

Abstract
Intracellular calcium (Ca2+) transients evoked by extracellular stimuli initiate a multitude of biological processes in living organisms. At the center of intracellular calcium release are the major intracellular calcium storage organelles, the endoplasmic reticulum (ER) and the more specialized sarcoplasmic reticulum (SR) in muscle cells. The dynamic release of calcium from these organelles is mediated by the ryanodine receptor (RyR) and the inositol 1,4,5-triphosphate receptor (IP3R) with refilling occurring through the sarco/endoplasmic reticulum calcium ATPase (SERCA) pump. A genetically encoded calcium sensor (GECI) called CatchER was created to monitor the rapid calcium release from the ER/SR. Here, the detailed protocols for the transfection and expression of the improved, ER/SR-targeted GECI CatchER+ in HEK293 and C2C12 cells and its application in monitoring IP3R, RyR, and SERCA pump-mediated calcium transients in HEK293 cells using fluorescence microscopy is outlined. The receptor agonist or inhibitor of choice is dispersed in the chamber solution and the intensity changes are recorded in real time. With this method, a decrease in ER calcium is seen with RyR activation with 4-chloro-m-cresol (4-cmc), the indirect activation of IP3R with adenosine triphosphate (ATP), and inhibition of the SERCA pump with cyclopiazonic acid (CPA). We also discuss protocols for determining the in situ Kd and quantifying basal [Ca2+] in C2C12 cells. In summary, these protocols, used in conjunction with CatchER+, can elicit receptor mediated calcium release from the ER with future application in studying ER/SR calcium related pathologies.
Keywords: Biochemistry, Issue 123, Calcium imaging, sarco-endoplasmic reticulum, genetically-encoded calcium indicator, calcium signaling, CatchER, fluorescence microscopy
Introduction
The spatio-temporal attributes of intracellular calcium (Ca2+) transients activate various biological functions1. These Ca2+ signaling events are triggered extracellularly through different stimuli and controlled intracellularly by the major Ca2+ storage organelle and by numerous Ca2+ pumps, channels, and Ca2+ binding proteins. Ca2+ transients can be significantly altered as a result of defects with signal modulation, leading to different diseases2. Because of the speed and intricacy of the Ca2+ signaling system, with the endo- (ER) and sarcoplasmic reticulum (SR) at the center, genetically-encoded Ca2+ probes that have been optimized for mammalian expression with fast kinetics are needed to observe global and local Ca2+ changes in different cells3.
The ER and the SR, its counterpart in muscle cells, are the major intracellular Ca2+ storage organelles and act as Ca2+ sinks that help to amplify the Ca2+ signal4. The ER/SR is an integral part in Ca2+ signaling with dual roles as a transmitter and receiver of signals5. The ryanodine receptor (RyR) and the inositol 1,4,5-triphosphate receptor (IP3R) are Ca2+ release receptors located on the membranes of the ER/ SR that are regulated by Ca2+ 6. Other agents directly or indirectly stimulate the function of these receptors. 4-chloro-m-cresol (4-cmc) is a potent agonist of the RyR, having a 10 fold higher sensitivity than caffeine for inducing SR Ca2+ release where both are regularly employed to study RyR-mediated Ca2+ release in healthy and diseased cells7. ATP increases IP3-mediated Ca2+ release through the IP3R8. ATP binds to the purinergic receptor P2YR, a G-protein coupled receptor (GPCR), triggering the production of IP3 that binds to the IP3R to release Ca2+ from the ER9,10. The sarco-endoplasmic reticulum calcium ATPase (SERCA) pump is a P-type ATPase pump, also located on the ER/SR membrane that reduces cytosolic Ca2+ and refills the ER/SR by actively pumping the ion into the ER/SR lumen11. Specific inhibitors of the SERCA pump include thapsigargin, from Thapsia garganica, and cyclopiazonic acid (CPA), from Aspergillus and Penicillium. CPA has a low affinity for the pump and reversibly blocks the Ca2+ access point12. Thapsigargin, on the other hand, irreversibly binds to the Ca2+ free pump at residue F256 in the M3 helix with nanomolar affinity11. Analyzing and quantifying the changes involved in Ca2+ stimulated events has been and remains a challenge. Since the ER/SR is the major subcellular Ca2+ containing compartment with a central function in the propagation of the Ca2+ signal, much work has been focused on understanding ER/SR Ca2+ signaling5.
