

Abstract
Neurotransmitter disruption is often a key component of diseases of the central nervous system (CNS), playing a role in the pathology underlying Alzheimer's disease, Parkinson's disease, depression, and anxiety. Traditionally, microdialysis has been the most common (lauded) technique to examine neurotransmitter changes that occur in these disorders. But because microdialysis has the ability to measure slow 1-20 minute changes across large areas of tissue, it has the disadvantage of invasiveness, potentially destroying intrinsic connections within the brain and a slow sampling capability. A relatively newer technique, the microelectrode array (MEA), has numerous advantages for measuring specific neurotransmitter changes within discrete brain regions as they occur, making for a spatially and temporally precise approach. In addition, using MEAs is minimally invasive, allowing for measurement of neurotransmitter alterations in vivo. In our laboratory, we have been specifically interested in changes in the neurotransmitter, glutamate, related to Alzheimer's disease pathology. As such, the method described here has been used to assess potential hippocampal disruptions in glutamate in a transgenic mouse model of Alzheimer's disease. Briefly, the method used involves coating a multi-site microelectrode with an enzyme very selective for the neurotransmitter of interest and using self-referencing sites to subtract out background noise and interferents. After plating and calibration, the MEA can be constructed with a micropipette and lowered into the brain region of interest using a stereotaxic device. Here, the method described involves anesthetizing rTg(TauP301L)4510 mice and using a stereotaxic device to precisely target sub-regions (DG, CA1, and CA3) of the hippocampus.
Keywords: Neuroscience, Issue 123, in vivo electrochemistry, microelectrode arrays, biosensors, Alzheimer's disease, glutamate, hippocampus, neurotransmitters, tau, rTg4510
Introduction
Measuring neurotransmitter alterations in the brain is an essential tool for neuroscientists studying diseases of the central nervous system (CNS) that are often characterized by neurotransmitter dysregulation. Though microdialysis in combination with high pressure liquid chromatography (HPLC/EC) has been the most widely used method to measure changes in extracellular neurotransmitter levels1,2,3,4, the spatial and temporal resolution of microdialysis probes may not be ideal for neurotransmitters, such as glutamate, that are tightly regulated in the extracellular space5,6. Because of the recent advances in genetics and imaging, there are additional methods that can be used to map glutamate in vivo. Using genetically encoded glutamate fluorescent reporters (iGluSnFR) and two-photon imaging, researchers are able to visualize glutamate release by neurons and astrocytes both in vitro and in vivo789. Notably, this allows for recording from a larger field of view and does not disrupt the intrinsic connections of the brain. While these new optical techniques allow for visualization of glutamate kinetics and measurement of sensory evoked responses and neuronal activity, they lack the ability to quantify the amount of glutamate in the extracellular space in discrete brain regions.
An alternative method is the enzyme-linked microelectrode array (MEA) that can selectively measure extracellular neurotransmitter levels, such as glutamate, through the use of a self-referenced recording scheme. The MEA technique has been used to study alterations in extracellular glutamate following traumatic brain injury10,11,12, aging13,14, stress15,16, epilepsy17,18, Alzheimer's disease19,20, and injection of a viral mimic21 and represents an improvement over the spatial and temporal limitations inherent in microdialysis. Whereas microdialysis restricts the ability to measure near the synapse22,23, MEAs have a high spatial resolution that allows for selective measures of extracellular glutamate spillover near synapses24,25. Second, the low temporal resolution of microdialysis (1 - 20 min) limits the ability to investigate the fast dynamics of glutamate release and clearance occurring in the millisecond to second range26. Because differences in the release or clearance of glutamate may not be evident in measures of tonic, resting glutamate levels, it may be essential that glutamate release and clearance be directly measured. MEAs allow for such measures due to their high temporal resolution (2 Hz) and low limits of detection (< 1 µM). Third, MEAs allow for examination of subregional variations in neurotransmitters within a particular brain region, such as the rat or mouse hippocampus. For example, using MEAs we can separately target the dentate gyrus (DG), cornu ammonis 3 (CA3) and cornu ammonis 1 (CA1) of the hippocampus, which are connected via a trisynaptic circuit27, to examine subregional differences in extracellular glutamate. Because of the size of microdialysis probes (1 - 4 mm length) and the damage caused by implantation28,29, subregional differences are difficult to address. Furthermore, the optical systems only allow stimulation through external stimuli, such as a whisker stimulation or light flicker, which does not permit subregional stimulation7. A final benefit of MEAs over other methods is the ability to study these subregions in vivo without disrupting their extrinsic and intrinsic connections.
Here, we describe how a recording system (e.g., FAST16mkIII) in combination with MEAs, consisting of a ceramic-based multisite microelectrode, can be differentially coated on the recording sites to allow for interfering agents to be detected and removed from the analyte signal. We also demonstrate these arrays can be used for amperometry-based studies of in vivo glutamate regulation within the DG, CA3, and CA1 hippocampal subregions of anesthetized rTg(TauP301L)4510 mice, a commonly used mouse model of Alzheimer's disease. In addition, we provide confirmation of the sensitivity of the MEA system to the fast dynamics of glutamate release and clearance by treating the mice with riluzole, a drug shown in vitro to decrease glutamate release and increase glutamate uptake30,31,32,33, and demonstrating these respective changes in vivo in the TauP301L mouse model.
Protocol
1. Coating the Microelectrode Array with Enzymes or Matrix Layer
- Preparing the protein matrix solution
- Weigh out 10 mg of bovine serum albumin (BSA) and transfer to the 1.5 mL microcentrifuge tube.
- Add 985 µL of DI water to the microcentrifuge tube containing BSA. Mix the solution by manual agitation (re-pipetting using 1,000 µL pipette ~ 3 times, until the BSA is dissolved). NOTE: Do not use vortex to mix the solution as doing so may introduce air into the solution. Also, set the pipette to a volume <1,000 µL (e.g. 700 µL) to avoid introducing air bubbles. The solution may be placed in a microcentrifuge if necessary.
- Once the BSA solution is dissolved, add 5 µL of 25% glutaraldehyde solution. Mix the solution by inverting the closed tube 3 - 5 times. The resulting solution is 1% BSA with 0.125% glutaraldehyde.
- Set aside the protein matrix solution for 5 min. The solution will appear light yellow in color.
- Preparing the desired enzyme solution (e.g., glutamate oxidase)
- Prepare the glutamate oxidase solution by adding filtered DI water to the lyophilized, purified enzyme (e.g., 50 µL DI water added to 25 units of glutamate oxidase to yield 0.5 U/µL). NOTE: Aliquot stock solution (e.g., 1 µL) and store the aliquots in appropriately sized containers (e.g., 500 µL microcentrifuge tubes) at -20 °C. Aliquots may be stored for up to 6 months under these conditions.
