

Abstract
Transmission Electron Microscopy (TEM) is an extraordinary tool for studying cell ultrastructure, in order to localize proteins and visualize macromolecular complexes at very high resolution. However, to get as close as possible to the native state, perfect sample preservation is required. Conventional electron microscopy (EM) fixation with aldehydes, for instance, does not provide good ultrastructural preservation. The slow penetration of fixatives induces cell reorganization and loss of various cell components. Therefore, conventional EM fixation does not allow for an instantaneous stabilization and preservation of structures and antigenicity. The best choice for examining intracellular events is to use cryofixation followed by the freeze-substitution fixation method that keeps cells in their native state. High-pressure freezing/freeze-substitution, which preserves the integrity of cellular ultrastructure, is the most commonly used method, but requires expensive equipment. Here, an easy-to-use and low-cost freeze fixation method followed by freeze-substitution for suspension cell cultures is presented.
Keywords: Cellular Biology, Issue 123, Plunge freezing, freeze-substitution, transmission electron microscopy, yeasts, parasites, bacteria, ultrastructure, immunolabeling
Introduction
Sample preparation is critical for the success of any electron microscopy study. Conventional EM fixation was the primary method for fixing tissue or cells for transmission electron microscopy (TEM)1. First, aldehydes and osmium tetroxide are used to chemically fix the material at room temperature. Then, the material is dehydrated with organic solvents, infiltrated, and embedded in epoxy resin. This method is dependent upon the penetration rate of fixatives into the cell. Consequently, the artifacts and extraction of cellular contents are usually observed2.
Cryofixation is clearly a better alternative for the preservation of cellular structures3, keeping them intact. The highest quality of TEM images4 in thin resin sections can be obtained using the cryofixation/freeze substitution method. The aim of this technique is to obtain vitrified biological samples without ice crystals formation or containing ice crystals small enough not to damage the ultrastructure of the cells. High-pressure freezing (HPF) and ultra-rapid cryofixation, which is also called plunge freezing (PF), are two methods to cryofix samples. HPF immobilizes molecules in the cell instantaneously and avoids the damage caused by conventional EM fixation. Several types of freezing machines and automatic substitution devices have been developed5. The freezing machines and consumables (liquid nitrogen, specimen carriers, etc.) are expensive, but they allow for producing high-quality electron micrographs6,7. PF is a technique that was used in the early 1950s and is described in the literature as simple and cheap8.During the PF procedure, the prepared sample is frozen at a rapid rate to obtain ice crystals size less than 3 to 5 nm. To this end, the sample is plunged into a liquid cryogen, such as ethane, propane, or an ethane-propane mixture. Since the 1950s, improvements of PF have been done to make this technique available to a greater number of users. High-pressure freezing is currently the only viable way to freeze a large variety of samples thicker than 50 µm (up to a thickness of 200 µm for disc-shaped samples)5, whereas PF is widely used to image small objects (<100 nm), such as macromolecular complexes suspended in a thin film of amorphous ice9. Larger samples, for example eukaryotic cells, can be cryofixed by PF, but requires specimen holders, such as capillary copper tubes or sandwich systems8,10,11.
Here, a rapid, easy-to-use and low-cost plunge freezing/freeze-substitution technology usable for various suspension cell cultures is presented.
Protocol
1. Preparation of Formvar Grid Film
NOTE: Perform chloroform manipulation under a fume hood using personal protective equipment (gloves, lab coat, glasses). Use 400 mesh electron microscopy copper grids. With other grid types (other mesh sizes, gold and nickel grids), the quality of freezing is worse.
Prepare a 100 mL solution of 0.3% formvar in chloroform. Allow it to dissolve overnight without agitation.
Put 400 mesh (number of holes per square inch)electron microscopycopper grids (diameter 3.05 mm) in a glass vial (height 3 cm and diameter 2 cm) containing acetone. Stir the glass vial with a circular movement of the hand. Remove the acetone with a plastic transfer pipette. Allow the solvent to evaporate for 5 min. Transfer the grids by pouring them onto a filter paper and allow them to dry for 5 min.
Polish a 76 x 26 mm2 glass slide by wiping it with a lint-free cloth until shiny/slippery.
Take a crystallizer (diameter 15 cm, height: 7 cm and capacity 1200 mL) and fill it to the brim with distilled water. Clean the water surface by passing a glass bar over the surface twice to remove dust.
Hold the glass slide between two fingers and dip it in the formvar solution for a few seconds. Hold it upright to dry under an upside-down beaker for 5 min.
When the film is dry, score the edges of the glass slide with a sharp razor blade to define the limits of the film to be dislodged from the slide.
- Take a deep breath and exhale heavily onto the slide to get a layer of water vapor on and under the film to render the film slightly opaque and enhance its visibility.