The creation of synthetic Ca2+ dyes helped to advance the field and practice of Ca2+ imaging. Although dyes, such as Mag-Fura-2, have been widely used to measure compartmentalized Ca2+ in different cells,13,14,15 they have limitations such as uneven dye loading, photobleaching, and the inability to be targeted to specific organelles. The discovery of the green fluorescent protein (GFP) and the advancement of fluorescent protein-based Ca2+ probes has propelled the field of Ca2+ imaging forward16. Some of the existing GECIs are Förster resonance energy transfer (FRET) pairs involving yellow fluorescent protein (YFP), cyan fluorescent protein (CFP), calmodulin and the M13 binding peptide17,18. Troponin C-based GECIs are also available as FRET pairs of CFP and Citrine and as single fluorophore probes19,20,21.Others, such as GCaMP2 and R-GECO are single fluorophore sensors involving calmodulin22,23. To overcome the limitations of narrow tuning of Kd's and cooperative binding associated with multiple Ca2+ binding sites found in their Ca2+ binding domains24, a novel class of calcium sensors was created by designing a Ca2+ binding site on the surface of the beta barrel in a chromophore sensitive location of enhanced green fluorescent protein (EGFP)25,26. This highly touted sensor, called CatchER, has a Kd of ~0.18 mM, a kon near the diffusion limit, and a koff of 700 s-1. CatchER has been used to monitor receptor-mediated ER/SR calcium release in different mammalian cell lines such as HeLa, HEK293, and C2C1225. Because of its fast kinetics, CatchER was used in flexor digitorum brevis (FDB) muscle fibers of young and old Friend Virus B NIH Jackson (FVB) mice to reveal that more Ca2+ remains in the SR after 2 s of depolarization in the FDB fibers of old mice compared to that of young mice27. To overcome its low fluorescence at 37 °C, which hinders its applications in calcium imaging of mammalian cells, we have developed an improved version of CatchER called CatchER+. CatchER+ exhibits enhanced fluorescence at 37 °C for better application in mammalian cells. Additional mutations were incorporated into CatchER to improve the thermostability and fluorescence at 37 °C28,29, to create CatchER+. CatchER+ exhibits a six-fold increase in its signal to noise ratio (SNR) over CatchER30.
Here, the protocols for the culture and transfection of HEK293 and C2C12 cells with CatchER+ and its application for monitoring ER/SR receptor-mediated calcium transients are presented. Representative results are shown for CatchER+ expressed in HEK293 cells treated with 4-cmc, CPA and ATP. We also provide a protocol for determining the in situ Kd of CatchER+ in C2C12 myoblast cells and quantification of basal [Ca2+].
Protocol
1. Slide Preparation
Place 22 mm x 40 mm glass microscope slides in 6 cm cell culture dishes, 1 slide per dish.
Expose each side of each slide as well as the 6 cm dish to ultraviolet (UV) light for 15-20 min to sterilize in a sterile hood.
Cover the dishes with slides inside with parafilm and store at 4 °C until ready to use.
2. Preparation of Media, Buffers, Solutions, and Reagents
Prepare 1 L of Hank's balanced salt solution (HBSS) in deionized water and add 10 mM HEPES, 5 mM NaHCO3, and 1 mM EGTA. Adjust to pH 7.2-7.3 with NaOH. Filter buffer with a 0.22 µm filter into an autoclaved bottle.
Prepare 900 mL of high glucose Dulbecco's Modified Eagle's Medium (DMEM) h in deionized water. Add 44 mM NaHCO3 and adjust the pH to 7.2-7.3 with HCl. Filter media with a 0.22 µm filter into an autoclaved bottle. Add 100 mL of fetal bovine serum (FBS) to the media to make the FBS concentration 10%. Store at 4 °C.
