

Abstract
SPHIRE (SPARX for High-Resolution Electron Microscopy) is a novel open-source, user-friendly software suite for the semi-automated processing of single particle electron cryo-microscopy (cryo-EM) data. The protocol presented here describes in detail how to obtain a near-atomic resolution structure starting from cryo-EM micrograph movies by guiding users through all steps of the single particle structure determination pipeline. These steps are controlled from the new SPHIRE graphical user interface and require minimum user intervention. Using this protocol, a 3.5 Å structure of TcdA1, a Tc toxin complex from Photorhabdus luminescens, was derived from only 9500 single particles. This streamlined approach will help novice users without extensive processing experience and a priori structural information, to obtain noise-free and unbiased atomic models of their purified macromolecular complexes in their native state.
Keywords: Biochemistry, Issue 123, structural biology, electron microscopy, electron cryo-microscopy, cryo-EM, TEM, single particle image processing, software development, single particle analysis, Tc toxin
Introduction
After the development of the direct electron detector technology, the remarkable progress in single particle cryo-EM is currently reshaping structural biology 1. Compared with X-ray crystallography, this technique requires only a small amount of protein material without the need for crystallization, while simultaneously posing fewer restrictions regarding purity of the sample and still allowing determination of structures at near-atomic resolution. Importantly, different compositions or states can now be computationally separated and structure determination of the different conformations can be carried out at unprecedented level of detail. Recently, density maps of challenging molecules could be produced at resolutions allowing de novo model building and thus deep understanding of their mode of action 2,3,4,5.
A wide variety of image processing software packages are available in the 3DEM (3D Electron Microscopy) community (https://en.wikibooks.org/wiki/Software_Tools_For_Molecular_Microscopy) and most of them are under continuous development. Near-atomic resolution has been achieved for proteins exhibiting various molecular weights and symmetries with several different software packages, including EMAN2 6, IMAGIC7, FREALIGN 8, RELION 9, SPIDER 10, and SPARX 11. Each package requires a different level of user expertise and provides a different level of user guidance, automation and extensibility. Moreover, whereas some programs provide complete environments to facilitate all steps of image analysis, others are designed to optimize specific tasks, such as the refinement of alignment parameters starting from a known reference structure. More recently, several platforms have been developed, including APPION 12 and SCIPION 13, that provide a single processing pipeline which integrates approaches and protocols from the different software packages listed above.
To contribute to the current development of cryo-EM, SPARX was re-developed into a new stand-alone and complete platform for single particle analysis, called SPHIRE (SPARX for High-Resolution Electron Microscopy). In order to increase accessibility of the technique for new researchers in the field and to cope with the large amount of data produced by modern fully-automated high-end electron microscopes, the processing pipeline was redesigned and simplified by introducing an easy-to-use Graphical User Interface (GUI) and automating the major steps of the workflow. Moreover, new algorithms were added to allow fast, reproducible and automated structure determination from cryo-EM images. Furthermore, validation by reproducibility was introduced in order to avoid common artifacts produced during refinement and heterogeneity analysis.
Although the program was extensively modified, its appreciated core features were maintained: straightforward open-source code, the modern object-oriented design and Python interfaces for all basic functions. Thus, it was not changed into a black box program, enabling users to study and easily modify the Python code, to create additional applications or modify the overall workflow. This is especially useful for non-standard cryo-EM projects.
Here we present a protocol for obtaining a near-atomic resolution density map from cryo-EM images using the GUI of SPHIRE. It describes in detail all steps required to generate a density map from raw cryo-EM direct detector movies and is not restricted to any particular macromolecule type. This protocol primarily intends to guide newcomers in the field through the workflow and provide important information about crucial steps of the processing as well as some of the possible pitfalls and obstacles. More advanced features and the theoretical background behind SPHIRE will be described elsewhere.
Protocol
NOTE: To follow this protocol, it is necessary to properly install SPHIRE on a system with an MPI installation (currently, a Linux cluster). Download SPHIRE and the TcdA1 dataset from http://www.sphire.mpg.de and follow the installation instructions: http://sphire.mpg.de/wiki/doku.php?id=howto:download. This procedure also installs EMAN2. SPHIRE currently uses EMAN2’s e2boxer for particle selection and e2display for displaying image files. For dose-weighted motion correction of the raw micrograph movies, SPHIRE uses unblur 14. Download the program and follow the installation instructions (http://grigoriefflab.janelia.org/unblur, Grigorieff lab). For interactive visualization of the resulting structures, the protocol will use the molecular graphics program Chimera 15 (https://www.cgl.ucsf.edu/chimera/download.html). A nice tutorial to get familiar with the features used throughout this protocol can be found here: https://www.cgl.ucsf.edu/chimera/data/tutorials/eman07/chimera-eman-2007.html. Instructions on how to submit a parallel job to a cluster from the SPHIRE GUI can be found here: http://sphire.mpg.de/wiki/doku.php?id=howto:submissions. The overall organization of the SPHIRE GUI and the major steps of the workflow performed throughout this protocol are illustrated in Figure 1.
1. PROJECT: Set Constant Parameter Values for This Project
Start the SPHIRE GUI application by typing "sphire &" and the ENTER key at a terminal window.
- Adjust the project-wide parameters (e.g., pixel size, particle radius and symmetry) in the respective input fields of the project settings page and then register these values for all subsequent steps of the workflow.
- Click on the "PROJECT" icon on the bottom right of the left panel to open the project settings page.
- Measure the longest axis of a particle using the e2display.py image interactive display tool, then enter the half of the particle size to "Protein particle radius". If the measurement is in Å, keep in mind to convert the unit to pixels using the pixel size (e.g., if a particle is 200 Å long and the pixel size is 1.2 Å/pixel, then the longest axis of the particle is 200/1.2 = ~166 pixels and the radius 166/2 = 83 pixels).