- Add 4 µL of the BSA/glutaraldehyde solution to the microcentrifuge aliquoted tube containing glutamate oxidase. Mix the solution by manual agitation (re-pipetting with a 10 µL pipette. The pipette should be set to volume < 5 µL). The resulting solution is 1% BSA, 0.125% glutaraldehyde and 0.1 U/µL glutamate oxidase. Use the solution for coating immediately. Note: Glutamate oxidase coating solution may only be used within 15 min. Although several MEAs may be coated with one prepared solution, it is recommended not to exceed the useable time window for glutamate oxidase. The number of electrodes that can be coated within 15 min vary. Generally, 4 to 6 electrodes can be coated at a time.
- Coating the MEA
- Typically, coat a pair of recording site(s) on a MEA with the desired enzyme (glutamate oxidase) and a different pair of recording site(s) ('sentinel' site(s)) is coated with the inactive protein-matrix.
- Use blunt-tip Hamilton microsyringes (e.g. 10 µL) to coat MEAs with L-glutamate oxidase or inactive protein matrix solution (BSA+glutaraldehyde). The procedures used for enzyme or inactive protein-matrix coatings are the same.
- Clean the syringes before and after use (3 rinses with DI water, 3 rinses with methanol, 5 rinses with DI water). NOTE: Dedicated syringes are used for applying coats of enzyme (glutamate oxidase) or protein-matrix. Do not use the same syringe for a different solution. It is recommended to perform protein-matrix coatings before enzyme coatings.
- Draw up glutamate oxidase or protein matrix using a 10 µL Hamilton syringe. Gently press the plunger to dispense a small bead of solution at the syringe tip (visualized using a dissecting microscope).
- Using the dissecting microscope to target the MEA recording sites, apply the solution to the appropriate recording sites by briefly contacting the recording sites with the solution droplet/bead, which represents one layer. Three layers are sufficient for both the protein-matrix and the enzyme coat. Thick layers can reduce the sensitivity of the MEA.
- Raise the solution droplet straight up and off the recordings sites (i.e. the syringe tip should not scrape across the MEA surface).
- Set a timer for 1 min. This is the minimum time requirement between coating applications to the recording site(s). Allow enzyme-coated MEAs to cure at 4 °C for 5-7 days.
- Record the number of coats applied, how long it took to apply the coatings, the electrode number, and a name and date in a laboratory notebook.
2. Electroplating with m-Phenylenediamine for Improved Selectivity
- Preparing m-Phenylenediamine dihydrochloride (mPD) solution NOTE: Plate the exclusion layer, mPD, to prevent the oxidation of dopamine and other large, interferent molecules (e.g., ascorbic acid), thereby improving the selectivity of the sensor for glutamate/H2O2 over other potentially-interferent molecules.
- Measure 50 mL of 0.05 M phosphate buffered saline (PBS) in a volumetric flask and transfer 50 mL of 0.05 M PBS to a larger (150 mL) Erlenmeyer flask.
- Using the tubing connected to a nitrogen tank (e.g., using the tubing connected a pressure ejector), attach a pipette tip (e.g. P200 tip) to the open end of the tubing. Place the tubing with the attached pipette tip in PBS solution and cover the solution loosely with parafilm. Turn on nitrogen and gently bubble solution for 20 min.
- Weigh 0.045 g of mPD. Caution: mPD is a potentially carcinogenic substance. Weighing of mPD should be performed using a scale contained within a fume hood. Wear gloves and mask when using mPD. Ensure the hood is functional and that the sash is pulled to the appropriate working level. Immediately clean surfaces that may have been contaminated with mPD to avoid permanent staining.
- After bubbling, transfer all of the mPD to the de-gassed PBS solution. Cover the flask opening with parafilm and gently swirl solution to dissolve mPD in solution. Avoid aggressive mixing or use of a vortex as doing so may introduce gasses into the solution.
- Transfer the solution to a 50-mL beaker. A stir bar is not necessary.
- Electroplating the electrode
- Obtain a reference electrode. Use reference electrodes dedicated to mPD plating only. Use a different reference electrode for calibration procedures.
- Position the plastic arm of the reference electrode holder over the beaker. Check for air bubbles in the glass reference electrode (may not be able to see). Gently flick the distal end of the reference electrode to remove any air bubbles at the tip.
- Place the reference electrode through the opening in the plastic arm and lower into the beaker containing PBS. Ensure that the reference electrode does not contact the bottom of beaker.
- Connect the reference electrode to the headstage (e.g. 2 pA/mV) and connect (e.g. DIP connector) the MEA to be mPD plated to the headstage.
- Lower the MEA into the beaker solution such that only the MEA tip is submerged in solution. Do not submerge the MEA tips in liquid beyond the black 'bubble' (the black insulation located above the MEA tip). Doing so may damage the MEA.
- Run a 'test' calibration to verify the functionality of the electrode channels and connection to the headstage. NOTE: Although hardware settings should reflect the equipment being used, details related to analyte/interferent type and the concentration do not have to be entered for the 'test' calibration. The recoding mode should be 'Amperometry' and the 'V-applied' should be -0.7 V. Verify typical recording for all channels.
- Cancel by selecting 'Cancel' when connectivity of all the recording sites is verified. It is not necessary to save the calibration data.
- Select the "Electroplating" icon on the desktop. The Electroplating Tool Menu will appear. Verify the correct settings are entered: P-P amplitude: 0.25 Offset: -0.5 Frequency: 0.05 Duration (min): 20
- Select the 'pause' button to begin plating. The elapsed time field should begin counting down.
- Upon completion (all time elapsed), select 'another MEA' in the popped up menu to begin mPD plating an additional MEA. Replace the plated MEA with an MEA to be mPD plated and repeat the electroplating procedure using the electroplating tool. NOTE: It is recommended to use the prepared mPD solution no more than 2 times or within a 1 h time period. Prepare a fresh solution to prepare more than 2 MEAs.
- Select 'exit software' to finish electroplating. Rinse mPD-plated electrode tips with DI water (to avoid a salt residue) and store (24 h) before calibrating.
- Remove the reference electrode from solution and rinse with DI water before placing into the appropriate storage solution (e.g. 3 M NaCl). Ensure the reference electrode tip is submerged in solution. Slowly lower glass reference electrode into the solution to avoid damaging the glass.
- Dispose of the mPD solution in an appropriately labeled hazardous waste container.
3. Calibrate the MEA for Glutamate Detection and Selectivity (Figure 1)21
Preparing the solutions needed for calibration. NOTE: Refer to Table 1.
Perform calibration under constant stirring (magnetic battery operated stirrer) at 37 °C. Turn on the heating pad or circulating water bath and obtain a small stir bar. Stir slowly but fast enough that when an addition is made (below), the new plateau is reached quickly.
Connect the headstage (2 pA/mV) to the recording control system and insert the MEA tip into 40 mL of stirred 0.05 M PBS (use a 50-mL beaker).
Connect the reference electrode. Flick the end to ensure there are no air bubbles.
Open the recording program and click "Calibration". Make sure the settings are correct (check that sentinel sites are selected as this may change with different electrodes), choose MEA number and press "Start".