- Immediately float the film onto the crystallizer water surface by touching the bottom of the slide with an angle of about 30°. Let the film release from the slide and peel it off onto the crystallizer water surface. Gently pull the film off with the tweezers, if necessary.
Gently lay the grids face down on the film floating on the water surface in the regions that are of the proper thickness (about 10 nm) and without wrinkles.
- Pick up the film by covering the grids with Joseph paper as they float on the crystallizer water surface.
- Holding the Joseph paper with one hand, touch one end of the Joseph paper to one end of the film. Wait until the Joseph paper is completely wet. Remove the Joseph paper with the grids stuck on.
Place the grids for 24 h in a covered Petri dish for final drying and storage prior to use at room temperature.
2. Preparation of Freeze-substitution Medium
NOTE: Osmium tetroxide (OsO4) and uranyl acetate are hazardous chemicals. Handle them in the fume hood while wearing appropriate personal protective equipment (PPE) including a lab coat and gloves. Follow the safety warnings for handling (OsO4) and uranyl acetate. Use O-ring cryovials to avoid the leakage of OsO4.
- Ultrastructural studies
- Put glass OsO4 ampoule in a glass flask.
- Break the ampoule by shaking the flask.
- Add an appropriate volume of acetone 100% to obtain a 4% final concentration.
- Stir and dispense 1.5 mL aliquots into 1.8 mL cryovials.
- Protein immunolabeling
- Place 0.05 g of uranyl acetate in a glass flask.
- Add 50 ml of 100% acetone.
- Stir the solution with a magnetic stirrer. When the solution becomes clear, filter it using a 0.2 µm filter.
- Dispense 1.5 mL aliquots into 1.8 mL cryovials. NOTE: Prepare the cryovials containing freeze-substitution medium prior to the plunge freezing process and keep them in -84 °C freezer. Invert the glass flask to throw away the OsO4 ampoule in the hazardous waste.
3. Preparation of Cells
NOTE: This step is critical for the success of the method. For all cell types, grow cells to obtain the indicated number of cells. Centrifuge in the indicated tube at the specified time and speed.
- To obtain the appropriate consistency of the pellets from different cell types, grow cells as follows:
- For Saccharomyces cerevisiae and Schizosaccharomyces pombe, inoculate yeast in 5 mL of minimal or complete liquid medium. Incubate overnight at 28 °C, rotating (180 rpm). Measure the optical density at 600 nm (OD600) of the overnight culture. Dilute yeast cells to an OD600 of 0.2 in 50 ml fresh selective medium. Continue to incubate cells at 28 °C, rotating, to obtain 1 x 109 yeast cells7.
- For Leishmania flagellum, inoculate parasites in 5 mL of AM medium with 7.5% FCS (fetal calf serum). Incubate overnight at 24 °C, rotating (180 rpm). Measure the OD600 of the overnight culture. Dilute parasite cells to an OD600 of 0.2 in 50 ml fresh selective medium. Continue to incubate cells at 24 °C, rotating, to obtain 5 x 108 parasite cells12.
- For Trypanosoma brucei, inoculate parasites in 5 mL of SDM-79 medium with 10% FCS and 30 µg/mL hygromycin. Incubate overnight at 27 °C, rotating (180 rpm). Measure the OD600 of the overnight culture. Dilute parasite cells to an OD600 of 0.2 in 50 mL fresh selective medium. Continue to incubate cells at 27 °C, rotating, to obtain 5 x 108 parasite cells13.
- For Escherichia coli, inoculate bacteria in 5 ml of DYT medium with 100 µg/mL ampicillin. Incubate overnight at 37 °C, rotating (180 rpm). Measure the OD600 of the overnight culture. Dilute bacteria cells to an OD600 of 0.2 in 50 ml fresh selective medium. Continue to incubate cells at 37 °C, rotating, to obtain 5 x 1010 bacteria cells14. NOTE: Following overnight incubation, cells in culture may be in either logarithmic or stationary growth phase. Very slow-growing cell strains may require incubation times longer than one night, or inoculation of a greater number of cells, as determined empirically.
For all cells types, transfer the culture medium containing the cells to 50 ml polypropylene tube and centrifuge for 3 min at 1,500 x g.
Remove the supernatant and resuspend the pellet of all cell types in 1 ml of the relevant cell culture medium.
Centrifuge all cell types in a microcentrifuge tube for 1 min at 3,900 x g. Completely remove the supernatant.
Keep the cells on ice.
4. Liquefaction of Propane
Note: Handle liquid nitrogen with care. Use personal protective equipment including cryogloves and googles. Propane is potentially explosive, so perform the liquefaction in a well-ventilated room or under a fume hood. No open flames are allowed.
