

Abstract
Flow cytometry has been used for the past 40 years to define and analyze the phenotype of lymphoid and other hematopoietic cells. Initially restricted to the analysis of a few fluorochromes, currently there are dozens of different fluorescent dyes, and up to 14-18 different dyes can be combined at a time. However, several limitations still impair the analytical capabilities. Because of the multiplicity of fluorescent probes, data analysis has become increasingly complex due to the need of large, multi-parametric compensation matrices. Moreover, mutant mouse models carrying fluorescent proteins to detect and trace specific cell types in different tissues have become available, so the analysis (by flow cytometry) of auto-fluorescent cell suspensions obtained from solid organs is required. Spectral flow cytometry, which distinguishes the shapes of emission spectra along a wide range of continuous wavelengths, addresses some of these problems. The data is analyzed with an algorithm that replaces compensation matrices and treats auto-fluorescence as an independent parameter. Thus, spectral flow cytometry should be capable of discriminating fluorochromes with similar emission peaks and can provide a multi-parametric analysis without compensation requirements.
This protocol describes the spectral flow cytometry analysis, allowing for a 21-parameter (19 fluorescent probes) characterization and the management of an auto-fluorescent signal, providing high resolution in minor population detection. The results presented here show that spectral flow cytometry presents advantages in the analysis of cell populations from tissues difficult to characterize in conventional flow cytometry, such as the heart and the intestine. Spectral flow cytometry thus demonstrates the multi-parametric analytical capacity of high-performing conventional flow cytometry without the requirement for compensation and enables auto-fluorescence management.
Keywords: Cellular Biology, Issue 123, Flow cytometry, spectral, auto-fluorescence, intestine, embryonic heart, multi-parametric analysis
Introduction
In the last few decades, flow cytometry (FCM) became a widely available analytical method essential for cell phenotyping studies. There has been a substantial increase in the available fluorescent dyes, particularly fluorochromes excited by the violet laser (405 nm) (e.g., Brilliant Violet and new Q-dot dyes). However, the growth of available fluorescent dyes increases the risk of overlapping emissions and requires labor-intensive compensation matrices. FCM became widely used to analyze cell suspensions from solid tissue, but the presence of auto-fluorescent cells limits the discrimination of specifically labeled populations.
The basic principles of spectral FCM are reported in detail in Futamura et al.1,2. Briefly, the spectral FCM used here (see the table of materials) is equipped with 405, 488, and 638 nm lasers. The spectral FCM captures all the emitted fluorescence as spectra in a 32-channel linear array PMT (32ch PMT) for 500 nm to 800 nm and 2 independent PMTs for 420 nm to 440 nm and 450 nm to 469 nm, respectively, which replace the conventional band-pass filters. The 488 and the 405/638 nm laser spots are spatially separated, while the 405 nm and 638 nm laser spots are co-linear. For each individual particle, the spectral FCM measures up to 66 channels of fluorescence data excited by the 405 nm and 488 nm lasers. When cells are excited by the 638 nm laser, the spectral FCM measures 58 channels of fluorescence data because a mask is inserted to prevent the 638 nm laser from shining into the PMT. Spectral FCM analyzes the acquired full spectrum data with an algorithm based on the weighted least squares method (WLSM), which enables the separation of overlapping fluorescent spectra. Spectra derived from single-stained and unstained samples are recognized as the basic reference spectra. Multi-stained samples are mathematically fitted and unmixed, and the spectrum of a sample with mixed fluorescent labels is decomposed into a collection of its constituent spectra. The unmixing, in spectral technology3,4, replaces the compensation that removes the signal from all detectors except the one measuring a given dye.
In this study, we combined and tested 19 fluorochromes in a single, 21-parameter analysis that characterized the major hematopoietic subsets found in the mouse spleen. Additionally, we demonstrated that the spectral cytometry can manage auto-fluorescent signal, thus improving the characterization of intestinal intra-epithelial lymphocytes and of the embryonic heart. Indeed, in these tissues, auto-fluorescence management allowed for the assignment of specific fluorescence to cells that would be excluded from the analysis in conventional FCM.
Protocol
All experiments were performed according to the Pasteur Institute Ethic Charter and the EU guidelines and were approved by the French Agriculture Ministry.
1. Cell Suspension Preparation from Adult Mouse Organs
- Isolation of splenocytes
- Euthanize adult mice by cervical dislocation. Make an incision at the abdomen midline and open the skin with scissors. Harvest the spleen with forceps.
- Crush the spleen between 2 glass microscope slides to dissociate the cells and dilute them in 5 mL of Hank's Balanced Salted Solution (HBSS) containing 1% fetal calf serum (FCS).
- Centrifuge for 10 min at 120 x g and 4 °C. Discard the supernatant.
- Resuspend the pellet in 5 mL of HBSS 1% FCS. Count the cells with a Neubauer chamber using a Trypan blue vital stain. NOTE: 80 million cells should be obtained from one adult spleen.
- Isolation of cells from the small intestine
- Euthanize adult mice by cervical dislocation. Using scissors, make an incision in the skin at the abdomen midline. Grasp the small intestine with forceps in one hand and use scissors to cut it at both ends: first, right after the stomach and then at the end of the small intestine, just before the large intestine. Flush the small intestine with 1x Dulbecco's Phosphate Buffered Saline (DPBS) using a 2 mL syringe with a needle.