Prepare 1 L of intracellular buffer (125 mM KCl, 25 mM NaCl, 10 mM HEPES, 0.2 mM CaCl2, 0.5 mM EGTA, 0.2 mM MgCl2) in deionized water, and adjust the pH to 7.25. The final [Ca2+] will be ~100 nM. Before use, add 0.5 mM ATP. Store at room temperature.
Prepare 1 L of Ringer's buffer (121 mM NaCl, 2.4 mM K2HPO4, 0.4 mM KH2PO4, 10 mM HEPES, and 1.2 mM MgCl2) in deionized water, and adjust the pH to 7.2. Store at room temperature. To use, aliquot 50 mL into a 50 mL tube and add 1.8 mM Ca2+ and 10 mM glucose.
Prepare 1 L of KCl rinse solution (125 mM KCl, 25 mM NaCl, 10 mM HEPES, 0.2 mM MgCl2) in deionized water, and adjust the pH to 7.25.
Dissolve 5 mg of ionomycin in 1 mL of dimethyl sulfoxide (DMSO). Aliquot 30 µL into microtubes and store at -20 °C.
Prepare the cyclopiazonic acid (CPA) stock by dissolving CPA into DMSO. Make sure the final percent of DMSO is ≤ 1% for the CPA added to the cells to prevent apoptosis. Prepare 20 mM 4-chloro-m-cresol (4-cmc) by dissolving into filtered H2O and letting it dissolve overnight by shaking at 37 °C. Prepare the ATP stock by dissolving in filtered H2O to 100 mM.
To determine the in situ Kd, prepare 15 mL each of 1 mM EGTA, 0.2 mM, 0.6 mM, 2 mM, 5 mM, 10 mM, 20 mM, 50 mM, and 100 mM Ca2+ in KCl buffer using a 100 mM EGTA stock and a 1 M CaCl2 stock dissolved in deionized water.
3. Cell Culture
Culture C2C12 myoblast and HEK293 cells according to ATCC protocols for each using 10 cm cell culture dishes in a sterile UV hood. NOTE: C2C12 cells should not be allowed to grow higher than ~70% confluence since the myoblasts will begin to differentiate into myotubes. Both HEK293 and C2C12 cells grow readily and should not be allowed to grow past 100% confluency to maintain cell integrity. Additionally, cells should not be passaged more than 10 times before using them for an experiment to preserve cell health.
4. Transfection of HEK293 Cells
Seed HEK293 cells onto sterilized 22 mm x 40 mm glass microscope slides in 6 cm dishes so they are ~70% confluent the day of transfection.
The next day, follow the manufacturer's protocol for the transfection reagent to transfect cells using 2 µg of CatchER+ cDNA and a 1:3 (weight/volume) DNA:transfection reagent ratio in the reduced serum media (e.g. Opti-MEM).
Empty the DMEM media from the dish, and add 3 mL of reduced serum media. Add DNA:transfection reagent mixture to the dish and incubate for 4-6 h at 37 °C.
After incubation, wash the cells with 5-6 mL of HBSS. Discard the HBSS from the dish and replace with 3 mL of fresh DMEM and incubate them at 37 °C for 48 h to allow expression of the GECI.
- Transfection of C2C12 myoblast cells
- For C2C12 myoblast cells, trypsinize cells by adding enough trypsin to cover the bottom of dish (1-2 mL). Place the dish in a 37 °C incubator for 2-6 min to allow the trypsin to loosen the cells from the bottom of the dish.
- Once the incubation is complete, remove the trypsin and aspirate the cells in 8 mL of DMEM. Pipette the cells thoroughly with the DMEM to evenly disperse and re-suspend te cells and seed them onto sterile coverslips in 6 cm cell culture dishes containing 3 mL of DMEM so that the final confluence is 60%.
- Transfect the cells the same day as outlined in step 4.2-4.3. Incubate for 24 h at 37 °C.