Put about 150 mL of liquid nitrogen in a polystyrene tray. Place a brass cup (height 3.6 cm, diameter 2 cm and thickness 1.5 mm) in liquid nitrogen to freeze it. Do not let the liquid nitrogen penetrate into the brass cup.
Wait until the bubbles subside to be sure that the brass cup is frozen.
Put the hose connected to the propane cylinder in contact with the brass cup wall.
Gently open the propane cylinder valve. NOTE: At this step, the propane liquefies in the brass cup.
Stop the liquefaction by closing the propane cylinder valve and rapidly removing the gas hose.
Fill the polystyrene tray with liquid nitrogen up to 5 mm from the top of the brass cup. Do not let the liquid nitrogen penetrate into brass cup.
5. Plunge Freezing of the Sample
Put about 200 mL of liquid nitrogen in polystyrene tray. Freeze little brass cups (height 1.5 cm diameter 2.5 cm and thickness 1 mm) by putting them in liquid nitrogen. Put one cup for one suspension cell sample. Be careful not to mix samples. To this end, put sample 1 in cup 1, sample 2 in cup 2, etc.
Freeze the tweezers by plunging their tip in liquid nitrogen.
Plunge a double edge dissecting needle in the pellet obtained in 3.5.
Using tweezers, take a 400 mesh copper microscopy grid coated with formvar prepared in section 1.
Using the dissecting needle, place a drop of the pellet obtained in 3.5 on the grid. Try different drop sizes (e.g. 2, 5 and 10 µL).
Very rapidly plunge the grid held by the tweezers into liquid propane generated in section 4 and stir the grid with circular movement for a few seconds.
Rapidly transfer the frozen grid with the tweezers to the little brass cup frozen in section 5.1. NOTE: For each sample, make at least 3 grids. Always freeze the tweezers before taking a copper grid.
6. Freeze-substitution
NOTE: Osmium tetroxide is potentially volatile, so use an air purifying respirator with vapor cartridges. Freezing the fixative in liquid nitrogen before plunge freezing is also a solution.
For ultrastructure studies, open the 1.8 mL cryovial tubes containing freeze-substitution medium prepared in section 2.1. For immunolocalization studies, open the 1.8 mL cryovial tubes containing freeze-substitution medium prepared in section 2.2. Place the cap on the cryovials.
Freeze the tweezers by plunging their tip in liquid nitrogen.
Rapidly remove the cap and transfer the frozen grids into the cryovials using the tweezers.
Put the cap back on the cryovials.
Repeat the operation for each grid of each sample.
Close all the caps and stir the cryovials with a circular movement of the hand to ensure the samples are in the freeze-substitution medium.
Place the cryovials in an airtight box for 3 days in a -84 °C freezer during the freeze-substitution process.
7. Sample Warming
- Ultrastructure studies
- Carry the cryovials in a polystyrene tray (to avoid a sudden rise in temperature) to a -30 °C freezer. Place them in the -30 °C freezer for 1 h.
- Place the cryovials in a -15 °C freezer for 2 h. Place the samples at 4 °C for 2 h and then for 30 min at room temperature.
- Remove the freeze substitution medium with a plastic transfer pipette. Add approximately 500 µL of 100% acetone.
- Stir the 1.8 ml cryovial with a circular movement of the hand and rapidly pour the acetone (1.5 mL) containing the grids in a glass vial (height 3 cm and diameter 2 cm).
- Rinse the same cryovial with 1 mL of 100% acetone to be sure to have transferred all the grids. Stir the cryovial with a circular movement of the hand and pour the content of the cryovial into a glass vial.
- Rinse each glass vial with 3 mL of 100% acetone 3 times for 10 min.
- Follow the embedding and inclusion procedures as described in reference15. Impregnate the sample with increasing concentrations of epoxy resin in acetone (25%, 50%, and 75%) and embed the sample with 100% epoxy resin in gelatin capsules.
- Immunolabeling studies
- Carry the 1.8 mL cryovials in a polystyrene tray (to avoid a sudden rise in temperature) to the -30 °C freezer.
- Place the cryovials for 2 h in a -30 °C freezer.
- In a -30 °C freezer, stir the cryovial with a circular movement of the hand and rapidly pour the freeze substitution medium in a polypropylene vial (height 5 cm and diameter 1.5 cm).
- Replace the freeze substitution medium with 2 ml of fresh freeze-substitution medium.
- Place the polypropylene vial for 2 h in a -30°C freezer.
- Rinse the polypropylene vial for 1 h with 2 mL of 100% acetone and three times for 1 h with 2 mL of 100% ethanol.