- Put the small intestine on a paper towel and carefully remove the Peyer's patches with a pair of sharp scissors.
- Use scissors to open the intestine on its long side by inserting one branch of the scissors inside the intestine. Cut the opened intestine into 1-cm pieces using sharp scissors.
- Incubate for 30 min at 37 °C under constant stirring in 30 mL of HBSS with 10% FCS.
- Place the cell suspension with the remaining non-dissociated fragments in a 50 mL plastic tube and vortex for 5 min at high speed. NOTE: There is no further dissociation of the tissue.
- Incubate for 10 min on ice to allow the large, non-dissociated fragments to pellet.
- Collect the supernatant and concentrate it by centrifugation for 7 min at 120 x g.
- Discard the supernatant and resuspend the pellet in 10 mL of HBSS with 1% FCS. Count cells with a Neubauer chamber using a Trypan blue vital stain. NOTE: ~60 million intra-epithelial cells (IELs) should be obtained from one adult small intestine.
- Panel design strategy
- Prepare the panels with antibodies directly coupled with fluorochromes. NOTE: All antibodies are previously titrated to obtain the optimal definition of the cell populations (the titration can vary with the antibody batch). The antibodies used in the 19-color panel to stain splenocytes are listed in Table 1.
- Construct multi-color panels for cytometry. As this can be a challenging task, use the following guidelines to optimize the panels:
- Check the spectral configuration and the fluorescent dyes that can be used on the instrument.
- Use the Brightness Index/Staining Index information provided by the manufacturer. NOTE: The Brightness Index/Staining Index is the relative indicator of fluorescence intensity versus background for each fluorochrome.
- Choose the antibodies following the rules below:
- Choose the brightest fluorochromes for proteins that are weakly expressed and the dimmest fluorochromes for more highly expressed proteins. NOTE: For example, CD45, CD4, and CD8 are highly expressed and can be used with dim fluorochromes.
- Start designing the panel by first choosing the antibodies with the fewest fluorescent dye possibilities.
- If possible, choose fluorescent dyes with the smallest overlap of emission spectra for markers expressed on the same cell types. NOTE: Tandems (e.g., PE-Cy5, PE-Cy7, and PerCP-Cy5.5) should be used with caution, as they are more susceptible to degradation by light exposure.
- Staining of cells for cytometry analysis
- Prepare one 5 mL tube with 1 x 106 splenocytes and one 5 mL tube with 1 x 106 intestinal cells and keep them unstained for use as negative controls.
- Prepare and label one 5 mL tube per sample and per panel according to Table 1. Prepare the mix of antibodies, according to Table 1, in HBSS 1% FCS.
- Transfer 1 x 106 cells – either splenocytes or intestinal cells – into each 5 mL tube. Add 2 mL of HBSS 1% FCS and centrifuge for 5 min at 4 °C and 120 x g. Discard the supernatant and resuspend the pellet in 50 µL of antibody mix.
- Label the splenocytes in 2 steps. First, use 50 µL of the mix containing the antibodies labeled with PE-Cyanine 5, PerCP-Cyanine 5.5, and PerCP (with Fc receptor blocking) in a 5 mL tube. After 20 min of incubation in the dark at 4 °C, add 50 µL of the mix containing all the other antibodies to a 5 mL tube. NOTE: This strategy limits steric hindrance.
- Incubate for 20 min in the dark at 4 °C.
- Add 2 mL of HBSS 1% FCS and centrifuge for 5 min at 4 °C and 120 x g. Discard the supernatant and resuspend the pellet in 200 µL of HBSS 1% FCS with 0.5 µg/mL propidium iodide (PI).
2. Preparation of Single-staining for Each Fluorochrome on Commercial Compensation Bead Microspheres (e.g., UltraComp eBeads), Hereafter Referred to as Beads
Prepare and label as many 5 mL tubes as the number of antibodies that are used in the panels. Place 1 drop of beads in each tube and add 1 µL of antibody per tube. NOTE: The beads used here (see the table of materials) contain stained (positive) and unstained (negative) beads. Antibodies are put in excess on the beads to ensure a bright signal and thus a clear spectral signature for each dye. This is required by the software to be able to make a precise unmixing.
Incubate for 20 min in the dark at 4 °C.
Add 2 mL of HBSS 1% FCS and centrifuge for 5 min at 120 x g. Discard the supernatant and resuspend the pellet in 100 µL of HBSS 1% FCS.
3. Cell Suspension Preparation from Embryonic Mouse Hearts
- Dissections of embryos
- Plan to obtain timed-pregnant females in advance by mating 2 females with one male (always in the male's cage) at the end of the day (between 17 and 19 h). The next morning, females with vaginal plugs are considered to be at 0.5 days post-coitum.
- Euthanize the E17.5 pregnant females by cervical dislocation. Ensure that the animal is unresponsive by firmly pressing on the mouse paw (toe-pinch reflex). Wet the abdomen with 70% ethanol to sterilize and to prevent the spread of mouse fur.
- Make an incision in the skin at the middle of the abdomen using scissors and pull the skin apart with fingers.