- Set "Particle box size" to at least 1.5 times of the particle size. Avoid window sizes containing large prime number. Also, remember that the 3D refinement algorithm currently requires an even numbered box size. NOTE: The window should include a margin to account for initial centering errors resulting from picking (the need to shift particles within the window) and for the sufficient background region outside of the particle boundary for a proper CTF correction (especially important for large defocus values16).
- Set "CTF window size" to that of "Particle box size". For projects with low contrast data, use a larger window to obtain smoother estimates of power spectra.
- Set "Point-group symmetry" of the complex (e.g., "C5"). If the symmetry of the target structure is not known, leave it at "C1" (asymmetric). However, if a specific high-order symmetry is identified later on during the processing, change this symmetry setting accordingly and repeat the steps after 2D alignment with ISAC.
- Set "Protein molecular mass" in kDa (approximate value will suffice). Press the "Register settings" button.
2. MOVIE: Align the Frames of Each Movie Micrograph to Correct the Overall Motion of the Sample
- For all movie micrographs, compute the x/y-shifts for all frames and then create their dose-unweighted and dose-weighted motion-corrected average (see Discussion). Note that the former is necessary only for the CTF estimation because the estimation does not perform well with the dose-weighted averages while the latter is used for all the other steps of the structure determination.
- Click on the "MOVIE" icon and then the "Micrograph Movie Alignment" button. Set "Unblur executable path" by selecting the executable file. Set "Input micrograph path pattern " by selecting a raw unaligned movie micrograph and replacing the variable part of the file names with the wildcard "*" (e.g., TcdA1_*.mrc). Specify the path for "Output directory".
- Set "Summovie executable path" by selecting the executable file.
- Set "Number of movie frames" to the number of frames in each movie micrograph. Set the "Microscope voltage" and "Per frame exposure" to the values used during data collection. (For example, if the overall dose is 60 e-/Å2 with 20 frames recorded without pre-exposure, the exposure for every frame is 60/20=3 e-/A2.) Press the "Run Command" button to align the frames of each movie micrograph. NOTE: This will automatically create two output directories containing dose-unweighted and dose-weighted motion-corrected average micrographs, respectively.
3. CTER: Estimate the Defocus and Astigmatism Parameters of the CTF
- Estimate the CTF parameters (defocus and astigmatism; the others are set by the user) for each dose-unweighted average micrograph.
- Click on the "CTER" icon and then the "CTF Estimation" button. To set "Input micrograph path pattern", select a dose-unweighted motion-corrected micrograph, then replace the variable part of the file names with the wildcard "*". Also, specify the path for "Output directory".
- Set "Amplitude contrast" to the value routinely used for the kind of data (ice thickness is a major factor) and microscope voltage in the laboratory (e.g., 10%). Typical values are in the 7 - 14% range17.
- Set "Microscope spherical aberration (Cs)" and "Microscope voltage" used during data collection.
- Set "Lowest frequency" and "Highest frequency" of search range for the CTF model fitting to 0.0285 and 0.285 Å-1 (40 - 4 Å), respectively. Press the "Run command" button to estimate the CTF parameters. NOTE: The CTF parameters will be automatically stored in the partres.txt file in the specified output directory. The CTF estimation of the 112 micrographs was calculated on 96 cores and finished after ~3 min on the Linux cluster used to obtain the representative results.
4. WINDOW: Extract Particles from the Dose-weighted Average Micrographs
- Pick particles manually or automatically from micrographs with e2boxer6 and create coordinate files, each containing a list of particle xy-coordinates within the associated micrograph.
- Click on the "WINDOW" icon and then the "Particle Picking" button. Press the "Run command" button to start e2boxer 6and pick the particles of each micrograph manually or automatically18 (see Discussion). Store the final particle coordinates for each micrograph in the EMAN1 file format (.box). Alternatively, import the coordinate files from other programs after converting them to the EMAN1 format.
- Create particle stacks by extracting particle images from the dose-weighted micrographs (in SPHIRE, particle stack is often simply called "stack").
- Press the "Particle Extraction" button. Specify "Input micrograph path pattern" by selecting a dose-weighted motion-corrected micrograph and then replacing the variable part of the file names with the wildcard "*" (e.g., TcdA1_*.mrc). Similarly, set "Input coordinates path pattern" by selecting a coordinates file (e.g., TcdA1_*.box). Specify the path for "Output directory".
- Set "CTF parameters source" by selecting the CTF parameter file (partres.txt produced in step 3.1). Press the "Run command" button.
- Combine the extracted particle image stacks into a single one.
- Click on the "Particle Stack" button. Specify the path to "Output virtual image stack" using a BDB file path format (e.g., "bdb:Particles/stack", where "Particles" points to the directory containing a BDB data base directory whose name is always EMAN2DB and "stack" refers to a particular image stack within this database). Specify "Input BDB image stack pattern" by selecting a directory starting with "mpi_proc" and then replacing the variable part of the directory names with the wildcard "*" (e.g., Particles/mpi_proc_000 to Particles/mpi_proc_* ). Press the "Run command" button.
5. ISAC: Classification of Particle Images in 2D
- Calculate 2D class averages by aligning particles and clustering them according to their 2D appearance. NOTE: The resulting 2D averages have an improved signal-to-noise ratio (SNR) compared to the individual particle images and are thus used to visually assess the quality and heterogeneity of the dataset, as well as to sort out undesirable images from the stack (e.g., ice crystals, carbon-edges, aggregates, fragments, and etc.)19. Moreover, they will be subsequently used to determine an initial 3D model.
- Click on the "ISAC" icon and then the "ISAC - 2D Clustering" button.Set "Input image stack" by selecting the stack file containing the extracted particles. Specify the path for "Output directory".
- Use 200 - 1000 for "Images per class". Choose the appropriate number considering the expected number of 2D classes (the total number of particles divided by the number of images per class). Adjust this parameter depending on the SNR and the size of the dataset. Increase the number of members per class in case the dataset is excessively noisy. Decrease the number when a low number of particles is available. NOTE: Due to memory limitations, for rather large datasets (>100,000 particles), split the full dataset into subsets, perform ISAC for each subset independently, and combine the results at the end. Detailed instructions for this processing scenario are given in http://www.sphire.mpg.de/wiki/doku.php.