- Follow the embedding and inclusion procedures as described in reference15. Impregnate the sample with increasing concentrations of acrylic resin (25%, 50%, and 75% in acetone) and embed the sample with 100% acrylic resin in gelatin capsules.
8. Visualization of Samples
Representative Results
In this article, an easy-to-use and low-cost plunge freezing method for ultrastructural (Figures 1 and Figure 2) and immunolabeling (Figure 3) studies is presented. We demonstrate that it is not necessary to have special equipment for the freeze-substitution procedure and that the warming procedure takes less than 6 h instead of the 24 h using a dedicated system.
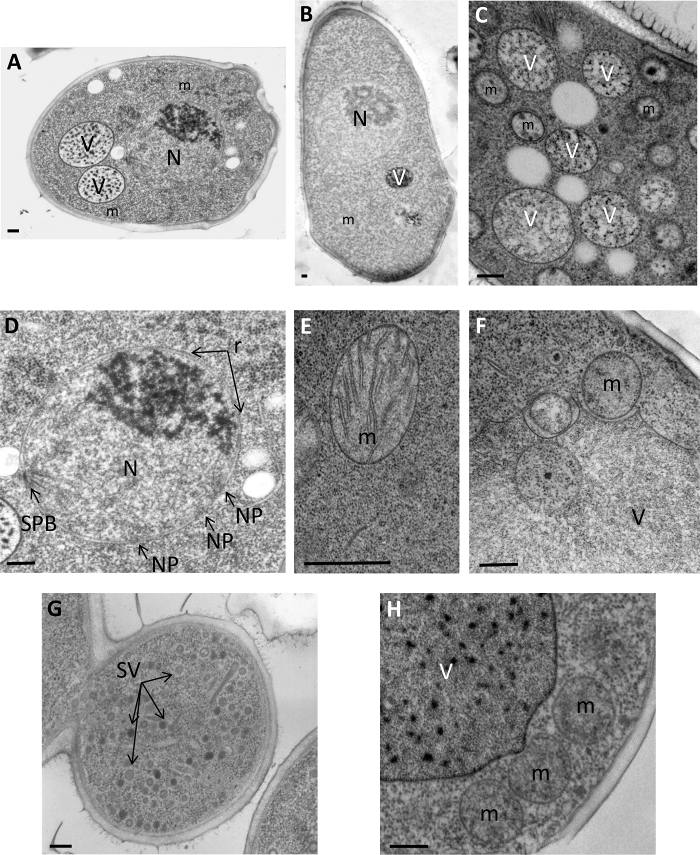
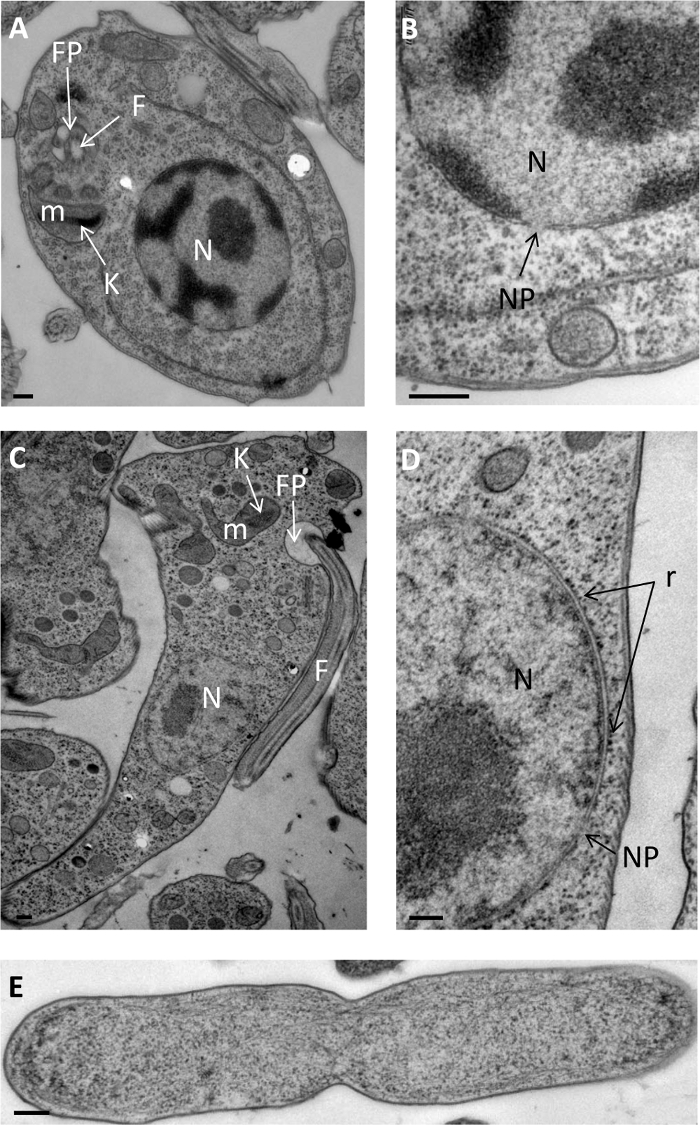
PF/freeze-substitution method resulted in the improved preservation of the intracellular structures of Saccharomyces cerevisiae (Figure 1A and 1D-H), Schizosaccharomyces pombe (Figure 1B andC), Leishmania flagellum (Figure 2 A and B), Trypanosoma brucei (Figure 2 C and D), and Escherichia coli (Figure 2E). The structures are not shrunken and the cytoplasm appears dense. Moreover, the membranes are smooth, which is proof of good preservation. All of the organelles, such as nucleus (Figure 1A, B D and Figure 2A-D), mitochondria (Figure 1A-C, E and H), and vacuoles (Figure 1A, C, F and H), can be perfectly identified. In the nucleus, a double membrane forming nuclear envelope, nuclear pore, spindle pole body, and ribosomes attached on the outer membrane of the nuclear envelope are distinctly visible (Figure 1D and Figure 2B, 2D). In the mitochondria, the internal membrane invaginations, called cristae (Figure 1E), are clearly visible. The vacuoles are critical organelles