- Open the abdominal cavity by pulling and making incisions in the abdominal muscle from the center towards the two sides of the abdomen using gross forceps and scissors. NOTE: Be careful not to damage the small intestine, which is immediately below the peritoneal muscle.
- Collect the uterine horns (with the embryos) by cutting both at the level of the uterine body and of the ovaries. Soak them in a 90 x 15 mm2 Petri dish with 50 mL of DPBS.
- Isolate each embryo by cutting between each decidua, followed by the uterus muscular wall and the visceral yolk sac with a fine forceps and scissors. Pull out the embryos intact. Finally, cut the umbilical cord.
- Cut the E17.5 embryos' heads with scissors (decapitation) before starting the dissection.
- Collection of hearts
- Soak the decapitated embryos in a 90 x 15 mm2 Petri dish containing a wet paper towel bedding in the dish bottom and 50 mL of HBSS with 1% FCS.
- Under a stereomicroscope, hold each embryo with gross forceps so that the ventral side is facing up.
- Carefully open the thoracic grid by cutting the right side of chest with scissors in order to prevent damage to the heart.
- Expose the heart by holding the rib cage with gross forceps.
- Pull apart the heart (still assembled with lungs and thymus) by holding the great vessels (containing the big vessels of in- and out-flow of the heart) and place them in a 35 x 15 mm2 Petri dish with 2 mL of HBSS 1% with FCS.
- Isolate the hearts from the surrounding organs and connective tissue with fine forceps and a scalpel.
- Wash the hearts in a new 35 x 15 mm Petri dish with 2 mL of HBSS 1% FCS and keep them on ice for 20 min to allow the heart to pump out the blood inside the chambers.
- Preparation of heart cell suspensions
- Pre-warm the 200 µg/mL collagenase in HBSS+/+ (hereafter referred to as enzymatic solution) to 37 °C. NOTE: Dilute the collagenase in HBSS+/+ (with Ca2+ and Mg2+ cations) to maximize the enzyme activity; collagenase activity is stable between batches.
- Replace the washing solution soaking the hearts (HBSS 1% FCS) with 1 mL of pre-warmed enzymatic solution.
- Under a stereomicroscope, mince the hearts into 1-mm3 pieces using fine forceps and a scalpel. Add another 1 mL of pre-warmed enzymatic solution and transfer the minced tissue to a 5 mL tube with a lid. NOTE: For an optimal digestion, place a maximum of 5 E17.5 hearts per 2 mL of enzymatic solution. If more hearts are digested at the same time, split them into different 5 mL tubes .
- Incubate the minced hearts for 15 min at 37 °C. NOTE: Lay down the 5 mL tubes (properly closed) in the incubator to spread the tissue along the solution.
- At the end of the incubation, homogenize the tissue in the same enzymatic solution by pipetting up and down approximately 20 times with the P1000 pipette. Put the 5 mL tube in the vertical position and let the remaining tissue fragment sediment (approximately 3 min).
- Collect the supernatant (containing the digested cells in suspension) in a 50 mL tube, add 2 mL of HBSS 10% FCS (the same volume as the volume of enzymatic solution added to the tissue fragments), and keep them on ice while continuing the digestion process. NOTE: The HBSS 10% FCS solution and the ice are important to inactive the enzyme action while the remaining tissue is digesting.
- Add 2 mL of pre-warmed enzymatic solution to the 5 mL tubes with the remaining tissue fragments and continue the digestion by repeating step 3.3.4 until no more tissue is observed. NOTE: Around 4 cycles of digestion are enough to completely dissociate the heart tissue (5 E17.5 hearts in 2 mL of enzymatic solution).
- Centrifuge the 50-mL tubes with cells in suspension for 10 min at 300 x g and discard the supernatant.
- Resuspend the cell pellet, add 1 mL of HBSS-/- 1% FCS, and filter the cell suspension though a 70 µm nylon mesh. Count the cells with a Neubauer chamber using a Trypan blue vital stain. NOTE: Resuspend the cells with HBSS-/- (without Ca2+ and Mg2+ cations) to minimize cell aggregation. Between 800,000 and 1,000,000 cells are obtained from one E17.5 heart.
- Staining cardiac cells with fluorescent antibodies NOTE: All antibodies are previously titrated to obtain optimal definition of the cell populations (the titration can vary with the antibody batch).The antibodies used in the heart panel are listed in Table 1.
- Distribute the cardiac cells in suspension through a 96-well plate (round bottom). Distribute them evenly throughout all needed wells (200 µL per well), short-spin centrifuge the 96-well plate for 1 min at 480 x g, and discard the supernatant. NOTE: All wells of the 96-well plate (round bottom) can be used.
- Resuspend the pellet and incubate the cardiac cells for 20 min in the dark at 4 °C with 100 µL of the cocktail of antibodies (i.e., CD45-PE, TER119-PE, CD31-Alexa 647, and Sca-1-PE-Cyanine5) diluted in HBSS-/- 1% FCS.
- Wash the cardiac cells from the excess of antibodies by adding 200 µL of HBSS-/- 1% FCS and short-spin centrifuging the cells for 1 min at 480 x g.
- Discard the supernatant and repeat the short-spin centrifugation (step 3.4.3).
- Resuspend the cells in 200 µL of HBSS-/- 1% FCS and transfer them to a 5 mL tube already containing 200 µL of HBSS-/- 1% FCS.