Allow 5 - 10 min for equilibration; begin once the baseline has stabilized (< 0.004 nA/min change).
Select "Baseline" and add 500 µL of 20 mM ascorbic acid (interferent; final [250 µM]) and select "Interferent" (AA is thought of as brain-wide interferent) once the current has reached a new, steady plateau, if any. Allow 0.5 - 1 min between additions. If the MEA is properly electroplated, almost no change should be observed.
Add 40 µL of 20 mM L-glutamate (final [20 µM]) and once the current has reached a new, steady plateau, mark 1st addition "Analyte". Repeat three times for a total of 3 glutamate additions.
Add 40 µL DA. Do not select "Analyte"; select "Test Substance" - DA may only be an interferent in areas containing DA.
Add 40 µL Peroxide. Do not select "Analyte"; select "Test Substance".
Click the "Stop" button once the calibration is finished.
- To determine the functionality of the MEA, click the calculation tab and record the following parameters in a lab notebook.
- For the MEA design (3,000 µm2) used in these calibrations, use a MEA with a slope of ≥ 0.004 nA/µM but lower slopes can be used for certain experiments. If there is less than a 10% difference in slope between enzyme coated and uncoated sites, then use these MEAs for evoked release only or clean/discard the MEA.
- Use a limit of detection (LOD) ≤ 1.5 µM. If the LOD is greater than 1.5 µM, then use these MEAs for evoked release and uptake only. Do not use these electrodes for recording tonic levels.
- Use a selectivity for the desired analyte (i.e., glutamate) over the interferent of > 20 arbitrary units. If selectivity is less than 20, then clean/discard the MEA.
- Use a linearity (R2) of the calibration curve of ≥ 0.90. If linearity is less than 0.90, then clean or discard MEA.
Save the file with the # of the MEA, date, and initials of the person calibrating.
4. Assemble the Micropipette
NOTE: Micropipettes (capillary glass) should have a tip with an internal diameter of 10 - 15 µm.
Place the micropipette centrally among all four platinum recording sites and mount 50-100 µm above the MEA using sticky wax and modeling clay.
To load the micropipette, use a 1 mL syringe filled with glutamate or KCl solution, a 0.22-µm sterile syringe filter, and a 97 mm long, 28-gauge pipette filler, backfill the micropipette. That is, insert the needle at the top of the micropipette and fill at the bottom of the micropipette (the end toward the MEA), making sure there are no bubbles.
Attach the micropipette to the pressure ejecting system at 2 - 20 psi for about 1 s.
5. Plate the Miniature Reference Electrode for In Vivo Use
Prepare the plating solution (50 g NaCl/90 mL 1 M HCl in a 100 mL beaker (plating bath)) and cut the bath electrode (~ 10 cm length of platinum (Pt) wire (~ 0.02" diameter)).
Cut a 10 - 15 cm long piece of Teflon coated silver wire (0.008" bare) and use a razor blade to scrape off ~ 1 cm of Teflon coating from each end of end of silver wire.
Solder a gold pin on one end and place one end of exposed silver wire into the plating bath.
Using a DC adapter (use only a DC adapter having an output of ~ 9 - 15 VDC max.), clamp the "red" (+) wire to the prepared silver wire (reference electrode) on the gold pin side. Clamp the "black" (-) wire to Pt bath cathode.
Plug in the DC adapter. Look for the correct plating process — bubbles should appear at the bath electrode; reference electrode turns a silver/gray color. CAUTION: Do not switch the leads [Black (-) vs Red (+)] — plating bath will be ruined if this occurs and fresh plating solution will need to be made. A bad solution will turn yellow in color: DO NOT USE! NOTE: The plating reaction takes 2-5 min generally: Ag0 + Cl- → AgCl + e-
- Test the reference electrode.
- Set a multimeter to the 100 - 200 mV DC setting.
- Place a benchmark (that is, a previously tested electrode known to be working) reference electrode in 3 M NaCl and test against the newly plated reference electrode. NOTE: If using a new reference electrode, soak it in 3 M NaCl before use (12 h recommended).
- Place the "red" (+) lead on the gold pin of the reference electrode to be tested. Place the "black" (-) lead on benchmark electrode. An acceptable readout range is ± 10 mV. 0 mV is ideal across the 2 reference electrodes.
6. General Animal Surgery for MEA Recordings
- Preparation for surgery
- Place surgery tools in disinfectant the night before.
- Remove the tools from the disinfectant and rinse thoroughly and place the tools on a sterile surgery pad. Obtain a heating pad.
- Calibrate two electrodes and attach the micropipette as described in procedures 3 and 4.
- Make the reference electrode as described in Section 5.
- Make 200 µM glutamate and 70 mM KCl. Refer to Table 1.
- Obtain the animal and record its weight on surgery sheets.
- Prepare the anesthesia and turn on the heating pad.
- Perform MEA surgery
- Using isoflurane (4% flow in oxygen), anesthetize the animal in an induction chamber until the respiratory rate has significantly declined and the animal does not respond to toe or tail pinch. Record respiratory rate and body temperature every 15 min.
- Place the animal in the stereotaxic device using the ear bars to stabilize the head. Make sure the animal is secure and the head does not move. Lower the isoflurane flow to 1.5 - 3% in oxygen. Ensure that the heating pad is properly positioned under the animal and set to 37 °C to provide supplemental heat. Failure to provide supplemental heat may result in profound hypothermia and premature death of the animal.
- Apply eye ointment using a sterile cotton applicator and record the respiratory rate and body temperature. Using a battery-powered trimmer, shave the top of the head. Use the small surgical scissors to remove the fur near the ears.
- At this point, put on the sterile surgery gloves. Apply iodine and then alcohol (three times) to the scalp.
- Using a scalpel, make an incision straight down the middle of the scalp and spread the skin using bull dog clamps. Take a sterile cotton tip to soak up any blood. Use hydrogen peroxide to facilitate the appearance of bregma and lambda.
- Check that the head is properly positioned by measuring the D/V and M/L coordinates of bregma and lambda. Coordinate changes should be zero if the head is on a flat plane. Adjust the head and ear bars as necessary to ensure a flat plane.
- Find bregma, and zero the coordinates. Using the desired coordinates, move to the left and right and make a mark using a permanent marker. Using a cauterizer/marker, make a large square around the mark with enough room to reach the desired region.
- Using a sterile rotary tool, drill around the square mark. Soak up any blood using a sterile cotton applicator. Thoroughly disinfect the drill bit prior to each use.
- Attach the MEA to the headstage and backfill the pipette with the first desired solution. Be sure leave a gap in the pipette without solution so that the solution being expelled can be examined. Attach the tubing to the glass micropipette.
- Turn on the nitrogen tank and the pressure ejector (psi=5, time=0.6 s). Test (press manual red button) to ensure the pipette is not clogged before beginning.