- Check the "Phase-flip" checkbox. Keep the default values for "Target particle radius" and "Target particle image size" in order to speed up the process by automatically shrinking all particle images with these settings. Press the "Run command" button to calculate the 2D class averages. NOTE: This step is computationally demanding and the running time increases significantly with the number of particles and classes as well as the target radius and image size. On a cluster with 96 processes, the 2D classification of ~10,000 particles finished after about 90 min.
- Display and visually inspect the resulting ISAC 2D averages to make sure that their quality is satisfactory (see Discussion).
- Press the "Display Data" button under "UTILITIES". Set "Input files" by selecting the file containing the ISAC 2D averages (class_averages.hdf produced in step 5.1). Press the "Run command" button to display the final reproducible and validated class averages delivered by ISAC.
- Create a new stack including only the particle members of the validated class averages.
- Press the "Create Stack Subset" button. Set "Input image stack" by selecting the same stack file as in the step 5.1.1. Set "ISAC averages" by selecting the ISAC 2D averages (class_averages.hdf produced in step 5.1). Specify the path for "Output directory". Press the "Run command" button.
6. VIPER: Calculate an Initial 3D Model
- Select a small set of the class averages (≥100 images) by deleting all bad class averages and identical views of the particle (see Discussion) and use them to calculate a reproducible initial model using VIPER. Remember that the selection should contain at least 60-80 high quality averages with ~200-500 members each.
- Click on the "VIPER" icon and then the "Display Data" button. Set "Input files" by selecting the ISAC 2D averages (class_averages.hdf produced in step 5.1). Press the "Run command" button.
- Press the mouse middle button somewhere on the graphics window of the e2display, and activate the "DEL " button in the pop-up window. Delete all bad class averages and identical views of the particle (see Discussion). Press the "Save" button to store the remaining 2D class averages to a new file.
- From the selected ISAC averages, generate an initial reference for the subsequent 3D refinement.
- Click on the "Initial 3D Model - RVIPER" button. Set "Input images stack" by selecting the screened class averages (produced in step 6.1). Specify the path for "Output directory".
- Make sure to use the same value for "Target particle radius" as ISAC step 5.1.3. Press the "Run command" button to generate a reproducible ab initio 3D model. NOTE: This step is computationally demanding and the running time increases significantly with number of averages and size of the particles. On a cluster with 96 processes, this job (~100 class averages) finished after ~15 min.
Check if the resulting 3D model is reasonable by taking into account the class averages and in addition its structural integrity (i.e. no disconnected parts and/or directional artifacts). To display the map, use the program Chimera15. At this point, perform a first comparison with a crystal structure of a homologous protein or a domain of the protein of interest if it exists (an example is shown in the section Representative Results).
- For the subsequent 3D refinement, generate an initial 3D reference and a 3D mask from an ab initio 3D model by removing its surrounding noise and rescaling it to match the original pixel size.
- Click on the "Create 3D Reference" button. Set "Input volume" by selecting the ab initio 3D model (average_volume.hdf produced in step 6.2). Specify the path for "Output directory".
- Set "Resample ratio source" by selecting the ISAC shrink ratio file (README_shrink_ratio.txt produced in step 5.1). Press the "Run command" button.
7. MERIDIEN: Refine the Initial 3D Volume
- Refine the 3D volume starting from the initial 3D model.
- Click on the "MERIDIEN" icon, then the "3D Refinement" button. Set "Input image stack" and "Initial 3D reference" by selecting the particle stack and the ab initio 3D model (produced in step 5.3 & 6.4, respectively). Specify the path for "Output directory".
- Set "3D mask" by selecting the 3D mask file (produced in step 6.4). Always use a 3D mask but, especially at an early stage of analysis, use a spherical mask or a soft-edged mask loosely fitted to the reference to avoid introducing bias of incorrect masking.
- Check the "Apply hard 2D mask" checkbox. Set "Starting resolution" to a cutoff frequency value between 20 - 25 Å. Keep in mind that a low-pass filter with this cutoff frequency will be applied to the initial 3D structure to reduce initial model bias.
- Check the specifications of cluster used for this process and then set "Memory per node" to the available memory in gigabytes. Press the "Run command" button to refine the 3D volume starting from the initial 3D model in a fully automated manner. NOTE: This procedure will divide the dataset into two halves, refine the two models independently and output two raw volumes, each from only half of the particles. It is computationally demanding and the running time will increase significantly with the number of particles. On this cluster, the meridian refinement finished after ~2.5 h running on 192 processes (~8,000 particles, 352 box size).
- Create a soft-edged 3D mask from the refined volume for the subsequent sharpening step.
- Click on the "Adaptive 3D Mask" button. Set "Input volume" by selecting one of the unfiltered half-volumes (produced in step 7.1). Specify the path for "Output mask".
- Set a value of "Binarization threshold". Use Chimera to make sure that, at this particular threshold, the noise is clearly outside the volume of interest in the solvent region of the unfiltered half maps and all densities of the protein are still connected to each other. Press the "Run command" button to create the soft-edge 3D mask. NOTE: The main body of the resulting mask (consisting from voxels whose values are >0.5) should tightly fit the particle structure but still enclose all densities of interest. The soft-edge fall-off should be at least 8-10 pixel wide.
- Merge the two unfiltered half-volumes obtained by the 3D refinement. Then, sharpen the merged volume by adjusting the power spectrum based on the modulation transfer function (MTF) of the detector, the estimated B-factor, and the FSC (Fourier Shell Correlation) estimate of the resolution.
- Select the "Sharpening" button. Set "First unfiltered half-volume" and "Second unfiltered half-volume" by selecting the corresponding files (vol_0_unfil.hdf and vol_1_unfil.hdf produced in step 7.1). Always use "B-factor enhancement". Typically, keep the default value in order to estimate the B-factor value from the input dataset using the range between the final resolution frequency and 10 Å. Alternatively, specify an ad-hoc value (e.g., -100).