because large ice crystals can develop easily inside them. As illustrated in Figure 1, vacuoles are perfectly preserved without ice crystal damage (Figure 1A, C, F, and H). Finally, as the ultrastructure is well preserved, different biological events can be observed and analyzed, such as autophagy (Figure 1C, F and H), mitochondrial morphology (Figure 1E), secretory vesicles (Figure 1G), and nuclear division (Figure 1A, Figure 2B, and 2C).
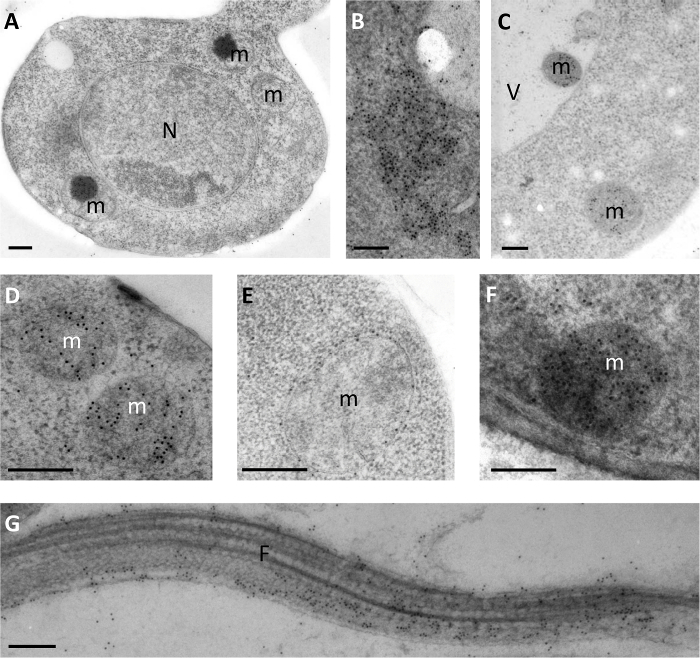
In situ localization of proteins with specific antibodies is one of the most powerful and popular techniques in cell biology. PF can be also used as a method to perform protein immunolocalization. This procedure preserves protein antigenicity and, as mentioned hereinabove, organelle identity. Different antibodies targeting different yeast proteins, such as anti-ATP synthase7,15 (Figure 3A, 3C and 3D), anti-GFP (Figure 3B), anti-porine16 (Figure 3E), and anti-fox217 (Figure 3F), have already been tested. It is also possible to perform these experiments on different organisms, such as Leishmania12.
Figure 1: TEM images of ultrastructural studies of yeastsSaccharomyces cerevisiae and Schizosaccharomyces pombe. (A) Overview of S. cerevisiae. (B) Overview of S. pombe. (C) Part of S. pombe cytoplasm. (D) High magnification of a nucleus. (E) Detail of a mitochondrion with cristae clearly visible. (F) Vacuolar morphology during autophagy. (G) Yeast bud with secretion vesicles inside. (H) Example of intimate contacts between vacuoles and mitochondria during the autophagy process. N, nucleus; V, vacuole; m, mitochondrion; SV, secretion vesicles; r, ribosomes. Scale bars = 200 nm. Please click here to view a larger version of this figure.