- Calibrate the micropipette. Start at 0 on the reticle and place a piece of blue tape on the headstage to indicate the start point. Turn on the digital reader and reset it to zero. Go down 1 mm (DV) and count the number of ticks the solution has moved on the reticle. 10 ticks should equal 1 mm (1 mm = 250 nL), so 1 tick = 25 nL.
- Place the reference electrode in a remote location from the MEA such that it is still in liquid contact with the brain to complete the circuit.
- Find bregma with the MEA attached and zero the digital reader. Move the MEA to the desired coordinates. Slowly lower the MEA until the tip touches the brain. Zero the DV coordinate and SLOWLY lower the MEA into the brain.
- On the recording program, click on the desired calibrated electrode and then click "Perform Experiment." The system will then start recording tonic levels for the analyte of interest while the animal is anesthetized.
- Zoom to an axis that will help in observing baseline and allow the electrode to baseline for 20 - 45 min.
- After the baseline is stable, set the pressure ejector to .6 s and 5 psi and press the red button on the pressure ejector to eject. Record the time and pressure as well as volume/ticks moved on the injection sheet (Figure 2). Make any notes that are necessary (i.e., larger peaks, dilution effects, clogged pipette, noisy baseline/o-scope).
- For glutamate injections, wait 3 - 5 min between injections. For KCl injections wait 1 - 2 min between injections. Inject using the same pressure and time at least 3 times. Change time and repeat. Try not to change pressure unless the micropipette is clogged.
- If the micropipette is clogged, increase the pressure up to 30 psi.
- Record from the desired brain regions, repeating on both sides of the brain using different solutions. Record a baseline in each new region for 20 min.
- Take the MEA with micropipette attached, rinse with DI water and soak in PBS overnight until all the blood is gone.
- Euthanize the animal and save the brain in a pre-labeled tube and store in the -80 °C freezer or keep one side for fixation. To euthanize the animal, overdose with isoflurane (inhalation). Perform decapitation using surgical scissors and dissect out the desired regions.
7. Cleaning Coated MEAs after Use
Do not clean MEAs if unused. If there is lint or dust on the surface, clean with methanol and let dry for 24 h before coating.
Turn on the water bath (80 °C) and soak the tip of the MEA for 30 min. Carefully wipe the tip of the electrode with a cotton tipped applicator to remove any debris that may be left on the electrode tip. Do not get the black "insulation portion" of the MEA wet.
Using three different sonicators, soak the tip of the MEA in (1) Citrisolv, a commercial solvent and clearing agent (2) isopropyl, and (3) DI water for 5 min. Carefully wipe the tip of the electrode with cotton tipped applicator to remove any debris that may be left on the electrode tip.
Examine the tips under a dissecting microscope to ensure the layers have been successfully removed.
8. Analysis
To analyze the data, first export the desired experiment by double-clicking the finished experiment and selecting "export data" from the file drop-down menu.
Save this data to the desired file location and open up the analysis program. Click "open" in the analysis program and choose the recently saved file.
Give the program a few moments to upload the file and then check experiment under the event markers tab. Under output, check the boxes for the desired parameters. For example, baseline, amplitude, peak area (area under the curve), Trise, and T80.
Click refresh and then analyze to export the data to a spreadsheet (this may take a few minutes). The program will give a prompt to save the file to the desired location. Once saved, the file can be edited in a spreadsheet.
Representative Results
While this technology can be used to measure alterations in glutamatergic signaling in many types of animal models, such as traumatic brain injury, aging, stress, and epilepsy, here we demonstrate how the MEA technology can be used to examine glutamatergic alterations in transgenic mouse model of human tauopathy19,20. The rTg(TauP301L)4510 mouse expresses the P301L mutation in tau associated with frontotemporal dementia and parkinsonism linked to chromosome 17, and is commonly used to study tau pathology associated with Alzheimer's disease, neurodegenerative tauopathies and frontotemporal dementia. We also demonstrate that glutamatergic signaling can be modified by pharmacological intervention. We treated TauP301L mice with riluzole, an FDA-approved disease-modifying drug for amyotrophic lateral sclerosis (ALS) that modulates glutamatergic signaling by stabilizing the inactivate state of voltage-gated sodium channels, leading to a decrease in glutamate release, and a potentiation of glutamate uptake via an increase in glutamate transporter expression, leading to increased glutamate clearance30,31,32,33.
We have chosen this particular dataset for demonstrative purposes for four reasons. First, this dataset demonstrates the temporal resolution of the system, as shown by our ability to measure the fast dynamics of transient release and uptake of glutamate. In addition, this preclinical animal model exhibits alterations in tonic glutamate, glutamate release, and glutamate clearance, allowing for an example of alterations in each measure. Second, utilizing this dataset, the high spatial resolution that allows for measurement of subregion differences in the DG, CA3, and CA1 of the hippocampus is demonstrated. Third, this data allows for demonstration of how pharmacological intervention can be used to mitigate glutamatergic alterations observed in preclinical animal models. Finally, because this preclinical model has alterations in glutamate release, the need for careful interpretation of glutamate clearance in the presence of altered glutamate release can be explained.
After reaching a stable baseline, as indicated by < 0.004 nA/min change in slope (10 - 20 min), we first measured tonic glutamate levels (µM) by averaging extracellular glutamate levels over 10 s prior to any application of solutions. We observed increased tonic glutamate levels in the CA3 and CA1 regions of TauP301L mice, and riluzole restored tonic glutamate levels to that of controls (Figure 3A). Next, KCl was locally delivered (via a micropipette) to each of the three sub-regions of one hemisphere every 2 - 3 min. After 10 reproducible signals, which is indicative of an intact glutamate neuronal system34, the results were averaged for each group and the average amplitude compared. Representative traces (Figure 3B) revealed increased glutamate release in TauP301L mice following application of KCl in all three subregions (only CA3 is shown), an effect that was rescued by riluzole treatment (Figure 3C).
The MEA was then moved to the opposite hemisphere and allowed to reach a stable baseline (10 - 20 min) before exogenous glutamate was applied to examine glutamate clearance (Figure 4). Varying volumes (50 - 250 nL) of 200 µM sterile-filtered glutamate solution were applied into the extracellular space every 2 - 3 min, and the net area under the curve (AUC) was used as a measure of glutamate uptake. This rapid application of glutamate into the extracellular space allows for mimicking of endogenous glutamate release and measurement of glutamate uptake. Because glutamate transporters exhibit Michaelis-Menten kinetics35, a range of volumes (50 - 250 nL) of exogenous glutamate is injected to expose differences in uptake. To do so, each animal receives 1 - 2 injections at 50 nL increments within the 50 - 250 nL range. Though one should always confirm that the volume injected per group per region is not statistically different at the p ≤ 0.05 level, the best way to ensure net AUC is related to uptake, and not the amount of glutamate applied or diffusion, is to ensure that both amplitude (Figure 4A) and Trise (a measure of diffusion from the point source (micropipette) to the MEA in Figure 4B) do not differ10,11,19,20. This allows for "peak matching" the amplitudes (Figure 4C) such that differences in net AUC are assumed to be due to differences in uptake and not the amount applied or diffusion. Peak matching is particularly important when animals have existing differences in glutamate release. Because a range of volumes is injected, the variance for both amplitude and Trise are often large and adding additional animals does not decrease the variance for these measures, as they are inherent to the design. Because the amplitude and Trise were similar among the groups in each subregion (Figure 4A, 4B), we assume that differences in net AUC (Figure 4C, 4D) are due to differences in glutamate uptake. Riluzole improved glutamate clearance compared to controls in all three subregions (Figure 4D). Finally, an MEA with an attached micropipette is used to locally apply a dye to confirm MEA placement after brain sectioning, as demonstrated in Figure 3D.