- Keep the default value for "Low-pass filter frequency" to apply an FSC-based filter.
- Set "User-provided mask" by selecting the 3D mask (produced in step 7.2). Remember that the reported resolution will be determined using FSC with this mask. Press the "Run command" button to sharpen the refined 3D volume.
- Generate the 3D angular distribution map from the projection directions of all particles estimated by the 3D refinement step above.
- Click on the "Angular Distribution" button. Set "Alignment parameter file" by selecting the file (final_params.txt produced in step 7.1), and press the "Run command" button.
Visually inspect the sharpened 3D model using Chimera. Make sure that the structure appears reasonable considering the achieved resolution (see Discussion).
Visually inspect the angular distribution using Chimera. Verify that the distribution covers approximately evenly the entire 3D angular space. Keep in mind that, for symmetric structures, the distribution is restricted within the unique asymmetric triangle.
8. SORT3D: Sort 3D Heterogeneity by Focusing on the Highly Variable Regions
- Calculate the 3D variability map from the particle stack used in the 3D refinement.
- Click on "SORT3D" icon and then "3D Variability Estimation" button. Set "Input image stack" by selecting the same screened particle stack given to the 3D refinement step 7.1.1. Specify the path for "Output directory".
- Keep the default value for "Number of projections". NOTE: The images from the angular neighborhood will be used to estimate the 2D variance at each 3D projection angle. The larger the number, the less noisy the estimate but the lower the resolution and rotational artifacts are more pronounced.
- Check the "Use CTF" checkbox. Press the "Run command" button.
- Use the 3D variability map to create a focus mask for the 3D clustering step below.
- Select the "Binary 3D Mask" button. Set "Input volume" by selecting the 3D variability map (produced in step 8.1). Specify the file path for "Output mask".
- Set "Binarization threshold" by using the output of the "Level" field in the "Volume Viewer" of Chimera. Press the "Run command" button.
- Sort particle images into homogeneous structural groups by focusing on structurally highly variable regions.
- Press the "3D Clustering - RSORT3D" button. Set "Input 3D refinement directory" by selecting the output directory of the 3D refinement (produced in step 7.1). Specify the path for "Output directory".
- Set "3D mask" by selecting the soft-edged 3D mask (produced in step 7.2). Set "Focus 3D mask" by selecting the binarized 3D variability map (produced in step 8.2).
- For large datasets, use at least 5,000-10,000 for "Images per group". Keep in mind that the program always keeps the number of images per group lower than this setting. Adjust the value by considering the expected number of 3D groups (the total number of particles divided by the "Images per group" value), the dataset, the SNR, and the degree of heterogeneity. Start with ~5-10 initial 3D groups, if a sufficient number of particles is available, unless a higher number of distinct structural states in the dataset is expected.
- Use at least 3,000-5,000 particles for "Smallest group size". Note that the program will disregard groups comprising a lower number of images than the setting of "Smallest group size". Press the "Run command" button to perform the 3D clustering. NOTE: RSORT3D is subdivided into two steps. The first "sort3d" step sorts out 3D heterogeneity. Then, it reconstructs the volumes of each homogeneous structural group using the 3D alignment parameters determined by the 3D refinement step above. The second "rsort3d" step finds out reproducible members of each group by carrying out a two-way comparison of the two independent sorting runs. Then, it reconstructs homogeneous structures using only the reproducibly assigned particles. On a cluster with 96 cores, this job (~8,000 particles, 352 box size) finished after about 3 h.
After the program has finished, use Chimera to select a homogeneous 3D group. Select the structure of the highest apparent resolution, typically associated with the most populous group. Make sure that the selected structure is visually reasonable by taking into account the 2D class averages and biological aspects of the protein of interest (see Discussion). If there are other volumes that have an almost identical structure at similar resolution, consider them as emerging from a single homogeneous 3D group.
- Perform a local refinement against the particle members of the most homogeneous 3D group (with the highest resolution).
- Click on the "Local Subset Refinement" button. Set "Subset text file path" by selecting the text file containing the particle IDs of the selected group (e.g., Cluster0.txt produced in step 8.3). Set "3D refinement directory" by selecting the output directory of the previous 3D refinement (produced in step 7.1).
- Set "Restarting iteration" to the one where the highest resolution is achieved in the previous 3D refinement. Press the "Run command" button to perform a local refinement of the selected population of particles.
Similar to step 7.2, create a soft-edged 3D mask from an unfiltered final half-volume reconstructed by the local subset refinement.
Similar to step 7.3, merge two unfiltered final half-volumes derived by the local subset refinement and sharpen the merged volume. However, do not filter the sharpened volume this time. NOTE: Should the heterogeneity analysis in step 8.4 indicate several distinct states at comparable resolution, one may want to refine all different states independently.
9. LOCALRES: Estimate the Local Resolution of the Final 3D Volume
- Estimate the local resolution of the 3D volume obtained from the homogeneous set of particles.
- Click on the "LOCALRES" icon and then "Local Resolution" button. Set "First half-volume" and "Second half-volume" by selecting the unfiltered final half-volumes of the local subset refinement (produced in step 8.5). Set "3D mask" by selecting the soft-edged 3D mask produced in step 8.6. Specify the file path for "Output volume".
- Keep the default value of 7 pixels for "FSC window size". Remember that this setting defines the size of window where the local-real-space correlation is computed; larger window sizes produce smoother resolution maps at the expense of local resolvability.
- Keep the default value 0.5 of "Resolution cut-off" for resolution criterion. NOTE: For each voxel, the program will report the local resolution as the frequency at which the local FSC drops below the selected resolution threshold. A threshold lower than 0.5 is not recommended, because lower correlation values have high statistical uncertainty. Therefore, the corresponding local resolution will vary strongly between voxels.
- For "Overall resolution", set the absolute resolution estimated in the sharpening after the local subset refinement (step 8.7). Press the "Run command" button to calculate the local resolution of the volume.
- Apply the 3D local filter to the volume sharpened after the local subset refinement by using the 3D local resolution map.