- Repeat step 4.4.
5. Preparation of Slide and Fluorescence Microscope
Turn on microscope and light source, then open the Simple PCI program. Allow the microscope to warm up for 15-20 min or until the charge-coupled device (CCD) camera cools to -65 °C. Prepare the chamber and slide with cells while waiting.
Cover the bottom of the low profile open diamond bath imaging chamber with a thin layer of sealant, using a cotton swab.
Take the slide containing transfected cells out of the incubator and gently rinse three times with 1 mL of Ringer's buffer with 10 mM glucose and 1.8 mM Ca2+ preheated to 37 °C.
Leave 1 mL of Ringer's buffer in the dish, to prevent cells from drying, and use forceps to take the slide out of the dish. Allow excess solution to absorb onto a laboratory tissue by touching the edge of the slide to a laboratory tissue. Then place onto the side of the chamber containing the grease with cells facing up towards grease and secure chamber onto the stage mount using a screwdriver.
Add 1 mL of Ringer's buffer to the chamber to prevent cells from drying while mounting the chamber onto the stage and completing the rest of the microscope setup. Once the vacuum suction hose is attached to the chamber, the final volume the chamber holds is ~450 µL.
Switch the microscope objective to the 40X oil immersion objective.
Use lens paper and lens cleaning solution to clean and dry the objective. Do not rub the objective. Dab gently until the surface is clean.
Add one drop of the immersion oil to the objective.
Place the mount onto the microscope stage and add the vacuum tip to the chamber. Once the vacuum suction hose is attached to the chamber and turned on, the final volume the chamber holds is ~450 µL. This final volume is level with the sides of the chamber. It is imperative for the chamber solution to be level, during the experiment, to prevent the solution from spilling down into the objective due to overfilling.
Raise the objective and use the coarse adjustment until the oil on the objective touches the bottom of the slide.
Using the bright field mode, adjust the gain to 175 and the exposure time to 0.03 s to focus the cells.
Once cells are focused, switch to fluorescence mode using 488 nm excitation. Change the exposure time to 0.07 s. Locate a field of view that has enough cells that are healthy with adequate fluorescence to image.
Once a field of view has been chosen, take pictures in fluorescence and bright field mode.
Circle a region of interest (ROI) in the cells or circle the whole cell to record the intensity from the chosen area.
6. Imaging Drug-induced Ca2+ Transients and In Situ Kd Calibration
Start intensity measurement with frame rate set at one every 5 s.
Allow the baseline intensity to stabilize for 20-30 frames (100-150 s). If needed, decrease the frames/s during this step to prevent photobleaching.
Calculate the amount of 4-cmc needed from a 20 mM stock based on the final chamber volume of ~450 µL to make a final concentration of 200 µM. Make dilutions of the stock as necessary.
Carefully take 60 µL of Ringer's buffer from the chamber and place in a microcentrifuge tube. Dilute the calculated amount of 4-cmc with this solution and add back to the chamber to evoke the intended response from the cells being imaged.
Add the calculated amount of 4-cmc to the microcentrifuge tube and mix by pipetting.
Gently add the solution over the field of view while simultaneously adding the event marker in the intensity measurement.
Allow time for the drug to bind to the receptor and subsequent signal decrease, then wash with 3-6 mL of Ringer's buffer with 1.8 mM Ca2+ and 10 mM glucose while simultaneously adding the event marker. Since the chamber volume is 450 µL and is connected to a vacuum, adding 3-6 mL of buffer ensures that the former solution containing the drug has been washed away and replaced with the newly added solution.
Repeat 6.1-6.7 for 100 µM ATP and 15 µM CPA, using a new slide of transfected cells for each assay.
Once the measurement is complete, end the data collection in the intensity measurement window in the Simple PCI program.
Take fluorescence and brightfield pictures of the cells.
Open the data file for the experiment. Here, re-circle the cells or regions of interest to re-calculate the intensity values, if needed.