- Before acquisition, add 400 µL of HBSS-/- 1% FCS with 0.5 µg/mL PI and filter the cell suspension though a 70 µm nylon mesh.
4. Acquisition of Labeled Splenic, Intestinal, and Cardiac Cells with the Spectral Flow Cytometer
Before acquisition of the data, automatically calibrate the spectral cytometer following the manufacturer's instructions. Perform the daily and the monthly quality control procedures each day of experiment.
When the cytometer is ready, start the preview of the samples containing the cell suspensions labeled with all antibodies. NOTE: Do not acquire.
Make sure that all lasers are on as required. Adjust the FSC gain such that all cells are visible in the respective plots. Gate the cell population and apply this gate to the two spectral plots corresponding to the 488-nm and to the 638/405-nm lasers. NOTE: When fluorochromes are excited by the 638-nm laser (i.e., when the 638-nm laser is on), there is a mask that shields light from 617-662 nm to prevent the 638 nm laser from shining into the PMT. This induces the loss of a part of the spectrum and therefore causes a lower separation of close dyes emitting in these wavelengths. However, the entire signal can be collected by switching off the 638-nm laser if it is not required for acquisition.
Adjust the 32-channel photo multiplier (PMT) voltage (%) such that all fluorochromes are within the range of the intensity displayed in the y-axis (1-106) in the two different spectral plots displaying 488 nm and 405/638 nm excitations. NOTE: Training is required to recognize the spectra of all fluorochromes used.
Start to acquire the samples. Click on "Preview" to visualize the cells on the screen and then click on "Acquire" to record the sample. Save up to 2 x 106 events from each sample.
After acquiring the stained samples, acquire the sample stained with the vital dye and then the compensation beads individually stained with all antibodies used. Click on “Preview” to visualize the cells on the screen and then on “Acquire” to record the samples. NOTE: Keep the same PMT voltage for all samples and controls (i.e., beads and unstained samples); 5,000 events are largely sufficient.
Finally, acquire the data from the unstained cell suspensions. Click on "Preview" to visualize the cells on the screen and then on "Acquire" to record the samples. NOTE: Acquiring the unstained samples at the end of the experiment minimizes the carryover of fluorescent cells.
Clean the cytometer following the manufacturer's instructions and close the experiment in the acquisition folder. NOTE: Only after closing the experiment will it be saved and available for analysis.
5. Analysis of the Data with Spectral FCM Software
In the "Color Palette" window of the "Analysis" tab, register all parameters present in the panel by adding each fluorochrome used and the corresponding marker (select from the available list or add a new parameter). Also, include the auto-fluorescence and viability parameters; all selected parameters will appear in the "unmixing" window.
- Select the first single stained sample (done with compensation beads) in the "Control" window under the "Tube List" of this experiment on the “Analysis” tab. In the worksheet, click on the "polygon design tool" and design a gate on the bead population in the FSC/SSC plot. Double-click on the top of gate to open a spectral or dot plot to visualize the gated beads in a daughter plot.
- Design one gate for the positive and one gate for the negative beads either on the spectrum or on the dot plot. Check the whole spectra corresponding to the two laser sets for each positive and negative fraction and eliminate outliers by successively gating and clicking on the gate to verify the daughter population. Enter the negative and positive gates in the "Unmixing" window.
- Repeat step 5.2 for each single-stained sample. NOTE: Be aware that the positive and negative gating strategy corresponds to the sample that is being analyzed, although the program allows for it to be placed in any sample. Instead of determining a negative fraction for each single-stained sample, use an unstained bead sample to set a universal negative.
Select the unstained cell sample in the "Tube List" and design gates for auto-fluorescent and for non-autofluorescent cells.
When all the negative and positive gates for all parameters, including auto-fluorescence and viability, are selected in the "Unmixing" window, click on "Calculate." Apply the unmixing to the samples to be analyzed.
- Analyze the data by designing gates and creating two-parameter plots.
- First, design a gate for SSC versus FSC and then remove dead cells by excluding all cells labeled with the viability marker (PI versus CD45). On the rest of the cells, design gates for all populations of interest following the strategy described for each cell suspension (Figures 1-3). NOTE: If necessary, adjust the compensation in the "reference spectrum adjuster." This is a tool that can be used after unmixing to re-adjust the median of the positive populations for each fluorochrome if the algorithm could not do it perfectly.
Representative Results
21-parameter spectral FCM panel to analyze splenocytes
Figure 1 shows the results obtained with the 19-fluorescent-antibody panel applied to splenic cells comprising different antibodies recognizing subsets of T, B, NK, dendritic, and myeloid cells, while CD45 labels all hematopoietic nucleated cells. The panel also includes a viability dye (PI), as well as the size (FSC) and granularity (SSC) parameters. Figure 1A shows the reference fluorochrome spectra. After the elimination of dead cells in the CD45+ gate (Figure 1B), the analysis shows that CD3+ T cells express the TcRβ chain and have the expected CD4/CD8 distribution and proportion of CD25+ cells in CD4 T cells. CD8 T cells show a normal distribution of CD44- and Ly49D-expressing cells. CD19+ B cells co-express B220 and MHCII, as well as IgM and IgD, typical of mature splenic B cells. A subset of NK1.1+ NK cells represents a small subset of the splenic lymphocytes that co-express Ly49D. The remaining cells comprise a small subset of CD11c+ dendritic cells with high levels of MHC II, some of which were also CD11b+. F4/80+ cells characteristic of tissue-resident macrophages co-express CD11b and the highest levels of CD44 found in the spleen. The remaining cells (CD19-Nk1.1-CD3-F4/80-CD11c-) could be subdivided by the expression of CD11b and Gr1, where the small subset of double-positive cells also express high levels of CD44, likely corresponding to progenitors known to be present at low frequencies in the spleen. A large fraction was negative for the three markers. Figure 2 shows the same analysis before adjusting the compensation.