Figure 2: TEM images of ultrastructural studies ofLeishmania amazonesis, Trypanosoma brucei and E. coli. (A) Overview of Leishmania. (B) Detail of Leishmania cytoplasm with nuclear pore and endoplasmic reticulum. (C) Overview of T. brucei. (D) High magnification of T. brucei nucleus. (E) Overview of E. coli cell. N: nucleus; V, vacuole; m, mitochondrion; NP, nuclear pore; F, flagellum; FP, flagellar pocket; K, kinetoplast; r, ribosomes. Scale bars = 200 nm. Please click here to view a larger version of this figure.
Figure 3: Examples of protein labeling using monoclonal and polyclonal antibody. (A) Overview of S. cerevisiae. (B) Immunolabeling of amyloid-β with anti-GFP antibodies. (C-D) Example of mitochondria labeling with anti-ATP synthase antibodies. (E) Porin localization in external membrane of a mitochondrion. (F) Fox2 protein is detected in mitochondrion by immunolabeling. (G) Localization of LdFlabarin in a longitudinal section of a flagellum of Leishmania amazonesis. All of the antibodies are detected using 10 nm gold polyclonal antibodies. N, nucleus; V, vacuole; m, mitochondrion. Scale bars = 200 nm. Please click here to view a larger version of this figure.
Discussion
TEM is a powerful method for the ultrastructural observation of organelles, cells, and tissues. Cryofixation/freeze-substitution is currently the best method for the preservation of both ultrastructure and protein antigenicity. Chemical fixatives penetrate and act very slowly thereby allowing structural rearrangements before the complete stabilization of the ultrastructure2. Conversely, cryofixation/freeze-substitution instantly stabilizes cellular structures4. However, cryofixation/freeze-substitution techniques include a number of constraints and difficulties thereby limiting their range of application. This concerns sample, freezing artifacts, realization of the technique, cost, and equipment. The high-pressure freezing/freeze-substitution technique has enabled a high degree of preservation of cellular details 6,7. In contrast, PF is already used for observing small objects suspended on thin films of amorphous ice9. Here, PF/freeze-substitution method for ultrastructural and immunolabeling studies provides high quality images of yeasts, parasites, and bacterial ultrastructure comparable to high pressure freezing images.
Fixation quality depends on cooling rates18. At high cooling rates, water is in the vitreous state and does not show visible crystalline structure at the electron microscopy level. The vitrification occurs solely in a very thin superficial layer (200 µm in general) and, therefore, good thickness freezing will gradually decrease from the surface of the specimen to the inner layers4. In plant cells, waxy coating or spaces filled with gas can slow the cooling rates. With PF, surprisingly, no decrease in the quality of freezing performance was observed. In all of the experiments, the percentage of well frozen cells is greater than 50%. This percentage varies between 50% to more than 90% and is dependent on various parameters (culture medium, physiological status of the cells, outdoor temperature, and moisture level). As stated above, the formation of ice crystals destroys the cell structure and ice crystals are particularly visible in vacuoles. In the PF/freeze substitution process, special attention must be paid to the freezing temperature. The freeze substitution step must not be performed above -82 °C, otherwise, the vitreous water will be transformed into crystalline water. To avoid the formation of ice crystal damage, cryoprotectant is currently used, but that leads to alterations in the ultrastructure of the cells19. Yeast, bacteria, and parasite culture mediums might contain a component that is cryoprotective agent. In this context, it was decided not to use additional cryoprotectant in order to be as close as possible to the actual cellular state.
The success of PF/freeze substitution depends on many critical steps during the development phase of the method. First, a critical point is to obtain the appropriate consistency of the pellets. The pellet obtained after centrifugation must be compact enough to be rolled out on an electron microscopy grid. If the pellet is too compact, the freeze-substitution solution cannot penetrate the drop, and the ultrastructure will not be well-preserved. Conversely, if the pellet is not compact enough, the leftover culture medium may develop ice crystals and impair the cell ultrastructure. To obtain the appropriate consistency of the pellets from the different cell types, it is necessary to respect the culture conditions to obtain the indicated number of cells and centrifugation parameters indicated in the protocol (see above) for all cell types. However, the amount of the material put on the copper grid is not critical. Different droplet sizes can be tested. Second, the grid type that is used as a support is important. Several gold, nickel, and copper grids with different mesh sizes were tested. The best results were obtained with a 400 mesh copper grid with thin bars. Third, with regards to the freezing step, a brass cup is preferred to liquefy the propane. Liquid propane in copper cup tends to freeze and, therefore, complicates the experiment. Finally, the manipulator must work quickly and in cold conditions to avoid accidental sample warm-up. If it happens, the sample must be discarded. For example, to prevent sample warming, the tweezers must be pre-cooled before using them and the freeze substitution medium must be prepared well ahead before the procedure. Chilled tweezers must also be used during the plunge freezing process becauseif the plunge freezing is realized without freezing the tweezers, a lot of smoke forms as the tweezers are plunged in the propane (due to the heat/cold exchange). This inevitably has an impact on the cooling rates. Tests with samples frozen with unfrozen tweezers appear not to be properly frozen.
Moreover, use of hazardous chemicals such as OSO4 and uranyl acetate in the freeze-substitution medium requires, during the preparation of solutions and freeze-substitution process, to work under the fume hood with adequate PPE. To prevent the leakage of OsO4 and inhalation of vapors, cryovials must have a hard O-ring to ensure that the tubes remain sealed during freeze-substitution process. To prevent unnecessary exposure to osmium vapor during the preparation of freeze-substitution medium, the OsO4 ampoule can be broken in a glass flask. The presence of glass shards in the resin block after inclusion can be avoided by using fine-tip transfer pipettes during acetones rinses and steps of embedding. Performing the inclusion step under a binocular loop and the realization of semi-thin sections also allows for verifying that there are no glass shards. When the experiments cannot be conducted under a fume hood (particularly in the first step of the freeze substitution process), use an air purifying respirator with vapor cartridges. The manipulator must also be careful when working in cold conditions with liquid nitrogen or during freezing steps. The use of gloves and glasses are recommended. Furthermore, propane is potentially explosive, so the liquefaction must be performed in a well-ventilated room or under a fume hood and no open flames are allowed.
Several articles showed that, on a wide variety of samples such as brain tissue, ivy leaf, or root tips of A. thaliana and N. benthamiana, the preservation of the ultrastructure is improved with the HPF technique compared to chemical fixation20,21. With PF, the experiments were performed on cells in suspension. Therefore, there is no indication of the success of this technique on larger specimens like tissues. Further experiments need to be conducted to address this point. However, the method has been successfully tested with filamentous fungus Podospora anserina that is not isolated cells but is rather of mycelium form22.
The method presented in this article represents a significant improvement over the current methods of chemical fixation or HPF. For instance, PF does not require expensive equipment or consumables in contrast to HPF23. Care must also be taken because the HPF technique uses a large amount of liquid nitrogen (about 80 L per experiment). With the technique proposed, one major advantage is that no important equipment is necessary23. Thus, the cost of an experiment is much lower (about one hundred times cheaper). Another advantage is that no particular expertise is needed because the technique is easy to use. Moreover, the sample processing of PF/freeze-substitution is much shorter than HPF/freeze-substitution due to the warming process that takes less than 6 h instead of 24 h24. It is not more time consuming than the conventional chemical fixation. The advantage is that the cellular ultrastructure is perfectly preserved with minimum artifacts and is much closer to the cells' original state. The application of this technology will certainly be of use to study different rapid biological events or to localize proteins at high resolution for cells grown in suspension and for samples of a few micrometers thickness, such as fungal hyphae22.
Disclosures
The authors declare that they have no conflicts of interest.
Acknowledgments
We express our gratitude to M. Bouchecareilh, E. Tétaud, S. Duvezin-Caubet, and A. Devin for their help and comments on the manuscript. We are grateful to the electronic imaging pole of Bordeaux Imaging Centre where the images were taken. This work was supported by Centre National de la Recherche Scientifique.
References
- Palade GE, Porter KR. Studies on the endoplasmic reticulum. I. Its identification in cells in situ. J Exp Med. 1954;100(6):641–656. doi: 10.1084/jem.100.6.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger E, Johansen R, Maeder M, Bohrmann B, Stauffer E, Villiger W. Artefacts and morphological changes during chemical fixation. J Microsc. 1992;168(Pt 2):181–201. doi: 10.1111/j.1365-2818.1992.tb03260.x. [DOI] [PubMed] [Google Scholar]
- Studer D, Humbel BM, Chiquet M. Electron microscopy of high pressure frozen samples: bridging the gap between cellular ultrastructure and atomic resolution. Histochem Cell Biol. 2008;130(5):877–889. doi: 10.1007/s00418-008-0500-1. [DOI] [PubMed] [Google Scholar]
- McDonald KL. Out with the old and in with the new: rapid specimen preparation procedures for electron microscopy of sectioned biological material. Protoplasma. 2014;251:429–448. doi: 10.1007/s00709-013-0575-y. [DOI] [PubMed] [Google Scholar]
- Studer D, Graber W, Al-Amoudi A, Eggli P. A new approach for cryofixation by high-pressure freezing. J Microsc. 2001;203(Pt 3):285–294. doi: 10.1046/j.1365-2818.2001.00919.x. [DOI] [PubMed] [Google Scholar]
- Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006;4(12):2311–2324. doi: 10.1371/journal.pbio.0040423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissová I, Salin B, Schaeffer J, Bhatia S, Manon S, Camougrand N. Selective and non-selective autophagic degradation of mitochondria in yeast. Autophagy. 2007;3(4):329–336. doi: 10.4161/auto.4034. [DOI] [PubMed] [Google Scholar]
- Gilkey JC, Staehling A. Advances in ultrarapid freezing for the preservation of cellular ultrastructure. Microsc Res Tech. 1986;3(2):177–210. [Google Scholar]
- Nogales E, Scheres SH. Cryo-EM: A Unique Tool for the Visualization of Macromolecular Complexity. Mol Cell. 2015;58(4):677–689. doi: 10.1016/j.molcel.2015.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba M. Electron microscopy in yeast. Methods Enzymol. 2008;451:133–149. doi: 10.1016/S0076-6879(08)03210-2. [DOI] [PubMed] [Google Scholar]
- Leunissen JLM, Yi H. Self-pressurized rapid freezing (SPRF): a novel cryofixation method for specimen preparation in electron microscopy. J Microsc. 2009;235(1):25–35. doi: 10.1111/j.1365-2818.2009.03178.x. [DOI] [PubMed] [Google Scholar]
- Lefebvre M, et al. LdFlabarin a new BAR domain membrane protein of Leishmania flagellum. PLoS One. 2013;8(9):1–12. doi: 10.1371/journal.pone.0076380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetaud E, et al. TbFlabarin, a flagellar protein of Trypanosoma brucei, highlights differences between Leishmania and Trypanosoma flagellar-targeting signals. Exp Parasitol. 2016;166:97–107. doi: 10.1016/j.exppara.2016.04.004. [DOI] [PubMed] [Google Scholar]
- Wasmer C, et al. Solid-state NMR spectroscopy reveals that E coli inclusion bodies of HET-s(218-289) are amyloids. Angew Chem Int Ed Engl. 2009;48(26):4858–4860. doi: 10.1002/anie.200806100. [DOI] [PubMed] [Google Scholar]
- Lefebvre-Legendre L, et al. Failure to assemble the alpha 3 beta 3 subcomplex of the ATP synthase leads to accumulation of the alpha and beta subunits within inclusion bodies and the loss of mitochondrial cristae in Saccharomyces cerevisiae. J Biol Chem. 2005;280(18):18386–18389. doi: 10.1074/jbc.M410789200. [DOI] [PubMed] [Google Scholar]
- Paumard P, et al. The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J. 2002;21(3):221–230. doi: 10.1093/emboj/21.3.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel F, et al. A Fox2-dependent fatty acid ß-oxidation pathway coexists both in peroxisomes and mitochondria of the ascomycete yeast Candida lusitaniae. PLoS One. 2014;9(12):1–27. doi: 10.1371/journal.pone.0114531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubochet J. The physics of rapid cooling and its implications for cryoimmobilization of cells. Methods Cell Biol. 2007;79:7–21. doi: 10.1016/S0091-679X(06)79001-X. [DOI] [PubMed] [Google Scholar]
- Meryman HT. Cryopreservation of living cells: principles and practice. Transfusion. 2007;47(5):935–945. doi: 10.1111/j.1537-2995.2007.01212.x. [DOI] [PubMed] [Google Scholar]
- Vanhecke D, Graber W, Studer D. Close-to-native ultrastructural preservation by high pressure freezing. Methods Cell Biol. 2008;88:151–164. doi: 10.1016/S0091-679X(08)00409-3. [DOI] [PubMed] [Google Scholar]
- Bobik K, Dunlap JR, Burch-Smith TM. Tandem high-pressure freezing and quick freeze substitution of plant tissues for transmission electron microscopy. J Vis Exp. 2014. p. e51844. [DOI] [PMC free article] [PubMed]
- Pinan-Lucarré B, Clavé C. Monitoring autophagy in the filamentous fungus Podospora anserina. Methods Enzymol. 2008;451:251–270. doi: 10.1016/S0076-6879(08)03218-7. [DOI] [PubMed] [Google Scholar]
- Follet-Gueye ML, Pagny S, Faye L, Gomord V, Driouich A. An improved chemical fixation method suitable for immunogold localization of green fluorescent protein in the Golgi apparatus of tobacco Bright Yellow (BY-2) cells. J Histochem Cytochem. 2003;51(7):931–940. doi: 10.1177/002215540305100708. [DOI] [PubMed] [Google Scholar]
- Jesus DM, Moussatche N, Condit RC. An improved high pressure freezing and freeze substitution method to preserve the labile vaccinia virus nucleocapsid. J Struct Biol. 2016;195(1):41–48. doi: 10.1016/j.jsb.2016.05.001. [DOI] [PubMed] [Google Scholar]