These data indicate that the MEA technology is capable of measuring neurotransmitter release within small subregions of the hippocampus24,25, unlike previous techniques (e.g., microdialysis), which only recorded from large sample areas and often damaged neuronal circuits28,29. In addition, the MEAs provide us with high temporal resolution to measure the fast kinetics of glutamate release and clearance25.
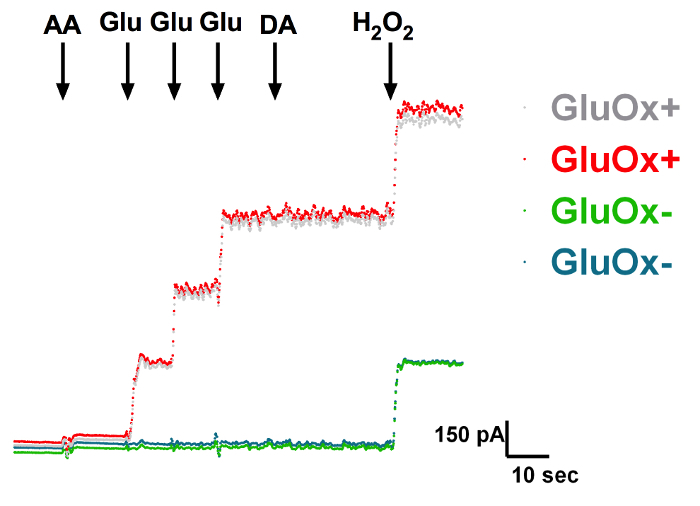 Figure 1: In Vitro Calibration of a Self-referencing Microelectrode Measuring the Change in Current (pA) on a Glutamate Oxidase Site (GluOx; red) vs. a Sentinel Site (Sent; blue). The interferents ascorbic acid (AA: 250 µM) and dopamine (DA: 2 µM) did not alter the current at either glutamate oxidase or sentinel sites. Addition of glutamate (Glu: 20, 40, 60 µM) produced a stepwise current increase on the glutamate oxidase site, but no change on the sentinel site. Hydrogen peroxide (H2O2: 8.8 µM) produced an increase in current on both sites. Sensitivity, slope, limit of detection, and R2 values were calculated after calibration. Please click here to view a larger version of this figure.
Figure 1: In Vitro Calibration of a Self-referencing Microelectrode Measuring the Change in Current (pA) on a Glutamate Oxidase Site (GluOx; red) vs. a Sentinel Site (Sent; blue). The interferents ascorbic acid (AA: 250 µM) and dopamine (DA: 2 µM) did not alter the current at either glutamate oxidase or sentinel sites. Addition of glutamate (Glu: 20, 40, 60 µM) produced a stepwise current increase on the glutamate oxidase site, but no change on the sentinel site. Hydrogen peroxide (H2O2: 8.8 µM) produced an increase in current on both sites. Sensitivity, slope, limit of detection, and R2 values were calculated after calibration. Please click here to view a larger version of this figure.
 Figure 2. A Sample Electrochemistry Worksheet. This worksheet includes columns for time of injection or injection number (TIME/TTL), brain coordinates (AP, mL, DV), the amount of pressure applied (nitrogen), the length of pressure (time), and the volume injected by the pressure ejector. Any notes, such as odd signals or clogging of the micropipette should be recorded in the notes column. Please click here to view a larger version of this figure.
Figure 2. A Sample Electrochemistry Worksheet. This worksheet includes columns for time of injection or injection number (TIME/TTL), brain coordinates (AP, mL, DV), the amount of pressure applied (nitrogen), the length of pressure (time), and the volume injected by the pressure ejector. Any notes, such as odd signals or clogging of the micropipette should be recorded in the notes column. Please click here to view a larger version of this figure.
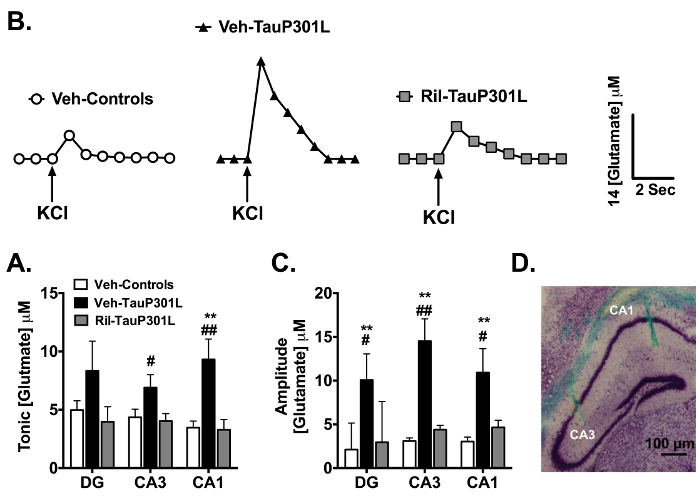 Figure 3. Extracellular Tonic and Potassium Chloride (KCl)-evoked Release of Glutamate in the DG, CA3, and CA1 Regions of the Hippocampus. (A) In the CA3 and CA1 regions of the hippocampus, tonic glutamate levels were significantly increased in vehicle-treated TauP301L mice (Veh-TauP301L), an effect attenuated by riluzole treatment. (B) Baseline-matched representative recordings of KCl-evoked glutamate release in the CA3 showed riluzole treatment (Ril-TauP301L) attenuated the significant increase in the amplitude of glutamate release observed in Veh-TauP301L mice. Local application of KCl (↑) produced a robust increase in extracellular glutamate that rapidly returned to tonic levels. (C) The significantly increased KCl-evoked glutamate release observed in Veh-TauP301L mice in the DG, CA3, and CA1 after local application of KCl was attenuated with riluzole treatment. (D) Cresyl violet-stained 20 µm section of hippocampus shows location of MEA tracks in CA3 and CA1 (Mean ± SEM; ** p < .01 Veh-Control vs. Veh-TauP301L, # p < .05 Ril-Tau-P301L vs. Veh-TauP301L, ## p < .01 Ril-Tau-P301L vs. Veh-TauP301L; n = 14 - 19/group). This figure reprinted with permission from John Wiley and Sons19,20. Please click here to view a larger version of this figure.