- Click on the "3D Local Filter" button. Set "Input volume" by selecting the sharpened but unfiltered 3D volume (produced in step 8.7). Similarly, set "Local resolution file" and "3D mask" (produced in step 9.1 and 8.6, respectively). Remember that the 3D mask defines the region where the local filtering will be applied. Specify the file path for "Output volume". Press the "Run command" button to apply the 3D local filter.
Use Chimera to visually inspect the final 3D model and the 3D local resolution map (produced in step 9,2 and 9.1, respectively). Select the "Surface color" option to color the 3D volume according to the local resolution. Keep in mind that the distribution of local resolution should be smooth (see Discussion).
Representative Results
The protocol described above was executed starting from 112 direct detector movies of the A component of the Photorhabdus luminescens Tc complex (TcdA1) 20,21,22. This dataset was recorded on a Cs-corrected electron cryo-microscope with a high-brightness field emission gun (XFEG), operated at an acceleration voltage of 300 kV. The images were acquired automatically with a total dose of 60 e-/Å-2 at a pixel size of 1.14 Å on the specimen scale. After alignment of the movie frames (Protocol Step 2), the resulting motion-corrected averages had isotropic Thon rings extending to high-resolution (Figure 2a). The individual particles were easily visible and well separated (Figure 2b). Particles were then picked using the swarm tool of e2boxer 18 (Protocol Step 4.1). In this case, an appropriate threshold was set using the more selective option (Figure 2c). The 112 digital micrographs yielded 9,652 particles. The majority of the extracted images (Protocol Step 4.2) contained well-defined particles and their box size was ~1.5 times larger than the particle size, as recommended (Figure 2d). Next, using ISAC, a 2D heterogeneity analysis was performed (Protocol Step 5). It yielded 98 class averages (Figure 3a). Using these 2D class averages, an ab initio model was calculated using VIPER (Protocol Step 6) at intermediate resolution (Figure 3b). This model shows excellent agreement with the crystal structure of TcdA1 previously solved at 3.9 Å resolution 22 (Figure 3c). This ab initio model was used as an initial template for the 3D refinement (MERIDIEN), yielding a 3.5 Å (0.143 criterion) reconstruction (Protocol Step 7) from only ~40,000 asymmetric units (Figure 4). This near-atomic resolution map was obtained within 24 h, using up to 96 CPUs for the steps of the workflow that benefit from multiple cores.
For the 3D variability analysis (Protocol step 8), only 2,000 particle images per group were used in step 8.3.3 (i.e. the process starts with 5 initial 3D groups) and 200 images for the smallest group size in step 8.3.4 due to the small number of particles (~10,000). The analysis revealed localized flexibility mainly at the N-terminal region of the complex that contains the His tag used for purification (Figure 5a). Indeed, twelve N-terminal residues and the His tag were not resolved in the previously published crystal structure of TcdA1 22 and this most probably disordered region remained unresolved in the present cryo-EM density, likely due to its flexibility. Additional variability was detected at the receptor-binding domains and the BC-binding domain (Figure 5a). Due to the overall satisfactory resolution of the structure and the rather small size of the dataset, this heterogeneity was decided to be tolerable and therefore a focused 3D classification 23 was not performed. Finally, the local resolution of the final density map was computed (Protocol step 9.1, Figure 5b) and the sharpened 3D map was locally filtered (Protocol step 9.2). A volume of this quality can be used for de novo model building using Coot 24 or any other refinement tool (Figure 6).
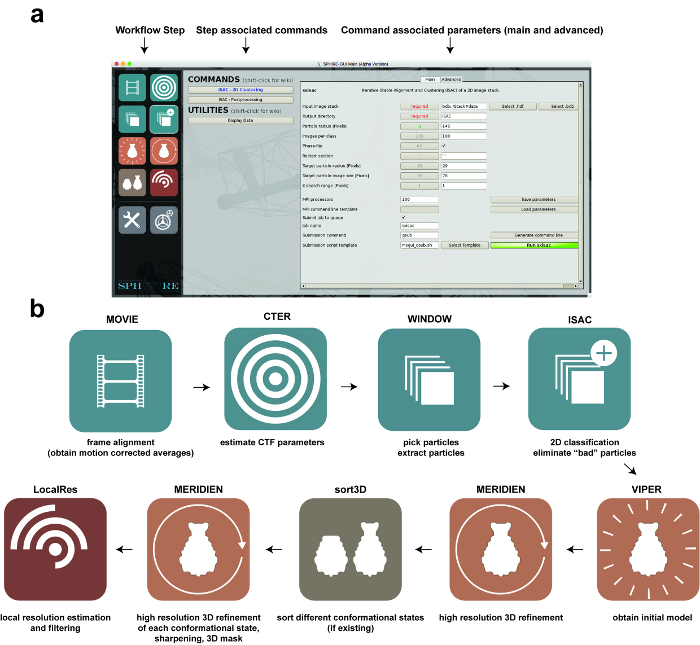 Figure 1: Image Processing using SPHIRE. (a) The GUI of the SPHIRE software package. A specific step of the workflow can be activated by selecting the respective pictogram on the left side of the GUI ("workflow step"). The commands and utilities associated with this step of the workflow will appear in the central area of the GUI. After selecting one of the commands, the respective parameters are shown on the right area of the GUI. Advanced parameters usually do not require modification of the preset default values. (b) Stages in the workflow of single particle image processing using the SPHIRE GUI. Please click here to view a larger version of this figure.
Figure 1: Image Processing using SPHIRE. (a) The GUI of the SPHIRE software package. A specific step of the workflow can be activated by selecting the respective pictogram on the left side of the GUI ("workflow step"). The commands and utilities associated with this step of the workflow will appear in the central area of the GUI. After selecting one of the commands, the respective parameters are shown on the right area of the GUI. Advanced parameters usually do not require modification of the preset default values. (b) Stages in the workflow of single particle image processing using the SPHIRE GUI. Please click here to view a larger version of this figure.