- To determine the in situ Kd in C2C12 myoblast cells, follow the protocol as outlined in section 4.2 and section 5 and put 1 mL of 0 mM Ca2+ Ringer's buffer in the chamber.
- Permeabilize the cells with 0.002-0.005% saponin in intracellular buffer for 15-30 s to empty the cells of any CatchER+ that may be in the cytosol. Wash the cells with 3-6 mL of 1 mL EGTA in KCl buffer with 10 µM ionomycin. Allow the intensity to decrease until a plateau is reached.
- Rinse with 3-6 mL of KCl buffer with 10 µM ionomycin and allow the intensity to stabilize, then add each Ca2+ solution detailed in step 2.7. Allow the intensity to reach a plateau between each addition.
To calibrate the sensor for basal [Ca2+] quantification, follow step 6.12. Use the EGTA and the 100 mM Ca2+ buffer from step 2.7 to get the minimum and maximum fluorescence intensity.
7. Data Processing
Download the spreadsheet file of the intensity measurement data.
Normalize the data for each cell to the baseline intensity (F/F0). Plot the normalized data versus time in any graphing program.
To calculate the % change, subtract the normalized peak value from 1 and multiply by 100. Calculate the average and standard deviation if multiple cells were imaged.
To calculate the signal to noise ratio, SNR, open up the image file saved from the experiment in an image processing software, such as ImageJ, and then measure the signal, transfected cells, and noise, background. Calculate the SNR using the equation SNR = signal/noise.
To calculate the in situ Kd from the collected fluorescence imaging data, average the intensity data, comprising the plateaus, after the addition of each Ca2+ concentration and EGTA (0 mM Ca2+). Calculate the fractional saturation (relative intensity) using θ = (F – Fmin)/(Fmax- Fmin), where θ is the relative intensity, F is the fluorescence at any point, Fmin is the minimum fluorescence intensity, and Fmax is the maximum fluorescence intensity, to see the change in the intensity of the probe with the addition of Ca2+ compared to the minimum intensity. Plot the relative intensity data against the [Ca2+] in any graphing and curve fitting program. Use the 1:1 binding equation θ = [MT]/(Kd + [MT]) translated for any graphing and curve fitting program to calculate the Kd. MT is the total metal concentration.
To quantify the basal calcium from the calibration outlined in 6.13, average the intensity data, comprising the plateaus, after the addition of 1 mM EGTA (Fmin) and 100 mM Ca2+ (Fmax). Use the calibration equation [Ca2+] = Kd([F – Fmin)/(Fmax – F)] to calculate the basal calcium.
Representative Results
This section will illustrate the results that were achieved using the previously described methods using the optimized ER/SR-targeted GECI CatchER+ to monitor changes in ER/SR Ca2+ through different receptor mediated pathways.
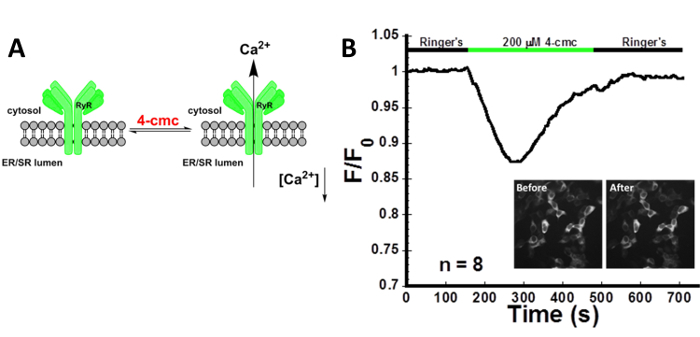
Figure 1 illustrates ER emptying through the RyR stimulated with 200 µM 4-cmc. 4-cmc is an agonist of the RyR. Addition of the drug induces a decrease in ER calcium as measured by the decrease in the CatchER+ fluorescence intensity. As marked, the protocol outlined here allows the visualization and measurement of receptor-mediated Ca2+ transients using CatchER+ which provides information about the changes in ER Ca2+.