Figure 3. Extracellular Tonic and Potassium Chloride (KCl)-evoked Release of Glutamate in the DG, CA3, and CA1 Regions of the Hippocampus. (A) In the CA3 and CA1 regions of the hippocampus, tonic glutamate levels were significantly increased in vehicle-treated TauP301L mice (Veh-TauP301L), an effect attenuated by riluzole treatment. (B) Baseline-matched representative recordings of KCl-evoked glutamate release in the CA3 showed riluzole treatment (Ril-TauP301L) attenuated the significant increase in the amplitude of glutamate release observed in Veh-TauP301L mice. Local application of KCl (↑) produced a robust increase in extracellular glutamate that rapidly returned to tonic levels. (C) The significantly increased KCl-evoked glutamate release observed in Veh-TauP301L mice in the DG, CA3, and CA1 after local application of KCl was attenuated with riluzole treatment. (D) Cresyl violet-stained 20 µm section of hippocampus shows location of MEA tracks in CA3 and CA1 (Mean ± SEM; ** p < .01 Veh-Control vs. Veh-TauP301L, # p < .05 Ril-Tau-P301L vs. Veh-TauP301L, ## p < .01 Ril-Tau-P301L vs. Veh-TauP301L; n = 14 - 19/group). This figure reprinted with permission from John Wiley and Sons19,20. Please click here to view a larger version of this figure.
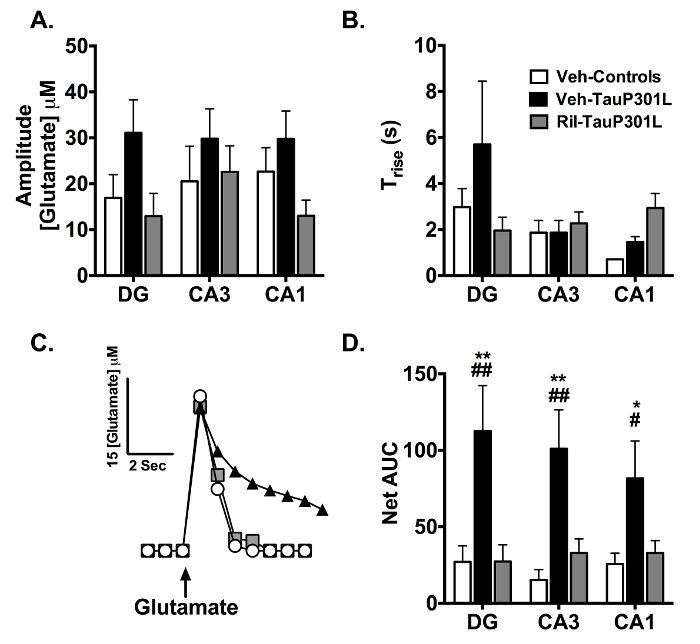 Figure 4. Glutamate Uptake Following Exogenous Glutamate Application in the DG, CA3, and CA1 Regions of the Hippocampus. (A) The amplitude of the locally-applied glutamate signal was similar among groups in each region. (B) Trise, an indicator of glutamate diffusion from the micropipette, was similar among the groups in each region. (C) Representative glutamate signals in the CA3 from local application of glutamate in Veh-Controls, Veh-TauP301L, and Ril-TauP301L mice. (D) Riluzole treatment reduced the significant increases in the net area under the curve (AUC) observed in Veh-TauP301L mice in all 3 regions of the hippocampus, indicating improved glutamate uptake in riluzole-treated TauP301L mice (Mean ± SEM; * p < .05 Veh-Control vs. Veh-TauP301L, ** p < .01 Veh-Control vs. Veh-TauP301L, # p < .05 Ril-Tau-P301L vs. Veh-TauP301L, ## p < .01 Ril-Tau-P301L vs. Veh-TauP301L; n = 13 - 15/group). This figure was reprinted with permission from John Wiley and Sons20. Please click here to view a larger version of this figure.
Figure 4. Glutamate Uptake Following Exogenous Glutamate Application in the DG, CA3, and CA1 Regions of the Hippocampus. (A) The amplitude of the locally-applied glutamate signal was similar among groups in each region. (B) Trise, an indicator of glutamate diffusion from the micropipette, was similar among the groups in each region. (C) Representative glutamate signals in the CA3 from local application of glutamate in Veh-Controls, Veh-TauP301L, and Ril-TauP301L mice. (D) Riluzole treatment reduced the significant increases in the net area under the curve (AUC) observed in Veh-TauP301L mice in all 3 regions of the hippocampus, indicating improved glutamate uptake in riluzole-treated TauP301L mice (Mean ± SEM; * p < .05 Veh-Control vs. Veh-TauP301L, ** p < .01 Veh-Control vs. Veh-TauP301L, # p < .05 Ril-Tau-P301L vs. Veh-TauP301L, ## p < .01 Ril-Tau-P301L vs. Veh-TauP301L; n = 13 - 15/group). This figure was reprinted with permission from John Wiley and Sons20. Please click here to view a larger version of this figure.
| 0.05 M PBS | 2 L DI water: |
| Store in glass beaker wrapped with tin foil. Bring pH to 7.4 with KOH if necessary. | NaH2PO4·H2O: 2.8 g Na2HPO4: 11.34 g Sodium Chloride: 11.68 g |
| 20 mM Ascorbic Acid | 50 mL DI water: |
| Make fresh daily. | Ascorbic acid: 0.18 g |
| 20 mM L-Glutamate | 50 mL DI water: |
| Make fresh weekly. | L-glutamate monosodium salt hydrate: 0.15 g |
| 2 mM Dopamine | Dopamine hydrochloride: 0.038 g |
| Make fresh monthly. | 0.1 M Perchloric acid: 10 mL Bring volume to 100 mL with DI water |
| 8.8 mM Hydrogen Peroxide | 50 mL DI water: |
| Make fresh weekly. | Hydrogen peroxide: 500 μL |
| 70 mM potassium chloride | 100 mL DI water: |
| Make fresh day of experiment. | Potassium chloride: 0.08 g Sodium chloride: 0.07 g Calcium Chloride: 0.004 g |
| 200 μM Glutamate | 20 mM glutamate: 100 μL |
| Make fresh day of experiment. | Sterile saline: 10 mL |
Table 1: Calibration Solutions.
Discussion
The MEA technique allows for measurement of fast kinetics of neurotransmitter release and uptake in vitro and in vivo. Hence, the technology produces a wide variety of data output including tonic neurotransmitter levels, evoked neurotransmitter release, and neurotransmitter clearance. However, because use of MEAs is a relatively complex procedure, there are numerous factors that may need to be optimized for successful use. For example, during calibration, one may note that there are no signal waveforms (oscilloscope screen) or that responses to stimulation are minimal, or absent. Likely due to a system malfunction, restarting the recording system, or computer, may resolve the issue. Additionally, the problem could be a connection error. Before calibrating, it is pertinent to double-check that the MEA and reference electrode connections are connected properly. The replacement of the DIP socket on the headstage may resolve MEA connection issues. If the MEA is not completely in solution or a poorly maintained reference electrode is used, these may also interfere with the recording during calibration. Slowed signal responses during calibration is another common issue, which may result from a thicker than needed BSA/glutaraldehyde mixture, or a slowly spinning stir bar. In the event that interferents are giving a signal during calibration, it is possible that there was an error in electroplating (e.g. did not electroplate long enough) the mPD exclusion layer. In such a case, repeating the electroplating procedure is necessary.