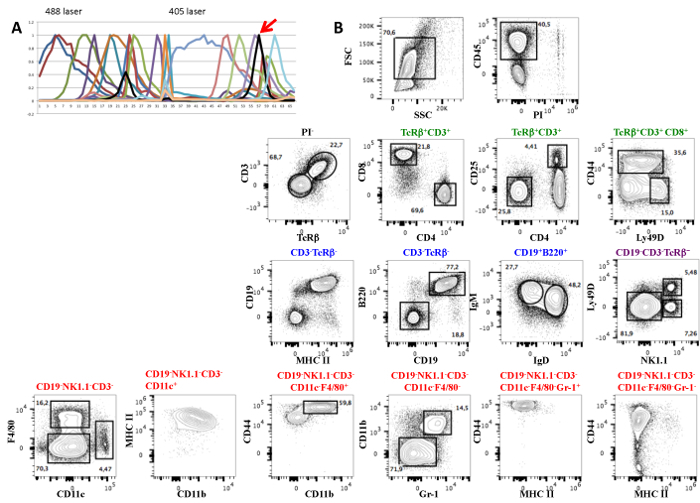 Figure 1: A 19-color Antibody Panel for the Analysis of Murine Spleen Cells. (A) Mouse splenocytes were stained with the previous antibody combination, to which CD45-evolve 655 was added. The reference spectra of all fluorochromes are shown. Fluorophore-labeled CD45 is marked by a red arrow. The fluorochromes are listed in Table 1. (B) The contour plots show the discriminative capacity of this multi-parametric analysis to separate the different populations of B, T, NK, dendritic, or myeloid cells. The analysis was done in conventional FCM software after unmixing deconvolution.
Figure 1: A 19-color Antibody Panel for the Analysis of Murine Spleen Cells. (A) Mouse splenocytes were stained with the previous antibody combination, to which CD45-evolve 655 was added. The reference spectra of all fluorochromes are shown. Fluorophore-labeled CD45 is marked by a red arrow. The fluorochromes are listed in Table 1. (B) The contour plots show the discriminative capacity of this multi-parametric analysis to separate the different populations of B, T, NK, dendritic, or myeloid cells. The analysis was done in conventional FCM software after unmixing deconvolution.
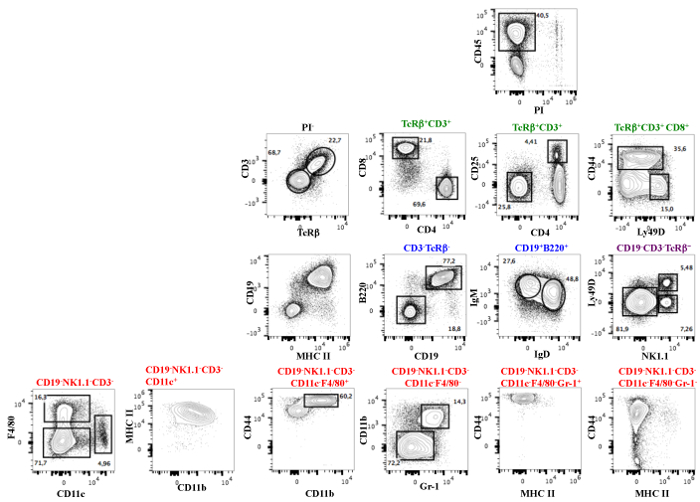 Figure 2: A 19-color Antibody Panel for the Analysis of Murine Spleen Cells. The same analysis as in Figure 1 was done before the adjustment of the compensation with the reference spectra adjuster.
Figure 2: A 19-color Antibody Panel for the Analysis of Murine Spleen Cells. The same analysis as in Figure 1 was done before the adjustment of the compensation with the reference spectra adjuster.
Small intestine intra-epithelial lymphocytes
Several T-cell populations are found in the intestine within the epithelial cell layer5,6. Conventional isolation of these subsets includes a density gradient that separates lymphocytes from epithelial cells. However, this preparation is delicate, time consuming, and results in cell loss. To improve quantification and to facilitate the experimental procedure, the potential of spectral FCM to discriminate populations of intestinal lymphocytes within a large fraction of epithelial cells was tested. After the mechanical disruption of the epithelial tissue, cell suspensions were stained with antibodies that label subsets of αβ and γδ T cells usually found in the intestinal epithelium. The cells were analyzed in two conventional cytometers and in spectral FCM.
