbon receptor mediates raloxifene-induced apoptosis in estrogen receptor-negative hepatoma and breast cancer cells. Cell Death Dis. 2014;5:e1038. doi: 10.1038/cddis.2013.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koch DC, Jang HS, O’Donnell EF, Punj S, Kopparapu PR, Bisson WH, Kerkvliet NI, Kolluri SK. Anti-androgen flutamide suppresses hepatocellular carcinoma cell proliferation via the aryl hydrocarbon receptor mediated induction of transforming growth factor-beta1. Oncogene. 2015;34:6092–6104. doi: 10.1038/onc.2015.55. [DOI] [PubMed] [Google Scholar]
- 39.O’Donnell EF, Kopparapu PR, Koch DC, Jang HS, Phillips JL, Tanguay RL, Kerkvliet NI, Kolluri SK. The aryl hydrocarbon receptor mediates leflunomide-induced growth inhibition of melanoma cells. PLoS One. 2012;7:e40926. doi: 10.1371/journal.pone.0040926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jin UH, Lee SO, Pfent C, Safe S. The aryl hydrocarbon receptor ligand omeprazole inhibits breast cancer cell invasion and metastasis. BMC Cancer. 2014;14:498. doi: 10.1186/1471-2407-14-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jin UH, Kim SB, Safe S. Omeprazole Inhibits Pancreatic Cancer Cell Invasion through a Nongenomic Aryl Hydrocarbon Receptor Pathway. Chem Res Toxicol. 2015;28:907–918. doi: 10.1021/tx5005198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frericks M, Meissner M, Esser C. Microarray analysis of the AHR system: tissue-specific flexibility in signal and target genes. Toxicol Appl Pharmacol. 2007;220:320–332. doi: 10.1016/j.taap.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 43.Yeste A, Takenaka MC, Mascanfroni ID, Nadeau M, Kenison JE, Patel B, Tukpah AM, Babon JA, DeNicola M, Kent SC, et al. Tolerogenic nanoparticles inhibit T cell-mediated autoimmunity through SOCS2. Sci Signal. 2016;9:ra61. doi: 10.1126/scisignal.aad0612. [DOI] [PubMed] [Google Scholar]
- 44.Yeste A, Nadeau M, Burns EJ, Weiner HL, Quintana FJ. Nanoparticle-mediated codelivery of myelin antigen and a tolerogenic small molecule suppresses experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2012;109:11270–11275. doi: 10.1073/pnas.1120611109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45*.Lawrence BP, Vorderstrasse BA. New insights into the aryl hydrocarbon receptor as a modulator of host responses to infection. Semin Immunopathol. 2013;35:615–626. doi: 10.1007/s00281-013-0395-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lawrence BP, Roberts AD, Neumiller JJ, Cundiff JA, Woodland DL. Aryl hydrocarbon receptor activation impairs the priming but not the recall of influenza virus-specific CD8+ T cells in the lung. J Immunol. 2006;177:5819–5828. doi: 10.4049/jimmunol.177.9.5819. [DOI] [PubMed] [Google Scholar]
- 47.Rouse M, Singh NP, Nagarkatti PS, Nagarkatti M. Indoles mitigate the development of experimental autoimmune encephalomyelitis by induction of reciprocal differentiation of regulatory T cells and Th17 cells. Br J Pharmacol. 2013;169:1305–1321. doi: 10.1111/bph.12205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Duarte JH, Di Meglio P, Hirota K, Ahlfors H, Stockinger B. Differential influences of the aryl hydrocarbon receptor on Th17 mediated responses in vitro and in vivo. PLoS One. 2013;8:e79819. doi: 10.1371/journal.pone.0079819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berg J, Mahmoudjanlou Y, Duscha A, Massa MG, Thone J, Esser C, Gold R, Haghikia A. The immunomodulatory effect of laquinimod in CNS autoimmunity is mediated by the aryl hydrocarbon receptor. J Neuroimmunol. 2016;298:9–15. doi: 10.1016/j.jneuroim.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 50.Kaye J, Piryatinsky V, Birnberg T, Hingaly T, Raymond E, Kashi R, Amit-Romach E, Caballero IS, Towfic F, Ator MA, et al. Laquinimod arrests experimental autoimmune encephalomyelitis by activating the aryl hydrocarbon receptor. Proc Natl Acad Sci USA. 2016;113:E6145–E6152. doi: 10.1073/pnas.1607843113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quintana FJ, Murugaiyan G, Farez MF, Mitsdoerffer M, Tukpah AM, Burns EJ, Weiner HL. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2010;107:20768–20773. doi: 10.1073/pnas.1009201107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nugent LF, Shi G, Vistica BP, Ogbeifun O, Hinshaw SJ, Gery I. ITE, a novel endogenous nontoxic aryl hydrocarbon receptor ligand, efficiently suppresses EAU and T-cell-mediated immunity. Invest Ophthalmol Vis Sci. 2013;54:7463–7469. doi: 10.1167/iovs.12-11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Di Meglio P, Duarte JH, Ahlfors H, Owens ND, Li Y, Villanova F, Tosi I, Hirota K, Nestle FO, Mrowietz U, et al. Activation of the aryl hydrocarbon receptor dampens the severity of inflammatory skin conditions. Immunity. 2014;40:989–1001. doi: 10.1016/j.immuni.2014.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goettel JA, Gandhi R, Kenison JE, Yeste A, Murugaiyan G, Sambanthamoorthy S, Griffith AE, Patel B, Shouval DS, Weiner HL, et al. AHR Activation Is Protective against Colitis Driven by T Cells in Humanized Mice. Cell Rep. 2016;17:1318–1329. doi: 10.1016/j.celrep.2016.09.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang Z, Jiang Y, Yang Y, Shao J, Sun X, Chen J, Dong L, Zhang J. 3,3′-Diindolylmethane alleviates oxazolone-induced colitis through Th2/Th17 suppression and Treg induction. Mol Immunol. 2013;53:335–344. doi: 10.1016/j.molimm.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 56.Furumatsu K, Nishiumi S, Kawano Y, Ooi M, Yoshie T, Shiomi Y, Kutsumi H, Ashida H, Fujii-Kuriyama Y, Azuma T, et al. A role of the aryl hydrocarbon receptor in attenuation of colitis. Dig Dis Sci. 2011;56:2532–2544. doi: 10.1007/s10620-011-1643-9. [DOI] [PubMed] [Google Scholar]
- 57.Jeong KT, Hwang SJ, Oh GS, Park JH. FICZ, a tryptophan photoproduct, suppresses pulmonary eosinophilia and Th2-type cytokine production in a mouse model of ovalbumin-induced allergic asthma. Int Immunopharmacol. 2012;13:377–385. doi: 10.1016/j.intimp.2012.04.014. [DOI] [PubMed] [Google Scholar]
- 58.Bae MJ, See HJ, Choi G, Kang CY, Shon DH, Shin HS. Regulatory T Cell Induced by Poria cocos Bark Exert Therapeutic Effects in Murine Models of Atopic Dermatitis and Food Allergy. Mediators Inflamm. 2016;2016:3472608. doi: 10.1155/2016/3472608. [DOI] [PMC free article] [PubMed] [Google Scholar]