During surgery, one anesthetic level may not be sufficient for all mice; some mice may require higher levels of anesthesia to "go under" and some mice may require less. It is essential to start at a low level of anesthesia and slowly raise it until the mouse is under light anesthesia. Deep anesthesia may be tested once the mouse is in the stereotaxic device by a tail or toe pinch. When choosing coordinates for implanting the MEA, it is also important to note and correct for possible brain atrophy that can occur in many neurodegenerative animal models36,37,38. In addition, it is imperative during surgery that blood does not accumulate on the MEA, as this can affect the recording properties of the MEA and result in slow temporal resolution or greatly minimized signal responses. If blood clots around the electrode, simply remove the MEA and rinse with saline.
During recording, one of the most common problems that can occur is that of a noisy signal. Interference from other electronic devices, such as auxiliary lighting, additional instruments, and drills are the mostly likely culprits, so it is best to avoid plugging them into the same electrical circuit and/or outlet as the recording system. Additionally, if the recording system is not grounded properly, one will observe oscillations on the oscilloscope of the recording system as well as noise stemming from all sites, and not just the recording sites. The recommended grounding mat under the recording system must be connected to a grounded pipe to avoid these issues. If it is determined that none of these are the error, the error may lie with the reference electrode, or the MEA itself. When encountering noise issues, using a new, unused MEA to verify the operation of the system can be a useful troubleshooting approach. If disconnecting or replacing the reference electrode results in little to no change, the reference electrode or half-cell may have malfunctioned. Changing the reference electrode may be the only way to resolve the problem. Once recording is complete, a final benefit of the MEA is the ability to clean and reuse them. MEAs can be re-used approximately three times as long as the MEA continues to perform adequately during calibration.
To conclude, though troubleshooting may occasionally be required, use of the MEA technique is advantageous for examining the fast release and clearance of neurotransmitter within desired sub-regions of the brain. The current studies focused on the use of the MEA technology in anesthetized TauP301L mice to take advantage of the MEA methodology for examining glutamate release and uptake in sub-regions of the hippocampal trisynaptic pathway. Future studies will address additional tonic and phasic issues in awake mice, as this has been very beneficial for linking behavioral events to the dynamic measures of tonic and phasic glutamate in awake rats and more recently, awake nonhuman primates. While this technology was developed for basic science studies, the future holds promise for the implementation of the recording technology for clinical research and intraoperative neurochemical monitoring for treatment and evaluation of brain disorders such as Parkinson's disease and epilepsy.
Disclosures
GG is the sole proprietor of Quanteon, LLC that makes the FAST-16 recording system used for glutamate measurements in this study. JEQ is a paid consultant for Quanteon.
Acknowledgments
This work was supported by the National Institute of General Medical Sciences (MNR; U54GM104942), NIA (MNR; R15AG045812), Alzheimer's Association (MNR; NIRG-12-242187), WVU Faculty Research Senate Grant (MNR), and WVU PSCOR Grant (MNR).
References
- Bito L, Davson H, Levin E, Murray M, Snider N. The concentrations of free amino acids and other electrolytes in cerebrospinal fluid, in vivo dialysate of brain, and blood plasma of the dog. J Neurochem. 1966;13(11):1057–1067. doi: 10.1111/j.1471-4159.1966.tb04265.x. [DOI] [PubMed] [Google Scholar]
- Cavus I, et al. Extracellular metabolites in the cortex and hippocampus of epileptic patients. Ann Neurol. 2005;57(2):226–235. doi: 10.1002/ana.20380. [DOI] [PubMed] [Google Scholar]
- Montgomery AJ, Lingford-Hughes AR, Egerton A, Nutt DJ, Grasby PM. The effect of nicotine on striatal dopamine release in man: A [11C]raclopride PET study. Synapse. 2007;61(8):637–645. doi: 10.1002/syn.20419. [DOI] [PubMed] [Google Scholar]
- Chefer VI, Thompson AC, Zapata A, Shippenberg TS. Overview of brain microdialysis. Curr Protoc Neurosci. 2009. Chapter 7 Unit 7.1. [DOI] [PMC free article] [PubMed]
- Hu S, Sheng WS, Ehrlich LC, Peterson PK, Chao CC. Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation. 2000;7(3):153–159. doi: 10.1159/000026433. [DOI] [PubMed] [Google Scholar]
- He X, et al. The association between CCL2 polymorphisms and drug-resistant epilepsy in Chinese children. Epileptic Disord. 2013;15(3):272–277. doi: 10.1684/epd.2013.0603. [DOI] [PubMed] [Google Scholar]
- Xie Y, et al. Resolution of High-Frequency Mesoscale Intracortical Maps Using the Genetically Encoded Glutamate Sensor iGluSnFR. J Neurosci. 2016;36(4):1261–1272. doi: 10.1523/JNEUROSCI.2744-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvin JS, et al. An optimized fluorescent probe for visualizing glutamate neurotransmission. Nat Methods. 2013;10(2):162–170. doi: 10.1038/nmeth.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefendehl JK, et al. Mapping synaptic glutamate transporter dysfunction in vivo to regions surrounding Abeta plaques by iGluSnFR two-photon imaging. Nat Commun. 2016;7:13441. doi: 10.1038/ncomms13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinzman JM, Thomas TC, Quintero JE, Gerhardt GA, Lifshitz J. Disruptions in the regulation of extracellular glutamate by neurons and glia in the rat striatum two days after diffuse brain injury. J Neurotrauma. 2012;29(6):1197–1208. doi: 10.1089/neu.2011.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas TC, Hinzman JM, Gerhardt GA, Lifshitz J. Hypersensitive glutamate signaling correlates with the development of late-onset behavioral morbidity in diffuse brain-injured circuitry. J Neurotrauma. 2011;29(2):187–200. doi: 10.1089/neu.2011.2091. Epub 2011 Dec 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinzman JM, et al. Diffuse brain injury elevates tonic glutamate levels and potassium-evoked glutamate release in discrete brain regions at two days post-injury: an enzyme-based microelectrode array study. J Neurotrauma. 2010;27(5):889–899. doi: 10.1089/neu.2009.1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens ML, Quintero JE, Pomerleau F, Huettl P, Gerhardt GA. Age-related changes in glutamate release in the CA3 and dentate gyrus of the rat hippocampus. Neurobiol Aging. 2009;32(5):811–820. doi: 10.1016/j.neurobiolaging.2009.05.009. Epub 2009 Jun 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickell J, Salvatore MF, Pomerleau F, Apparsundaram S, Gerhardt GA. Reduced plasma membrane surface expression of GLAST mediates decreased glutamate regulation in the aged striatum. Neurobiol Aging. 2006;28(11):1737–1748. doi: 10.1016/j.neurobiolaging.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Hascup ER, et al. An allosteric modulator of metabotropic glutamate receptors (mGluR(2) ) (+)-TFMPIP, inhibits restraint stress-induced phasic glutamate release in rat prefrontal cortex. J Neurochem. 2012;122(2):619–627. doi: 10.1111/j.1471-4159.2012.07784.x. Epub 02012 Jun 07713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford EC, Pomerleau F, Huettl P, Stromberg I, Gerhardt GA. Chronic second-by-second measures of L-glutamate in the central nervous system of freely moving rats. J Neurochem. 2007;102(3):712–722. doi: 10.1111/j.1471-4159.2007.04596.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matveeva EA, et al. Reduction of vesicle-associated membrane protein 2 expression leads to a kindling-resistant phenotype in a murine model of epilepsy. Neuroscience. 2011;202:77–86. doi: 10.1016/j.neuroscience.2011.11.055. Epub 2011 Dec 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matveeva EA, et al. Kindling-induced asymmetric accumulation of hippocampal 7S SNARE complexes correlates with enhanced glutamate release. Epilepsia. 2012;53(1):157–167. doi: 10.1111/j.1528-1167.2011.03345.x. Epub 02011 Dec 0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunsberger HC, Rudy CC, Batten SR, Gerhardt GA, Reed MN. P301L tau expression affects glutamate release and clearance in the hippocampal trisynaptic pathway. J Neurochem. 2015;132(2):169–182. doi: 10.1111/jnc.12967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunsberger HC, et al. Riluzole rescues glutamate alterations, cognitive deficits, and tau pathology associated with P301L tau expression. J Neurochem. 2015;135(2):381–394. doi: 10.1111/jnc.13230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunsberger HC, et al. Peripherally restricted viral challenge elevates extracellular glutamate and enhances synaptic transmission in the hippocampus. J Neurochem. 2016. [DOI] [PMC free article] [PubMed]
- Obrenovitch TP, Urenjak J, Zilkha E, Jay TM. Excitotoxicity in neurological disorders--the glutamate paradox. Int J Dev Neurosci. 2000;18(2-3):281–287. doi: 10.1016/s0736-5748(99)00096-9. [DOI] [PubMed] [Google Scholar]
- Hillered L, Vespa PM, Hovda DA. Translational neurochemical research in acute human brain injury: the current status and potential future for cerebral microdialysis. J Neurotrauma. 2005;22(1):3–41. doi: 10.1089/neu.2005.22.3. [DOI] [PubMed] [Google Scholar]
- Burmeister JJ, Gerhardt GA. Self-referencing ceramic-based multisite microelectrodes for the detection and elimination of interferences from the measurement of L-glutamate and other analytes. Anal Chem. 2001;73(5):1037–1042. doi: 10.1021/ac0010429. [DOI] [PubMed] [Google Scholar]
- Burmeister JJ, et al. Improved ceramic-based multisite microelectrode for rapid measurements of L-glutamate in the CNS. J Neurosci Methods. 2002;119(2):163–171. doi: 10.1016/s0165-0270(02)00172-3. [DOI] [PubMed] [Google Scholar]
- Diamond JS. Deriving the glutamate clearance time course from transporter currents in CA1 hippocampal astrocytes: transmitter uptake gets faster during development. J Neurosci. 2005;25(11):2906–2916. doi: 10.1523/JNEUROSCI.5125-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene JG, Borges K, Dingledine R. Quantitative transcriptional neuroanatomy of the rat hippocampus: evidence for wide-ranging, pathway-specific heterogeneity among three principal cell layers. Hippocampus. 2009;19(3):253–264. doi: 10.1002/hipo.20502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borland LM, Shi G, Yang H, Michael AC. Voltammetric study of extracellular dopamine near microdialysis probes acutely implanted in the striatum of the anesthetized rat. J Neurosci Methods. 2005;146(2):149–158. doi: 10.1016/j.jneumeth.2005.02.002. Epub 2005 Mar 2005. [DOI] [PubMed] [Google Scholar]
- Jaquins-Gerstl A, Michael AC. Comparison of the brain penetration injury associated with microdialysis and voltammetry. J Neurosci Methods. 2009;183(2):127–135. doi: 10.1016/j.jneumeth.2009.06.023. Epub 2009 Jun 2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azbill RD, Mu X, Springer JE. Riluzole increases high-affinity glutamate uptake in rat spinal cord synaptosomes. Brain Res. 2000;871(2):175–180. doi: 10.1016/s0006-8993(00)02430-6. [DOI] [PubMed] [Google Scholar]
- Gourley SL, Espitia JW, Sanacora G, Taylor JR. Antidepressant-like properties of oral riluzole and utility of incentive disengagement models of depression in mice. Psychopharmacology (Berl) 2011;219(3):805–814. doi: 10.1007/s00213-011-2403-4. Epub 2011 Jul 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frizzo ME, Dall'Onder LP, Dalcin KB, Souza DO. Riluzole enhances glutamate uptake in rat astrocyte cultures) Cell Mol Neurobiol. 2004;24(1):123–128. doi: 10.1023/B:CEMN.0000012717.37839.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli E, Funicello M, Rauen T, Gobbi M, Mennini T. Riluzole enhances the activity of glutamate transporters GLAST, GLT1 and EAAC1. Eur J Pharmacol. 2008;578(2-3):171–176. doi: 10.1016/j.ejphar.2007.10.023. Epub 2007 Oct 2025. [DOI] [PubMed] [Google Scholar]
- Day BK, Pomerleau F, Burmeister JJ, Huettl P, Gerhardt GA. Microelectrode array studies of basal and potassium-evoked release of L-glutamate in the anesthetized rat brain. J Neurochem. 2006;96(6):1626–1635. doi: 10.1111/j.1471-4159.2006.03673.x. Epub 2006 Jan 1625. [DOI] [PubMed] [Google Scholar]
- Kane RL, Martinez-Lopez I, DeJoseph MR, Vina JR, Hawkins RA. Na(+)-dependent glutamate transporters EAAT1, EAAT2, and EAAT3) of the blood-brain barrier. J Biol Chem. 1999;274(45):31891–31895. doi: 10.1074/jbc.274.45.31891. [DOI] [PubMed] [Google Scholar]
- Ramsden M, et al. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L) J Neurosci. 2005;25(46):10637–10647. doi: 10.1523/JNEUROSCI.3279-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39(3):409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Zang DW, et al. Magnetic resonance imaging reveals neuronal degeneration in the brainstem of the superoxide dismutase 1 transgenic mouse model of amyotrophic lateral sclerosis. Eur J Neurosci. 2004;20(7):1745–1751. doi: 10.1111/j.1460-9568.2004.03648.x. [DOI] [PubMed] [Google Scholar]