Figure 3 shows the results after following a similar gating strategy, with elimination of dead cells gating on PI-negative cells. The presence of auto-fluorescent cells is obvious in all conventional cytometers and in spectral cytometry when the auto-fluorescence manager is inactivated (top panels, red arrows), resulting in a poor discrimination of the living cells. In all plots, cells within the FSC-A/SSC-A lymphocyte gate are shown in blue and cells with larger scatters (epithelial cell gate) are shown in red. Both conventional instruments were unable to resolve the CD3+ T-cell subset (in blue) (turquoise arrow) from the remaining cells. Therefore, TcRβ- cells (green arrow) contaminated the CD3+TcRγδ- population, a biologically improbable subset never detected after the separation of the lymphocytes from epithelial cells. In contrast, in spectral FCM after auto-fluorescence management, the analysis of the different T-cell subsets was not impaired by the presence of epithelial cells, the populations were well-separated, and there were no TcR-negative cells in the CD3+ gate.
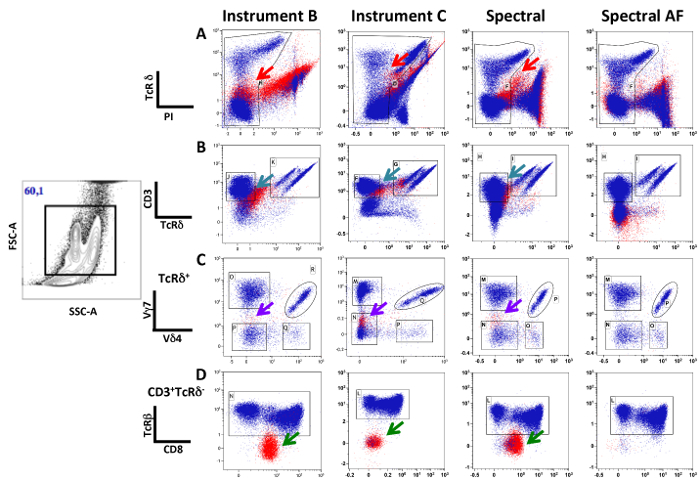 Figure 3: The Presence of Auto-fluorescent Cells Does not Impair the Detection of Intra-epithelial Lymphocytes in Spectral FCM. Small intestinal cells, including epithelial cells and lymphocytes, were stained with antibodies recognizing TcRδ, PI, TcRβ Cy7-APC CD3 (panels B), Vγ7 APC, Vδ4 Cy7-PE (panels C), and CD8 FITC (panels D). PI was added in the FACS buffer before analysis. The acquisition of the data was done sequentially in the three instruments after the appropriate quality control. Doublets were eliminated in FSC-H/FSC-W. The analysis was done in conventional FCM software. Plots show the different steps of the gating strategy. Cells within the FSC-A/SSC-A lymphocyte gate are labeled in blue, while cells within the intestinal epithelial cell gate are labeled in red. Arrows point to different population distortions and auto-fluorescence. Plots in C show the analysis of data obtained in spectral FCM without autofluorescence management in the unmixing, whereas D shows the same data after unmixing with autofluorescence management.
Figure 3: The Presence of Auto-fluorescent Cells Does not Impair the Detection of Intra-epithelial Lymphocytes in Spectral FCM. Small intestinal cells, including epithelial cells and lymphocytes, were stained with antibodies recognizing TcRδ, PI, TcRβ Cy7-APC CD3 (panels B), Vγ7 APC, Vδ4 Cy7-PE (panels C), and CD8 FITC (panels D). PI was added in the FACS buffer before analysis. The acquisition of the data was done sequentially in the three instruments after the appropriate quality control. Doublets were eliminated in FSC-H/FSC-W. The analysis was done in conventional FCM software. Plots show the different steps of the gating strategy. Cells within the FSC-A/SSC-A lymphocyte gate are labeled in blue, while cells within the intestinal epithelial cell gate are labeled in red. Arrows point to different population distortions and auto-fluorescence. Plots in C show the analysis of data obtained in spectral FCM without autofluorescence management in the unmixing, whereas D shows the same data after unmixing with autofluorescence management.
Embryonic heart
Conventional FCM showed a major population of auto-fluorescent cells (black arrow) that was excluded from the analysis because it fluoresced in the CD45 and TER119 dump channel (PE), marking hematopoietic cells. In contrast, this auto-fluorescent population was not present as such in spectral FCM and was comprised in the CD45-TER119- subset (Figure 4). To show that these cells were cardiac and not endothelial or hematopoietic, expressing low levels of CD45, TER119, or CD31, the expression of transcripts specific to cardiomyocytes were quantified on sorted cells. High levels of cardiac muscle troponin T (TNNT2)7 and atrial light chain-2 (MYL7)8 transcripts were found in the auto-fluorescent population.