As depicted in Figure 2, the addition of 15 µM CPA, to reversibly block the SERCA pump, produces a large decrease in the fluorescence intensity that forms a plateau. As the function of the SERCA pump is inhibited, the ER Ca2+ release channels are still actively releasing Ca2+ into the cytosol causing the fluorescence intensity to decrease. The cells recover after washing CPA away with Ringer's buffer containing 1.8 mM Ca2+ and 10 mM glucose. These results confirm the ability of CatchER+ to monitor Ca2+ transients specific to the SERCA pump.
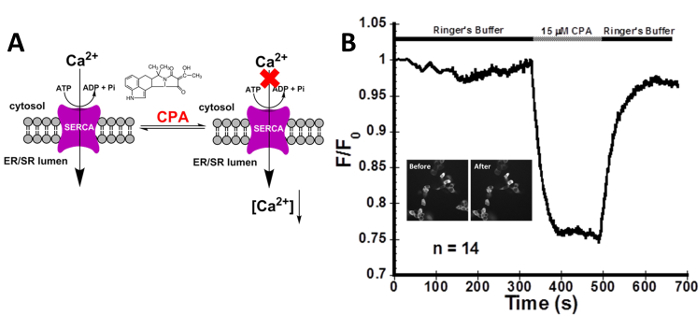
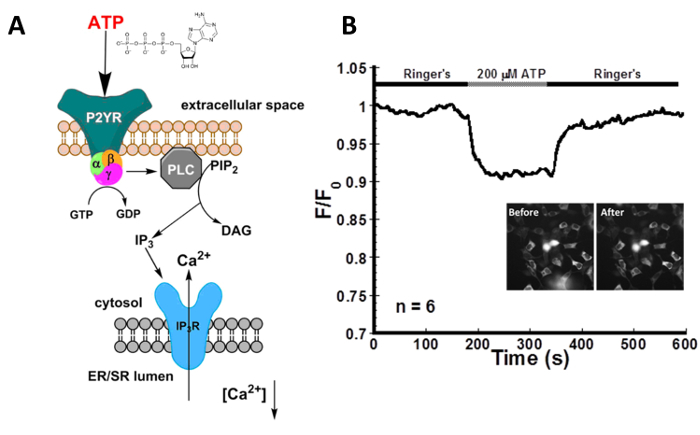
Figure 3 shows representative data for the indirect activation of the IP3R with 200 µM ATP in HEK293 cells transfected with CatchER+. ATP binds to the P2Y receptor, a G-protein coupled receptor, which generates IP3 that activates Ca2+ release through the IP3R. Although the change is small, the addition of 200 µM ATP produces a visible decrease in signal intensity. Washing the cells with Ringer's buffer containing 1.8 mM Ca2+ and 10 mM glucose allows the ER to refill and the signal to return to the baseline. These results highlight the ability of CatchER+ to monitor IP3R-mediated calcium release through indirect stimulation.
In Figure 4, CatchER+ was transfected into C2C12 myoblast cells to determine the in situ Kd. The data was collected as outlined in the protocol. C2C12 myoblast cells were permeabilized with 0.005% saponin for 15-30 s then washed with 1 mM EGTA in KCl buffer containing 10 µM ionomycin. The various Ca2+ concentrations were prepared in KCl buffer containing 10 µM ionomycin and were added in a stepwise manner. The data was normalized and fitted using the 1:1 binding equation. The average Kd was 3.1 ± 1.4 mM for 7 cells. The weak affinity of CatchER+ allows it to sense Ca2+ in high Ca2+ organelles like the ER or SR. To determine the basal [Ca2+] in the SR, C2C12 cells were permeabilized with 0.005% saponin for 15-30 s, washed with KCl buffer, and then washed with 1 mM EGTA in KCl buffer containing 10 µM ionomycin to obtain Fmin. After washing the EGTA away with KCl buffer a maximum, Fmax, of 100 mM Ca2+ with 10 µM ionomycin was added. The basal Ca2+ was calculated to be 0.4 ± 0.2 mM.
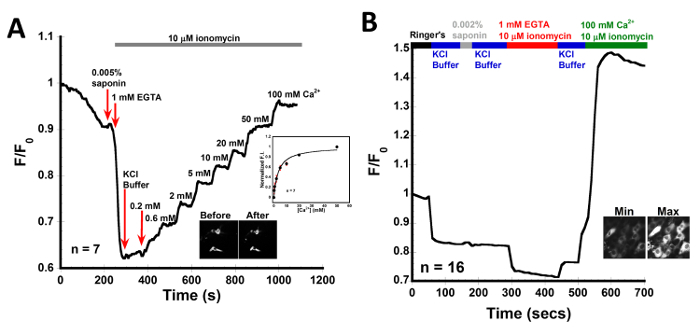
Figure 1: Stimulation of RyR using 4-cmc in HEK293 cells. (A) Depiction of the activation of RyR with 4-cmc. (B) Intensity recording of HEK293 cells transfected with CatchER+, localized in the ER, treated with 200 µM 4-cmc. Stimulation of the RyR with 4-cmc causes a decrease in the normalized fluorescence intensity that directly correlates to a decrease in [Ca2+]ER. n refers to the number of cells imaged. The average signal decrease was 12.0 ± 2.2%. The presence of 1.8 mM Ca2+ in the Ringer's buffer facilitated refilling of the ER reflected in the intensity recovery to baseline. Inset pictures show the cells before and after treatment with 4-cmc. SNR was calculated to be 4.3 ± 0.8. Please click here to view a larger version of this figure.
Figure 2: Decrease in [Ca2+]ER using the reversible SERCA pump inhibitor CPA in HEK293 cells. (A) The inhibition of the SERCA pump by CPA. (B) Live imaging of HEK293 cells transfected with CatchER+, localized in the ER, treated with 15 µM CPA. Because Ca2+ can still flow through the IP3R and RyR, blocking the SERCA pump with CPA causes a decrease in the normalized fluorescence intensity that directly correlates to a decrease in [Ca2+]ER. n refers to the number of cells imaged. Inset pictures are HEK293 cells transfected with CatchER+ before and after treatment. The average signal decrease was 21.0 ± 0.3%. The presence of 1.8 mM Ca2+ in the Ringer's buffer facilitated refilling of the ER reflected in the intensity recovery to baseline. SNR was calculated to be 5.5 ± 0.8. Please click here to view a larger version of this figure.
Figure 3: ATP decreases [Ca2+]ER in HEK293 cells. (A) Schematic of the indirect activation of IP3R with ATP through the purinergic receptor P2YR. P2YR is a GPCR that generates IP3 upon ATP binding.The IP3 activates the IP3R, stimulating calcium release from the ER. (B) Real time imaging of HEK293 cells transfected with CatchER+, localized in the ER, treated with 200 µM ATP. Indirect stimulation of the IP3R via the P2Y receptor with ATP causes a decrease in the normalized fluorescence intensity that directly correlates to a decrease in [Ca2+]ER. n refers to the number of cells imaged. The average signal decrease was 6.0 ± 3.0%. SNR was calculated to be 6.7 ± 2.7. Please click here to view a larger version of this figure.
Figure 4: In situ Kd of CatchER+ in C2C12 myoblast cells. Representative trace for normalized intensity collected from permeabilized C2C12 myoblast cells transfected with CatchER+. Cells were permeabilized with 0.005% saponin in intracellular buffer for 15-30 s. EGTA and Ca2+ solutions were prepared in KCl buffer, n refers to the number of cells imaged. (A) Inset fluorescence images are representative of cells before and after treatment. 0.3, 0.6, 2, 5, 10, 20, 50, and 100 mM Ca2+ was added to permeabilized C2C12 myoblast cells in the presence of 10 µM ionomycin to get a Kd of 3.1 ± 1.4 mM. (B) Inset fluorescence images are representative of cells at minimum and maximum values, after addition of 1 mM EGTA and 100 mM Ca2+ with 10 µM ionomycin each, respectively. The basal Ca2+ was calculated to be 0.4 ± 0.2 mM. