

Abstract
The craniotomy is a commonly performed procedure to expose the brain for in vivo experiments. In mouse research, most labs utilize a small craniotomy, typically 3 mm x 3 mm. This protocol introduces a method for creating a substantially larger 7 mm x 6 mm cranial window exposing most of a cerebral hemisphere over the mouse temporal and parietal cortices (e.g., bregma 2.5 - 4.5 mm, lateral 0 - 6 mm). To perform this surgery, the head must be tilted approximately 30° and much of the temporal muscle must be retracted. Due to the large amount of bone removal, this procedure is intended only for acute experiments with the animal anesthetized throughout the surgery and experiment.
The main advantage of this innovative large lateral cranial window is to provide simultaneous access to both medial and lateral areas of the cortex. This large unilateral cranial window can be used to study the neural dynamics between cells, as well as between different cortical areas by combining multi-electrode electrophysiological recordings, imaging of neuronal activity (e.g., intrinsic or extrinsic imaging), and optogenetic stimulation. Additionally, this large craniotomy also exposes a large area of cortical blood vessels, allowing for direct manipulation of the lateral cortical vasculature.
Keywords: Neuroscience, Issue 123, craniotomy, cranial window, imaging, mouse, middle cerebral artery, cortical activity, brain
Introduction
The craniotomy is a standard procedure used by neuroscientists to reveal a portion of the brain. Since the dawn of electrophysiology, the craniotomy has allowed unprecedented breakthroughs in the field of neuroscience. Dense mapping of the cerebral cortex with electrodes has led to experiments testing hypotheses and theories based on these maps. We have recently entered a new era where the craniotomy is being utilized for in vivo imaging of cortical blood flow1,2,3 and neurovascular architecture4, enabling real time visualization of cortical activity within the exposed areas5,6,7. Although many studies use craniotomies combined with in vivo optical imaging techniques to study the structure and function of cortical neurons, glia, and cortical vasculature8,9, further investigations are limited by small areas of exposed cortex (but see10).
The purpose of this protocol is to provide a method for creating a large lateral craniotomy, exposing the cerebral cortex from the midline to the squamosal bone, and extending beyond bregma and lambda. This large craniotomy enables simultaneous viewing of the association cortices (retrosplenial, cingulate, and parietal), primary and secondary motor, somatosensory, visual, and the auditory cortex. This method has been previously coupled with voltage sensitive dye imaging (VSDI) to investigate how multiple cortical areas interact with one another during spontaneous and stimulus-induced cortical activity5,11,12. The most challenging aspects of this procedure include positioning the head of the animal, fixing the head plate, and avoiding hemorrhage while separating the temporal muscle from the parietal bone. Care must also be taken during the drilling and skull removal processes as the skull curves at an oblique angle.
Protocol
The following protocol follows the University of Lethbridge Animal Care Committee (ACC) guidelines, and is conducted in accordance with the standards of the Canadian Council on Animal Care (CCAC).
1. Preparation
For prolonged study periods, autoclave all opened surgical supplies and ensure that sterility is maintained throughout the surgery. If multiple surgeries are required, autoclave between surgeries.
Ensure there is plenty of brain buffer on hand (at least 50 mL). The solution is comprised of 134 mM sodium chloride, 5.4 mM potassium, 1 mM magnesium chloride hexahydrate, 1.8 mM calcium chloride dihydrate, and 5 mM HEPES sodium, pH balanced to 7.4 with 5 M hydrogen chloride.
- Place the mouse in an induction chamber and anesthetize with 3 - 4% isoflurane. Follow with 1.0 - 2.0% isoflurane for maintenance during the surgery. Further reduce to as low as 0.4 – 0.8% during imaging5,13,14,15, provided proper anesthesia is maintained. These low levels of anesthesia require vigilant and frequent monitoring of the mouse (about every 5 min) to ensure it remains areflexic to painful stimuli. NOTE: Dehydration can become problematic due to the high surface to volume ratio of the mouse and exacerbated by prolonged use of isoflurane.
- Use subcutaneous injections of saline, 0.1 mL per 10 g body weight, every 1 - 2 h. When adequately hydrated, the mouse will urinate once every 1 - 2 h.
Closely monitor the mouse to ensure consistent anesthesia throughout surgery and imaging. Do not leave the mouse unattended and take care that it never regains consciousness.
Transfer the anesthetized mouse to the head-holder set-up and place on a thermo-regulating heating pad set to 37 °C. Secure the upper teeth in a teeth holder.
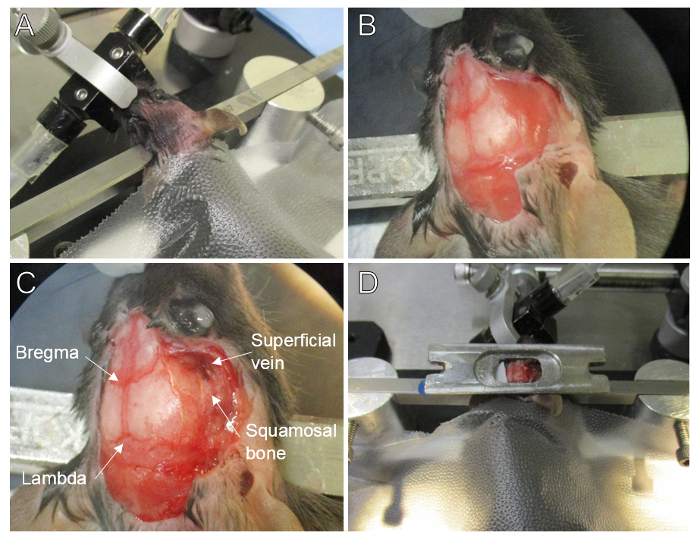
Rotate the mouse's head towards the left approximately 30° to expose the right lateral side of the head and secure the mouse's head with the blunt end of ear bars (Figure 1A).
Apply ophthalmic ointment to prevent corneal drying.
To reduce cerebral edema, inject dexamethasone (4 mg/kg) intramuscularly.
Wipe the skin over the surgical area with cotton swabs dipped in 4% chlorhexidine (3 times) and 70% ethanol (3 times). Use each cotton swab only once.
Don surgical gloves and cover the animal with adhesive plastic wrap. Provide local anesthetic by injecting of lidocaine (8 - 10 mg/kg; 2% epinephrine) subcutaneously over the craniotomy site. Wait 3 - 5 min for the drug to be absorbed into the tissue.
2. Removing the Skin and Retracting the Muscle from the Skull
Perform nearly all of these procedures while viewing the skull under a dissecting microscope (e.g., 0.7 - 4.5X power, depending on the situation).
Lift up the skin 1 mm left of the midline (just behind the ear) with forceps and make a small horizontal incision with surgical scissors.
Make a 5 - 6 mm lateral cut towards the right ear, and then cut towards the rostral end of the head.
At the initial incision point, insert the scissors and cut 10 mm rostrally.
Cut the skin around the right ear and near the right eye to expose the right side of the skull and temporal muscle. Ensure that the widest part of the exposed area is at least 7 mm. Trim the skin further if the surgical area needs to be extended.
Fix the skin around the incision by putting a few drops of butyl cyanoacrylate glue between the skull and the skin. Ensure the tissue has been previously dried with cotton tipped applicators or rolled tissues to increase the efficacy of the adhesive.
Using a cotton swab, rub the surface of the skull in a circular motion to remove the periosteum from the skull. Ensure that none remains by drying the skull completely.
Using spring scissors and forceps, separate the temporal muscle from the skull; cut and retract the muscle laterally until reaching the squamosal bone (Figure 1C). Take extreme care not to damage the superficial temporal vein that runs along the level of the squamosal bone near the eye, otherwise hemorrhaging might occur.
Control bleeding with gel foam pre-soaked in brain buffer. For serious hemorrhaging use a heat cauterizer. Drop butyl cyanoacrylate glue onto the bleeding sites following treatment with the heat cauterizer. Ensure the area is dried beforehand by using cotton tipped applicators or rolled tissues to increase the efficacy of the adhesive.
3. Craniotomy
NOTE: The surgeon must remain diligent during removal of the skull and dura to avoid unnecessary complications. Troubleshooting steps are included should complications arise.
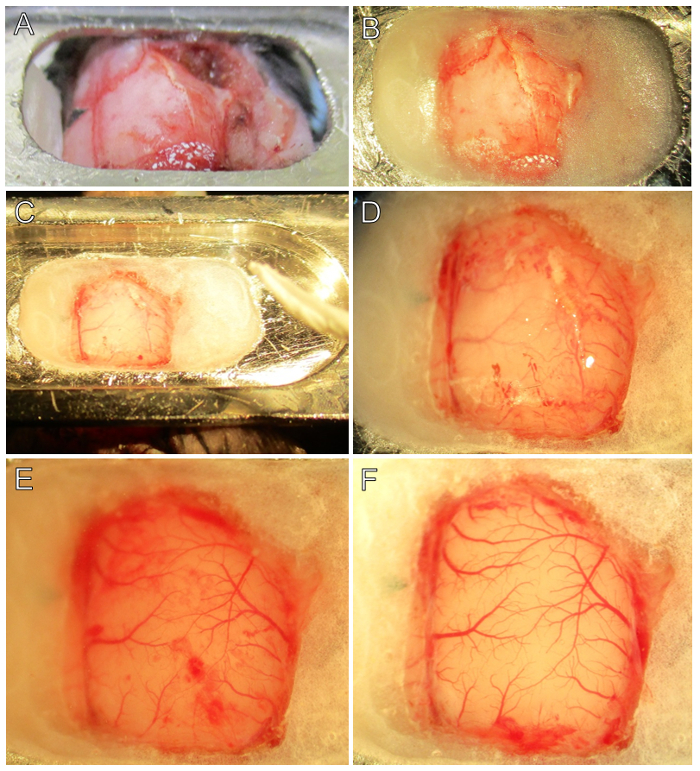
Mark the location of bregma either with a fine tip marker or cut a small triangular piece tape and point a corner at bregma (Figure 2A) to orient the underlying brain regions.
Prior to affixing the head plate ensure the skull is completely dry. The skull will quickly air dry after application of the gel foam soaked in brain buffer, if more drying is needed, use cotton tip swabs. This step is crucial for head plate fixation (Figure 1B). NOTE: If there is periosteum left on the skull, or if the skull is not dry before gluing on the head plate, it will likely detach. If this happens, gently remove the head plate and start over. Bleeding may occur during this process; allow a few minutes for it to clot, and then gently remove. This process is not recommended to be repeated more than twice.
Apply ethyl cyanoacrylate glue around the bottom edge of the head plate16, and glue the head plate over the craniotomy area (Figure 1D & Figure 2A).
- Fill the opening space between the skull and head plate with dental cement leaving only the craniotomy area exposed. Wait for the dental cement to dry and harden, typically 5 - 10 min (Figure 2B).
- Once the cement is set, briefly fill the well with brain buffer and allow to soak for 3 - 5 min. Use a rolled tissue to remove brain buffer before drilling (Figure 2C).
Outline the surgical area by lightly scoring the surface of the skull with a dental drill. Use a pneumatic drill (set to maximum of 20 PSI), with a FG ¼ burr, and controlled with a variable speed foot pedal.
Gently trace the drill along the original scoring to deepen it, ensuring the drill does not penetrate through the skull into the brain (Figure 2D). Take turns every few minutes between drilling and dabbing the skull surface with moistened rolled tissues. This will reduce heating and drying of the skull from mechanical friction and prolonged exposure. NOTE: The skull will quickly air dry after application of the wet gel foam. If more drying is needed, use cotton tip swabs. Caution: The skull is uneven in thickness. For example, the parietal-temporal ridge is the thickest area, while skull regions near the midline and squamosal landmarks are relatively thin.
During drilling, periodically check for buckling of the skull by gently pressing on it with forceps or the non-moving drill bit. When the bone begins to buckle, stop drilling and immerse the entire window in brain buffer. NOTE: If blood rushes out of an area, it may suggest that the dura has been damaged. If this is the case, place a semi-wet gel foam over the area and try to soak up the blood while gently applying pressure to the gel foam with a cotton tip swab.
Wait for at least 5 min before skull removal to soften the bone and to reduce the chance of the dura sticking to the bone, making the skull removal process easier.
Perform the skull removal process while the skull is submerged in brain buffer. NOTE: If a portion of the skull remains stubbornly attached, a #11 scalpel blade can be used to gently score the skull. Take extreme care to not puncture the blade through the skull and into the brain.
Beginning from the anterior edge, gently pry the loose skull from the dura using forceps. NOTE: If a small amount of bleeding occurs during the skull removal process, remove the buffer with a transfer pipette or syringe, and then replace with new buffer.
Once the bone is loose and "floating" on the dura, firmly grip the bone with forceps and lift the bone from the dura. Ensure the bone never penetrates into the brain.
- To control bleeding, roll the corner of a tissue into a point and remove most of the buffer from the cranial well. Quickly apply gel foam, pre-soaked in buffer, to the bleeding area while adding very light pressure with a cotton tip swab. NOTE: The bleeding usually comes from the edge of the bone or the surface of the dura; both cases are normal and bleeding will quickly stop if no major blood vessels are damaged. If bleeding continues, blood may fill the entire window, forming a clot sheet over the imaging area.
- To remove the clot sheet, carefully pick up pieces of clotted blood from the imaging area while leaving the blood clot intact around the source of the ruptured blood vessel. Take care to not remove the blood clot from the bleed source as this may cause even more blood loss. Irrigate the surface of the brain with brain buffer to wash away any blood.
- Take care to avoid touching the delicate brain tissue or adding foreign material to the brain; repeat until bleeding has stopped, for approximately 2 - 5 min (Figure 2E). NOTE: At this point, the craniotomy is ready for preparing the cranial window (see step 5). If needed, remove the dura before implanting the cranial window (see step 4).
4. Dura Removal
NOTE: Dura removal requires extreme care and may take over 15 min.
Draw away excess buffer from the craniotomy. While maintaining a moist surface, grab a small piece of dura with forceps and gently tear the dura.
Use forceps and spring scissors to gently tear and cut away the dura.
Drop more brain buffer on the brain surface to float the dura and help it separate from the brain. Continue until all dura is removed from the cranial window site. When performed correctly the brain will appear to be very clean with distinct blood vessels and no blemishes (compare Figure 2E with 2F). Caution: Some areas of the dura are attached to small arterioles on the surface of the brain (e.g., near the midline proximal to the parietal association area), and removal of such can rupture the arteriole. In such cases, it may be better to leave a small piece of dura intact over top of the arteriole. The VSD may not penetrate that small area, but this is preferred to having major bleeding.
Fix the brain surface in agarose as soon as possible to minimize movement from pulsation and to prevent further swelling (see step 5).
5. Preparing Cranial Window
Spray the cover glass with 70% ethanol and use an air canister to gently dry. Ensure the glass is completely clean with no spots or dust present.
Prepare 1.3% agarose by heating 200 mg of agarose powder dissolved in 15 mL of brain buffer. Set the microwave to high and heat for 10 - 15 s at a time, gently stirring in between, until all agar is dissolved. Note: Ensure no bubbles or particles are present, as these will interfere with imaging.
Place a thermometer in the hot agarose and cool the agarose down to just above solidifying temperature (~ 40 °C). NOTE: Running cool water over the outside of the agarose container may speed the cooling process. Gently stir continuously to ensure no bubbles or particulates are present.
Immediately before applying the agar, remove the brain buffer from the cranial well. Draw up the agarose with a transfer pipette and drop the agarose directly on the brain. Quickly place the cover slip over the surface and fasten the cover slip with agarose drops on the corners.
6. Euthanasia
NOTE: In our experience, this procedure takes experienced surgeons at least 3 - 4 practice surgeries to attain greater than 90% success rate. Less experienced surgeons may require even more practice. During the craniotomy or durotomy, the brain may sustain damage, such as if the drill punches through the bone into the brain. Some minor damage at the edge of craniotomy may be permissible. However, if the brain does not look "clean" with bright red undamaged blood vessels and white cortex, the experiment may need to be terminated. Examples of poor preparations include those with dead blood vessels, or when the cortex is marked with torn or damaged blood vessels. If any of these signs are present, the experiment will unlikely yield high quality data. Whether the surgery/experiment was successful or not, humanely euthanize the mouse at the completion of the experiment.
Deeply anesthetize with at least 3.5% isoflurane. Then, give an intraperitoneal injection of sodium pentobarbital at 300 mg/kg. Ideally, if the needle is inserted into the liver, death will be very rapid (< 1 min).
If perfusion is required, verify that the animal is deeply anesthetized before proceeding (see step 1.3).
Alternatively, if perfusion is not required, wait for a minimum of 5 min and then verify that the mouse is dead. Confirm the absence of respiration, heartbeat, as well as a lack of pain withdrawal and corneal reflexes. Also, observe for pale blue/white coloring of extremities and darkening of the blood vessels over the cortex.
Representative Results
To study the interactions between cortical areas within a single hemisphere, we used a large craniotomy extending across the sagittal sinus and 5 - 6 mm lateral. This cranial window included primary (motor, somatosensory, visual, auditory), secondary (motor, visual), and association (retrosplenial, cingulate, parietal association) cortices of right brain hemispheres (Figure 3A). For this work we used voltage sensitive dye (VSD) imaging, which reflects changes in the membrane potential3. This protocol would also be useful for other extrinsic (e.g., calcium17 and glutamate18 imaging) or intrinsic imaging experiments. When stimulating the hindlimb, forelimb, whiskers, visual, or auditory system of lightly anesthetized mice using 0.5% isoflurane, we observed consensus patterns of cortical depolarization (Figure 3B). Consistent with previous studies5,19,20,21,22, we found that brief tactile stimulation of C2 barrel cortex led to activation of primary somatosensory areas, as well as "islands" of responses within functionally related areas. For example, the primary motor cortex (M1) or secondary representation of somatosensory cortex (S2; Figure 3Bi). A single 1 ms tone pip (25 kHz) stimulation led to the activation of primary auditory cortex (A1) approximately 20 ms after auditory stimulation (Figure 3Bii). Over the next few milliseconds, the depolarization spread across the auditory cortex and passed to neighboring secondary somatosensory cortex. Approximately 25 ms after tone onset, a secondary cortical depolarization would emerge, located 1.0 ± 0.2 mm medial and 1.9 ± 0.1 mm posterior relative to bregma (n = 9 mice). This is approximately the location of the parietal association area (ptA). The VSD signal then propagated to the midline area where other cortical association areas are located including retrosplenial (RS) and cingulate cortex (CG). Therefore, auditory stimulation led to the activation of two separate focal areas, from which traveling waves of VSD depolarization spread to a larger area within midline cortex. Focal stimulation of the contralateral eye with a 1 ms green LED pulse, led to activation of the primary visual cortex within 40 ms (Figure 3Bv). This primary activation of the visual cortex was followed by: (1) Spatial expansion of VSD depolarization into neighboring areas located medially, laterally, and anterior to the initial activated area; (2) Depolarization of a second medial cortical region approximately 50 ms after stimulation (n = 8 experiments) located along sagittal suture. This was similar to sensory stimulations of forelimb (Figure 3Biii), hindlimb (Figure 3Biv), C2 whisker, or audition. Evoked VSDI responses from sensory stimulation of the forelimb, hindlimb or audition initially activated the respective primary sensory cortices, followed by an anisotropic spread of activity, as well as midline activation of cortex around 20 - 40 ms after stimulation. This result was similar to responses from visual and whisker stimulation. The propagation of sensory-evoked activity along these midline routes and frequent activation of same regions by spontaneous activity6 may suggest that these regions are the central hub of the connection core of the mouse cortex, in which sensory information may integrate with spontaneous cortical activity.
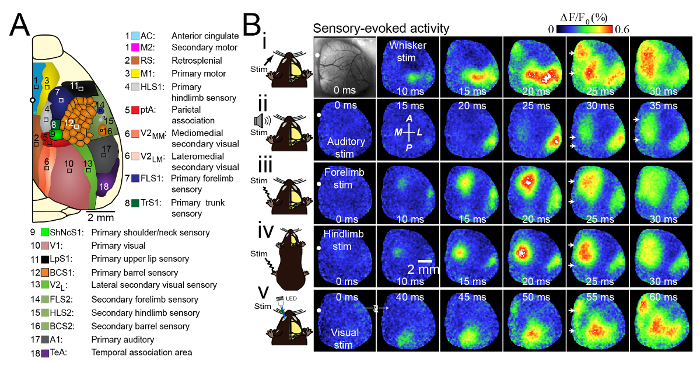
Figure 1. Surgical Setup and Preparation. (A) Mouse head is shaved, cleansed, rotated approximately 30° for lateral exposure, and secured with the blunt end of ear bars. Isoflurane anesthetic is delivered via a nose piece and teeth holder. The mouse is covered with self adhesive plastic wrap for increased sterility and warmth. (B) Close-up showing skin and periosteum removed from the parietal skull plate, the temporal muscle is untouched. (C) The temporal muscle is removed exposing the temporal plate and squamosal bone, note the superficial vein is undamaged. (D) Prior to fixation, the head plate is positioned into the correct location with wax. Please click here to view a larger version of this figure.
Figure 2. Step-by-step Surgical Procedure. (A) The head-plate is attached to the skull with ethyl cyanoacrylate glue at the anterior and posterior locations. Note the location of bregma (black piece of triangular tape). (B) The cranial window is prepared by applying thickened dental cement between the head-plate and skull. Note that bregma and the squamosal landmarks remain visible. (C) Following drying of cement, brain buffer is added to soften the skull and to prevent adhesion of the dura. A rolled tissue will help removing brain buffer prior to drilling. (D) The edges of the craniotomy have been scored. Note the vasculature is more easily seen through the thinned bone near the dental cement edges. (E) The parietal and temporal skull plates have been removed and the dura is visible. Note blemishes of blood on the dura from minor bleeding, which is normal. Careful examination under 2 - 4X magnification will reveal two layers of blood vessels, one in the dura and the other in the pia. (F) The dura is removed revealing a pristine cortex. Pial vasculature is bright red with no blemishes present. Note the stray pieces of white colored dura at the edges of the cranial window. In this example, there was a minor dural bleed at the posterior portion of the cranial window, which quickly clotted. Please click here to view a larger version of this figure.
Figure 3. Unique and Consensus Activation Patterns during Multiple Forms of Sensory Stimulation. (A) Schematic of the unilateral craniotomy showing the imaged cortical regions. (B) Photomicrograph of the wide unilateral craniotomy with bregma indicated by a white circle in each image. Patterns of cortical activation are shown in a mouse anesthetized with isoflurane (0.5%) after (i) stimulation (stim) of the contralateral C2 whisker, (ii) auditory stimulation, (iii) contralateral forelimb stimulation, (iv) contralateral hindlimb stimulation and (v) visual stimulation of the contralateral eye with a light-emitting diode (LED). There was midline activation after all forms of sensory stimulation (white arrows) at 10 - 25 ms after primary sensory cortex activation. The responses are the mean of 20 trials. The image second from the left in the second row (ii) indicates the anterior (A), posterior (P), medial (M) and lateral (L) directions. Modified with permission from Mohajerani, et al., 2013. Please click here to view a larger version of this figure.
Discussion
This innovative protocol for a large cranial window enables simultaneous imaging over the temporal and parietal areas of the cerebral cortex. Combined with optical imaging, it can help to reveal neural dynamics within cortical areas during spontaneous and stimulus-induced activity. This expansive craniotomy also exposes a large extension of the cortical vasculature network, including the proximal end of the middle cerebral artery (MCA), enabling in vivo imaging of blood flow and direct manipulation of lateral vessels for ischemic models. This technique will be of great use for recently developed lines of mice expressing voltage and calcium indicator proteins23. These mice offer the practical advantage of bypassing the need for incubating voltage sensitive dyes on the cortex. These extrinsic dyes take time to adequately penetrate the brain tissue (~ 60 - 90 min) and are limited by their mild toxicity. Large craniotomies have also been previously utilized to study the developing rat brain with VSDI11. Newborn rats have a much larger head and is comparable in size with adult mice. This affords researchers with a unique opportunity to study developmental problems in neuroscience, albeit not with transgenic mice.
The main limitations of this method are the inability for chronic experiments. The curvature of the skull makes the drilling process more challenging and time consuming than smaller craniotomies. For this large craniotomy, it is vital to position the head with the central suture and squamosal landmarks to be parallel to the focusing plane of the lens. While some distortion of the brain is expected from the curvature of the brain, these are overcome by focusing into the superficial layers of the cortex. This problem is further alleviated by obtaining numerous repetitions of stimulation and averaging. In summary, our large craniotomy technique is widely applicable for the study of current problems in neurobiology.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by a Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant #40352, Campus Alberta for Innovation Program Chair, Alberta Alzheimer Research Program to MHM, and NSERC CREATE in BIF doctoral fellowship and AIHS postgraduate fellowship to MK. We thank Pu Min Wang for the development of this protocol and for surgical training, and Behroo Mirza Agha and Di Shao for husbandry.
References
- Sigler A, Mohajerani MH, Murphy TH. Imaging rapid redistribution of sensory-evoked depolarization through existing cortical pathways after targeted stroke in mice. Proc Natl Acad Sci U S A. 2009;106(28):11759–11764. doi: 10.1073/pnas.0812695106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AY, et al. Two-photon microscopy as a tool to study blood flow and neurovascular coupling in the rodent brain. J Cereb Blood Flow Metab. 2012;32(7):1277–1309. doi: 10.1038/jcbfm.2011.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinvald A, Hildesheim R. VSDI: a new era in functional imaging of cortical dynamics. Nat Rev Neurosci. 2004;5(11):874–885. doi: 10.1038/nrn1536. [DOI] [PubMed] [Google Scholar]
- Blinder P, Shih AY, Rafie C, Kleinfeld D. Topological basis for the robust distribution of blood to rodent neocortex. Proc Natl Acad Sci U S A. 2010;107(28):12670–12675. doi: 10.1073/pnas.1007239107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohajerani MH, et al. Spontaneous cortical activity alternates between motifs defined by regional axonal projections. Nat Neurosci. 2013;16(10):1426–1435. doi: 10.1038/nn.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohajerani MH, McVea DA, Fingas M, Murphy TH. Mirrored bilateral slow-wave cortical activity within local circuits revealed by fast bihemispheric voltage-sensitive dye imaging in anesthetized and awake mice. J Neurosci. 2010;30(10):3745–3751. doi: 10.1523/JNEUROSCI.6437-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippert MT, Takagaki K, Xu W, Huang X, Wu JY. Methods for voltage-sensitive dye imaging of rat cortical activity with high signal-to-noise ratio. J Neurophysiol. 2007;98(1):502–512. doi: 10.1152/jn.01169.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld T, Kerschensteiner M. In vivo imaging of the diseased nervous system. Nat Rev Neurosci. 2006;7(6):449–463. doi: 10.1038/nrn1905. [DOI] [PubMed] [Google Scholar]
- Kerr JN, Denk W. Imaging in vivo: watching the brain in action. Nat Rev Neurosci. 2008;9(3):195–205. doi: 10.1038/nrn2338. [DOI] [PubMed] [Google Scholar]
- Aronoff R, et al. Long-range connectivity of mouse primary somatosensory barrel cortex. Eur J Neurosci. 2010;31(12):2221–2233. doi: 10.1111/j.1460-9568.2010.07264.x. [DOI] [PubMed] [Google Scholar]
- McVea DA, Mohajerani MH, Murphy TH. Voltage-sensitive dye imaging reveals dynamic spatiotemporal properties of cortical activity after spontaneous muscle twitches in the newborn rat. J Neurosci. 2012;32(32):10982–10994. doi: 10.1523/JNEUROSCI.1322-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweetnam D, et al. Diabetes impairs cortical plasticity and functional recovery following ischemic stroke. J Neurosci. 2012;32(15):5132–5143. doi: 10.1523/JNEUROSCI.5075-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin YQ, et al. In vivo field recordings effectively monitor the mouse cortex and hippocampus under isoflurane anesthesia. Neural Regeneration Research. 2016;11(12):1951–1955. doi: 10.4103/1673-5374.197136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PS, et al. Comparison of stimulus-evoked cerebral hemodynamics in the awake mouse and under a novel anesthetic regime. Scientific Reports. 2015;5:12621. doi: 10.1038/srep12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyweriga M, Mohajerani MH. Optogenetics: Methods and Protocols. In: Kianianmomeni A, editor. Methods in Molecular Biology. Vol. 1408. Humana Press Inc; 2016. pp. 251–265. [DOI] [PubMed] [Google Scholar]
- Grutzendler J, Gan WB. Imaging in neuroscience and development : a laboratory manual. Cold Spring Harbor Laboratory Press; 2005. [Google Scholar]
- Vanni MP, Murphy TH. Mesoscale transcranial spontaneous activity mapping in GCaMP3 transgenic mice reveals extensive reciprocal connections between areas of somatomotor cortex. J Neurosci. 2014;34(48):15931–15946. doi: 10.1523/JNEUROSCI.1818-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, et al. Resolution of High-Frequency Mesoscale Intracortical Maps Using the Genetically Encoded Glutamate Sensor iGluSnFR. J Neurosci. 2016;36(4):1261–1272. doi: 10.1523/JNEUROSCI.2744-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AW, Mohajerani MH, LeDue JM, Wang YT, Murphy TH. Mesoscale infraslow spontaneous membrane potential fluctuations recapitulate high-frequency activity cortical motifs. Nat Commun. 2015;6:7738. doi: 10.1038/ncomms8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim DH, et al. In vivo Large-Scale Cortical Mapping Using Channelrhodopsin-2 Stimulation in Transgenic Mice Reveals Asymmetric and Reciprocal Relationships between Cortical Areas. Front Neural Circuits. 2012;6 doi: 10.3389/fncir.2012.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferezou I, et al. Spatiotemporal dynamics of cortical sensorimotor integration in behaving mice. Neuron. 2007;56(5):907–923. doi: 10.1016/j.neuron.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Mohajerani MH, Aminoltejari K, Murphy TH. Targeted mini-strokes produce changes in interhemispheric sensory signal processing that are indicative of disinhibition within minutes. Proc Natl Acad Sci U S A. 2011;108(22):E183–E191. doi: 10.1073/pnas.1101914108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, et al. Transgenic mice for intersectional targeting of neural sensors and effectors with high specificity and performance. Neuron. 2015;85(5):942–958. doi: 10.1016/j.neuron.2015.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]