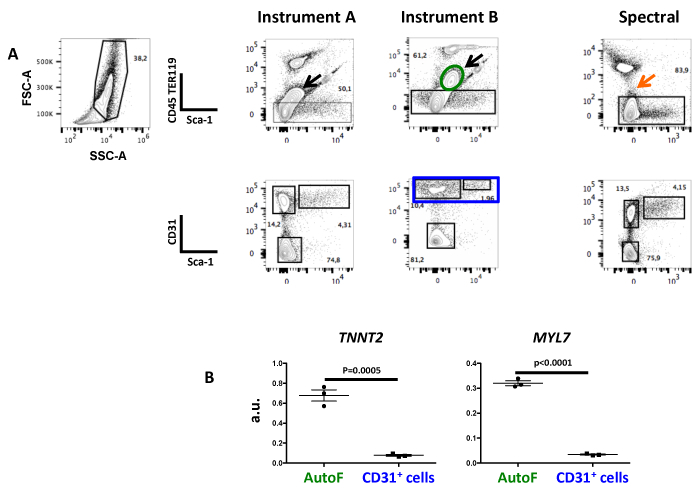 Figure 4: Auto-fluorescent Cells in E17.5 Cardiac Cell Suspensions are Properly Included in the Negative Cell Fraction in Spectral FCM. (A) Cell suspensions from fetal hearts were stained with anti-TER119 and CD45-PE, Sca-1-PECy5, and CD31-APC antibodies and sequentially acquired in the three instruments. Black arrows show auto-fluorescent cells in the two conventional cytometers, while these cells are not detected as auto-fluorescent in spectral cytometry and can thus be analyzed for specific fluorescent staining. (B) Auto-fluorescent cells (within the elliptical gate in green), but not CD31+ cells (rectangular gate in blue, negative control), showed the expression of cardiac troponin (TNNT2)7 and atrial light chain-2 (MYL7)8 transcripts. a.u. – arbitrary units relative to the GAPDH house-keeping transcript. Data presented as mean value ± SEM.
Figure 4: Auto-fluorescent Cells in E17.5 Cardiac Cell Suspensions are Properly Included in the Negative Cell Fraction in Spectral FCM. (A) Cell suspensions from fetal hearts were stained with anti-TER119 and CD45-PE, Sca-1-PECy5, and CD31-APC antibodies and sequentially acquired in the three instruments. Black arrows show auto-fluorescent cells in the two conventional cytometers, while these cells are not detected as auto-fluorescent in spectral cytometry and can thus be analyzed for specific fluorescent staining. (B) Auto-fluorescent cells (within the elliptical gate in green), but not CD31+ cells (rectangular gate in blue, negative control), showed the expression of cardiac troponin (TNNT2)7 and atrial light chain-2 (MYL7)8 transcripts. a.u. – arbitrary units relative to the GAPDH house-keeping transcript. Data presented as mean value ± SEM.
| Excitation laser | Fluorochrome | Specificity | Clone | |
| 488 nm | BB515 | CD8 | 53-6.7 | |
| 488 nm | FITC | Ly49D | 4E5 | |
| 488 nm | PE | F4/80 | 6F12 | |
| 488 nm | PE-CF594 | NK1.1 | PK136 | |
| 488 nm | PE-Cy5 | CD44 | IM7 | |
| 488 nm | PE-Cy7 | CD11b | M1/70 | |
| 488 nm | PerCP-Cy5.5 | IgM | R6-60.2 | |
| 488 nm | PerCP | Gr-1 | RB6-8CS | |
| 488 nm | AF532 | CD3 | 17A2 | |
| 488 nm | Propidium Iodide | Viability | ||
| 405 nm | V450 | IgD | 11-26c.2a | |
| 405 nm | BV421 | CD11c | HL3 | |
| 405 nm | BV510 | CD19 | 1D3 | |
| 405 nm | BV570 | TCRb | H57-597 | |
| 405 nm | BV605 | CD4 | RM4-5 | |
| 405 nm | BV650 | CD45R/B220 | RA3-6B2 | |
| 405 nm | BV711 | I-A/I-E | M5/114 | |
| 405 nm | BV786 | CD25 | PC61 | |
| 405 nm | eVolve 655 | CD45 | 30-F11 | |
| N/A | purified | CD16/CD32 | 2.4G2 | Fc-receptor block |
| List of Antibodies used in cardiac cell suspension | ||||
| 638 nm | Alexa Fluor 647 | CD31 | MEC13.3 | |
| 488 nm | PE | CD45 | 30-F11 | |
| 488 nm | PE-Cy5 | Sca-1 | D7 | |
| 488 nm | PE | Ter119 | TER-119 |
Table1: List of Antibodies Used in the 19-color Panel and in Cardiac Cell Suspension.
Discussion
Conventional FCM is based on the detection of photons emitted after the excitation of fluorescent probes. The fluorescence emission of one fluorochrome detected in a detector designed to measure the signal from another fluorochrome induces physical overlap. This spillover among emission spectra needs to be corrected by compensations.
Spectral FCM and data processing by the unmixing deconvolution algorithm allows for the combination of fluorochromes with close emission peaks without additional compensation, provided that they have different spectral shapes. The algorithm used for the analysis treats auto-fluorescence as an independent parameter, subtracting non-specific fluorescence from solid tissue cell suspensions.
A panel of 19 fluorochromes was assembled to analyze immune cells using only two lasers (488 nm and 405 nm). Indeed, when the 638 nm laser was on, a section of the 32-channel PMT corresponding to the excitation wavelength of this laser was not available. To get the maximum detection capacity of the PMT, the red laser was switched off. Mouse splenic cells were analyzed, allowing for the detection of more than 12 different cell populations, some of which comprised 1% or less of the hematopoietic cells.