 Figure 2: Motion Correction and Particle Extraction. (a, b) Typical high-quality, low-dose, drift-corrected digital micrograph recorded at a defocus of 1.7 µm. Note the isotropic Thon rings extending to a resolution of 2.7 Å in the power spectrum (a) and the well discernible particles in the 2D image (b). (c) Particle selection using e2boxer. Green circles indicate selected particles. (d) Typical raw particles extracted from the dose-weighted micrograph. Scale bars = 20 nm. Please click here to view a larger version of this figure.
Figure 2: Motion Correction and Particle Extraction. (a, b) Typical high-quality, low-dose, drift-corrected digital micrograph recorded at a defocus of 1.7 µm. Note the isotropic Thon rings extending to a resolution of 2.7 Å in the power spectrum (a) and the well discernible particles in the 2D image (b). (c) Particle selection using e2boxer. Green circles indicate selected particles. (d) Typical raw particles extracted from the dose-weighted micrograph. Scale bars = 20 nm. Please click here to view a larger version of this figure.
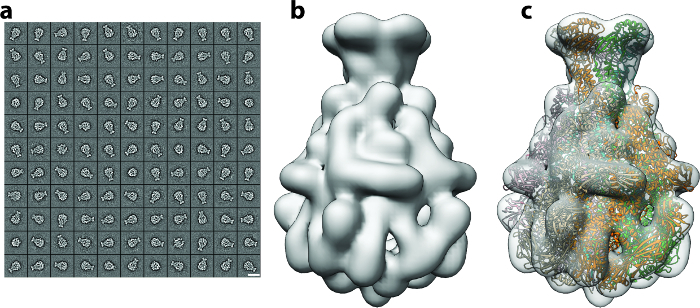 Figure 3: 2D Clustering and Initial Model Generation. (a) Gallery of 2D class averages, with the majority representing side views of the particle. Scale bar = 20 nm. (b) Ab initio 3D map of TcdA1 obtained using RVIPER from the reference-free class averages. (c) Rigid-body fitting of the TcdA1 crystal structure (ribbons) (pdb-id 1VW1) into the initial cryo-EM density (transparent gray). Please click here to view a larger version of this figure.
Figure 3: 2D Clustering and Initial Model Generation. (a) Gallery of 2D class averages, with the majority representing side views of the particle. Scale bar = 20 nm. (b) Ab initio 3D map of TcdA1 obtained using RVIPER from the reference-free class averages. (c) Rigid-body fitting of the TcdA1 crystal structure (ribbons) (pdb-id 1VW1) into the initial cryo-EM density (transparent gray). Please click here to view a larger version of this figure.
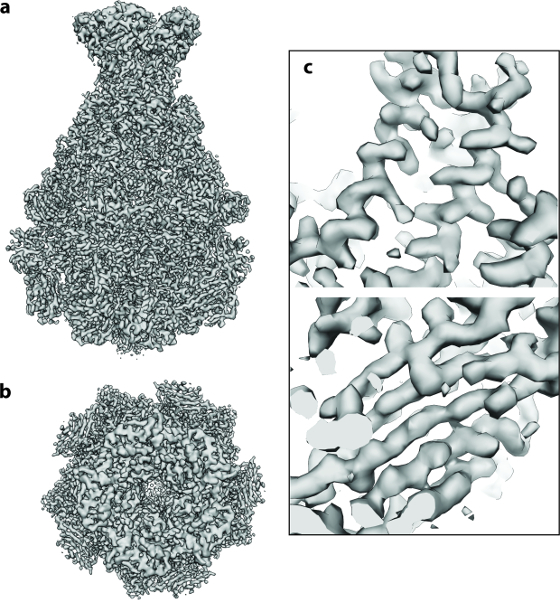 Figure 4: Cryo-EM 3D Structure of TcdA1. (a, b) Final 3.5 Å density map of TcdA1 computed using ~9,500 particle images: (a) side and (b) top view. (c) Representative areas of the cryo-EM density for an α-helix and a β-sheet. Please click here to view a larger version of this figure.
Figure 4: Cryo-EM 3D Structure of TcdA1. (a, b) Final 3.5 Å density map of TcdA1 computed using ~9,500 particle images: (a) side and (b) top view. (c) Representative areas of the cryo-EM density for an α-helix and a β-sheet. Please click here to view a larger version of this figure.
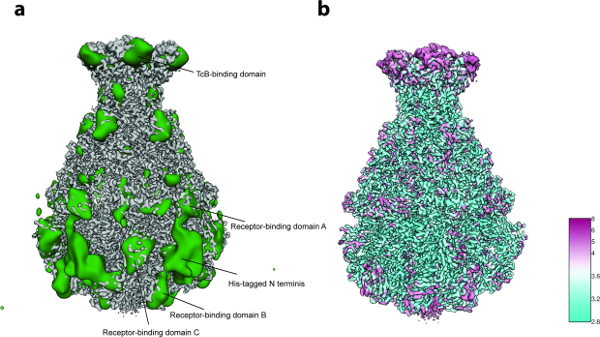 Figure 5: Variability Analysis and Local Resolution. (a) Surface of the sharpened TcdA1 cryo-EM map (gray) and the variability map (green). For better clarity, the variability map was low-pass filtered to 30 Å. (b) Surface rendering of the TcdA1 sharpened cryo-EM map colored according to local resolution (Å). Note the topological agreement between areas of high variability and low local resolution. Please click here to view a larger version of this figure.
Figure 5: Variability Analysis and Local Resolution. (a) Surface of the sharpened TcdA1 cryo-EM map (gray) and the variability map (green). For better clarity, the variability map was low-pass filtered to 30 Å. (b) Surface rendering of the TcdA1 sharpened cryo-EM map colored according to local resolution (Å). Note the topological agreement between areas of high variability and low local resolution. Please click here to view a larger version of this figure.
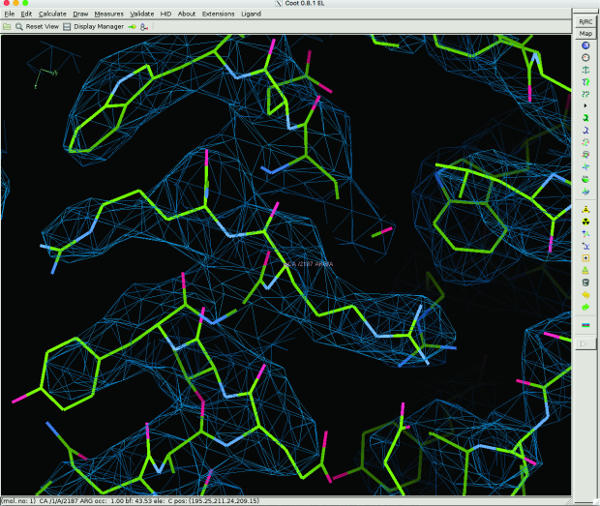 Figure 6: 3D Model Building of TcdA1 using Coot. Representative regions of the cryo-EM density and the atomic model are shown for an α-helix. The atomic model was built de novo using Coot. Please click here to view a larger version of this figure.
Figure 6: 3D Model Building of TcdA1 using Coot. Representative regions of the cryo-EM density and the atomic model are shown for an α-helix. The atomic model was built de novo using Coot. Please click here to view a larger version of this figure.
Discussion
Single particle cryo-EM has shown a rapid development in the recent years and delivered numerous atomic resolution structures of macromolecular complexes of major biological significance25. In order to support the large number of novice users that are currently entering the field, we developed the single particle image analysis platform SPHIRE and present here a walk-through protocol for the entire workflow including movie alignment, particle picking, CTF estimation, initial model calculation, 2D and 3D heterogeneity analysis, high-resolution 3D refinement and local resolution estimation and filtering.
The protocol described here is intended as a short guide to 3D structure determination using cryo-EM micrographs of the protein of interest and with the help of computational tools provided by the stand-alone GUI of SPHIRE.
The main feature of the workflow is that most of the procedures need to be run only once, since they rely on the concept of validation by reproducibility19 and do not require parameter tweaking. This automatic validation mechanism is a main advantage of SPHIRE over other software packages since the results tend to be objective as well as reproducible and, most importantly, obtainable at an acceptable computational cost. The pipeline provides in addition a wealth of diagnostic information for experienced users to conduct further independent validation and assessment with own methods. Nevertheless, a novice user who has at least elemental theoretical background in structural biology and electron microscopy should be able to obtain near-atomic resolution structures using own data and the automated validation procedures.
However, obtaining a near-atomic resolution structure is not always straightforward and the result will highly depend on the quality of the sample and the input data. For the procedures presented here, it is assumed that a sufficient number of high quality unaligned raw EM movies are available, with their averages showing clearly discernible homogeneous and randomly orientated single particles. In general, there are no restrictions regarding symmetry, size or overall shape of the molecule, but a low molecular weight can be a limiting factor, especially when the protein has a featureless globular shape. Usually, analysis of larger, well-ordered particles with high point-group symmetry is less demanding. Therefore, it is strongly recommend for novice users to run the present protocol first with a well-characterized cryo-EM dataset. Either the SPHIRE tutorial data (http:/sphire.mpg.de) or one of the EMPIAR submitted datasets (https://www.ebi.ac.uk/pdbe/emdb/empiar/) with raw movies are a good starting point.
When processing own data, it is very likely that some datasets or some of the images will not satisfy certain quality criteria. In this context, in addition to the automated stability and reproducibility checks, performed by the program for major steps of the workflow, it is still recommend for users to visually inspect the results at certain "checkpoints" of the protocol, especially if the final reconstruction is not satisfactory.
The first visual inspection can be done at the micrograph level after the movie alignment (Protocol step 2) and the CTF estimation (Protocol step 3). The resulting motion-corrected averages should show clearly discernible and well-separated single particles and their power spectra should show clearly discernible, isotropic Thon rings. The spatial frequency to which they are visible defines, in most cases, the highest resolution to which the structure can in principle be ultimately determined. Examples of a motion-corrected average of sufficient quality and its power spectrum are shown in the section "Representative results". Outlier images that might have a negative impact on the final result can be removed with the help of SPHIRE's Drift and CTF assessment GUI tools (http://sphire.mpg.de/wiki/doku.php).
With regard to particle screening, the crucial step in the SPHIRE pipeline is the 2D classification using ISAC (Protocol step 5.2). Here, the user should control that the reproducible 2D class averages identified automatically by the program adopt a range of orientations sufficient to quasi-evenly cover the angular space. If the quality of the class averages is not satisfactory (noisy and/or blurry images) and/or the number of reproducible class averages is very low, consider improving the auto-picking quality, optimizing dataset imaging or sample preparation. In most cases, it is not possible to calculate a reliable reconstruction from a dataset that does not yield good 2D class averages. Examples of high quality 2D class averages are shown in the section "Representative results".
At least 100 class averages are required to obtain a reliable initial 3D model using RVIPER in an automated manner (Protocol step 6.1). For this step, the user should select the averages with the highest quality and include as many different orientations of the particle as possible. The quality of the initial model is critical for the success of the subsequent high-resolution 3D refinement.
In other software packages, 3D classification is sometimes performed to remove "bad" particles8,9. However, in SPHIRE most of these particles are automatically eliminated already during 2D classification using ISAC. Thus, it is recommended to perform the computationally intensive step of 3D sorting only if the reconstruction and the 3D variability analysis indicate heterogeneity of the dataset.
Most importantly, the user should always carefully inspect the resulting 3D volumes carefully (Protocol step 9.3), and confirm that the features of the respective density agree well with the nominal resolution. At a resolution of <9 Å, rod-like densities corresponding to α-helices become visible. At a resolution <4.5 Å, densities corresponding to strands in β-sheets are normally well separated and bulky amino acids become visible. A high-resolution map (<3 Å) should show clearly discernible side chains, thus allowing building of an accurate atomic model.
Results obtained to date demonstrate that, with the help of SPHIRE's automated reproducibility tests and minimal visual inspections, the present protocol is generally applicable to any type of single particle cryo-EM project. Representative results of each processing step are shown for the reconstruction of the TcdA1 toxin of Photorhabdus luminescens 21, which has been solved to near-atomic resolution. Density maps of similar quality can be used to construct reliable atomic models by de novo backbone tracing as well as reciprocal or real-space refinement, and thus provide a solid structural framework for the understanding of complex molecular mechanisms.
ACCESION CODES:
The coordinates for the EM structure and the unprocessed movies have been deposited in the Electron Microscopy Data Bank and the Electron Microscopy Pilot Image Archive under accession numbers EMD-3645 and EMPIAR-10089, respectively.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
We thank D. Roderer for providing us TcdA1 micrographs. We thank Steve Ludtke for his ongoing support of EMAN2 infrastructure. This work was supported by funds from the Max Planck Society (to S.R.) and the European Council under the European Union's Seventh Framework Programme (FP7/2007-2013) (grant no. 615984) (to S.R.) and grant from the National Institutes of Health R01 GM60635 to P.A.P.).
References
- Nogales E. The development of cryo-EM into a mainstream structural biology technique. Nature Methods. 2016;13(1):24–27. doi: 10.1038/nmeth.3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M, Cao E, Julius D, Cheng Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature. 2013;504(7478):107–112. doi: 10.1038/nature12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X-C, Yan C, et al. An atomic structure of human γ-secretase. Nature. 2015;525(7568):212–217. doi: 10.1038/nature14892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecken JVD, Heissler SM, Pathan-Chhatbar S, Manstein DJ, Raunser S. Cryo-EM structure of a human cytoplasmic actomyosin complex at near-atomic resolution. Nature. 2016;534(7609):724–728. doi: 10.1038/nature18295. [DOI] [PubMed] [Google Scholar]
- von der Ecken J, Müller M, Lehman W, Manstein DJ, Penczek PA, Raunser S. Structure of the F-actin-tropomyosin complex. Nature. 2015;519(7541):114–117. doi: 10.1038/nature14033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Peng L, et al. EMAN2: An extensible image processing suite for electron microscopy. Journal of Structural Biology. 2007;157(1):38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- van Heel M, Harauz G, Orlova EV, Schmidt R, Schatz M. A new generation of the IMAGIC image processing system. Journal of Structural Biology. 1996;116(1):17–24. doi: 10.1006/jsbi.1996.0004. [DOI] [PubMed] [Google Scholar]
- Grigorieff N. FREALIGN: high-resolution refinement of single particle structures. Journal of Structural Biology. 2007;157(1):117–125. doi: 10.1016/j.jsb.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Scheres SHW. RELION: implementation of a Bayesian approach to cryo-EM structure determination. Journal of Structural Biology. 2012;180(3):519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh TR, Gao H, et al. SPIDER image processing for single-particle reconstruction of biological macromolecules from electron micrographs. Nature Protocols. 2008;3(12):1941–1974. doi: 10.1038/nprot.2008.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohn M, Tang G, et al. SPARX, a new environment for Cryo-EM image processing. Journal of Structural Biology. 2007;157(1):47–55. doi: 10.1016/j.jsb.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Lander GC, Stagg SM, et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. Journal of Structural Biology. 2009;166(1):95–102. doi: 10.1016/j.jsb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Rosa-Trevìn JM, Quintana A, et al. Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. Journal of Structural Biology. 2016;195(1):93–99. doi: 10.1016/j.jsb.2016.04.010. [DOI] [PubMed] [Google Scholar]
- Grant T, Grigorieff N. Measuring the optimal exposure for single particle cryo-EM using a 2.6 Å reconstruction of rotavirus VP6. eLife. 2015;4:06980. doi: 10.7554/eLife.06980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, et al. UCSF Chimera?A visualization system for exploratory research and analysis. Journal of Computational Chemistry. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Penczek PA, Fang J, Li X, Cheng Y, Loerke J, Spahn CMT. CTER-rapid estimation of CTF parameters with error assessment. Ultramicroscopy. 2014;140:9–19. doi: 10.1016/j.ultramic.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank J. Three-Dimensional Electron Microscopy of Macromolecular Assemblies. Oxford University Press; 2006. [Google Scholar]
- Woolford D, Ericksson G, et al. SwarmPS: rapid, semi-automated single particle selection software. Journal of Structural Biology. 2007;157(1):174–188. doi: 10.1016/j.jsb.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Yang Z, Fang J, Chittuluru J, Asturias FJ, Penczek PA. Iterative Stable Alignment and Clustering of 2D Transmission Electron Microscope Images. Structure/Folding and Design. 2012;20(2):237–247. doi: 10.1016/j.str.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatsogiannis C, Merino F, et al. Membrane insertion of a Tc toxin in near-atomic detail. Nature Publishing Group. 2016. [DOI] [PubMed]
- Gatsogiannis C, Lang AE, et al. A syringe-like injection mechanism in Photorhabdus luminescens toxins. Nature. 2013;495(7442):520–523. doi: 10.1038/nature11987. [DOI] [PubMed] [Google Scholar]
- Meusch D, Gatsogiannis C, et al. Mechanism of Tc toxin action revealed in molecular detail. Nature. 2014;508(7494):61–65. doi: 10.1038/nature13015. [DOI] [PubMed] [Google Scholar]
- Penczek PA, Frank J, Spahn CMT. A method of focused classification, based on the bootstrap 3D variance analysis, and its application to EF-G-dependent translocation. Journal of Structural Biology. 2006;154(2):184–194. doi: 10.1016/j.jsb.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta crystallographica. Section D, Biological crystallography. 2010;66:486–501. doi: 10.1107/S0907444910007493. Pt 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway E. The revolution will not be crystallized: a new method sweeps through structural biology. Nature. 2015;525(7568):172–174. doi: 10.1038/525172a. [DOI] [PubMed] [Google Scholar]