To test the capacity of spectral FCM to manage auto-fluorescence, two different situations that can impair the analysis were tested: one where lymphocytes were contaminated with epithelial cells, and one where auto-fluorescent cardiac cells were analyzed. In contrast with conventional FCM, spectral cytometry allows for the discrimination of all lymphocyte subsets, even when they represent minor subsets.
The controversy around the regenerative capacity of adult cardiomyocytes9,10,11 is in part due to the absence of a strategy to discriminate and isolate distinct subsets of viable cells in the heart. A multi-parametric analysis defining unique combinations of markers to discriminate the different cardiac subsets might enable the detection of rare subsets because of large numbers of cells that can be analyzed. Fetal hearts (E17.5) were analyzed, and a large fraction of auto-fluorescent cells, which would have escaped detection in conventional FCM, was integrated in the non-hematopoietic viable cardiac cell compartment in spectral FCM.
A recurrent problem of this method is the requirement for good-quality fluorescent labeled antibodies. This is particularly critical when using tandem dyes, where a component of the tandem is dominant. For example, BV786 and Cy7-PE can be difficult to distinguish from BV421 and PE, respectively.
FCM based on spectrometry provides analytical power higher than that obtained with conventional FCM, without the need for compensation matrices. This manuscript provides evidence that there is an alternative approach that demonstrates a high analytical capacity, the absence of complex compensation matrices, the unimpaired discrimination of cell subsets due to auto-fluorescence, and no additional financial commitments that still required for mass spectrometry FCM. Spectral FCM thus appears to be a method of choice for the analysis of cells obtained from a wide variety of solid tissues usually not amenable to conventional FCM and can be used in tumor, developmental, and stem cell biology.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We acknowledge the technical and theoretical contributions in spectral cytometry of K. Futamura, who also critically revised the manuscript. We are also indebted to C. Ait-Mansour, P.-H. Commere, A. Bandeira, and P. Pereira for critically reading the manuscript and for endless technical advice and support. We also thank P. Pereira for the gift of the anti Vγ7-APC and Vδ4-Biotin labeled antibodies. We aknowledge the Centre d’Enseignements from Pasteur Institute for welcoming and supporting the filming logistics. This work was supported by the Pasteur Institute, Institut National de la Santé et de la Recherche Médicale (INSERM); the Swiss National Science Foundation and Pasteur Bourse Roux (S.S.); Fundação para a Ciência e a Tecnologia - SFRH/BD/74218/2010 (M.V.); and Université Paris Diderot, the Agence Nationale de la Recherche (ANR) project Twothyme, the ANR, and Program REVIVE (Investment for the Future) (A.C.).
References
- Futamura K, et al. Novel full-spectral flow cytometry with multiple spectrally-adjacent fluorescent proteins and fluorochromes and visualization of in vivo cellular movement. Cytometry A. 2015;87(9):830–842. doi: 10.1002/cyto.a.22725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmutz S, Valente M, Cumano A, Novault S. Spectral Cytometry Has Unique Properties Allowing Multicolor Analysis of Cell Suspensions Isolated from Solid Tissues. PLoS One. 2016;11(8):e0159961. doi: 10.1371/journal.pone.0159961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roederer M. Multiparameter FACS analysis. Curr Protoc Immunol. Chapter 5, Unit 5.8. 2002. [DOI] [PubMed]
- Perfetto SP, Chattopadhyay PK, Roederer M. Seventeen-colour flow cytometry: unravelling the immune system. Nat Rev Immunol. 2004;4(8):648–655. doi: 10.1038/nri1416. [DOI] [PubMed] [Google Scholar]
- Grigoriadou K, Boucontet L, Pereira P. T cell receptor-gamma allele-specific selection of V gamma 1/V delta 4 cells in the intestinal epithelium. J Immunol. 2002;169(7):3736–3743. doi: 10.4049/jimmunol.169.7.3736. [DOI] [PubMed] [Google Scholar]
- Guy-Grand D, et al. Origin, trafficking, and intraepithelial fate of gut-tropic T cells. J Exp Med. 2013;210(9):1839–1854. doi: 10.1084/jem.20122588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoinne A, Cauliez B. [Cardiac troponin I and T: specific biomarkers of cardiomyocyte] Rev Med Interne. 2004;25(2):115–123. doi: 10.1016/s0248-8663(03)00218-2. [DOI] [PubMed] [Google Scholar]
- Staudt DW, Liu J, Thorn KS, Stuurman N, Liebling M, Stainier DY. High-resolution imaging of cardiomyocyte behavior reveals two distinct steps in ventricular trabeculation. Development. 2014;141(3):585–593. doi: 10.1242/dev.098632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrami AP, et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114(6):763–776. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- Keith MC, Bolli R. "String theory" of c-kit(pos) cardiac cells: a new paradigm regarding the nature of these cells that may reconcile apparently discrepant results. Circ Res. 2015;116(7):1216–1230. doi: 10.1161/CIRCRESAHA.116.305557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente M, Nascimento DS, Cumano A, Pinto-do-Ó P. Sca-1+ cardiac progenitor cells and heart-making: a critical synopsis. Stem Cells Dev. 2014;23(19):2263–2273. doi: 10.1089/scd.2014.0197. [DOI] [PMC free article] [PubMed] [Google Scholar]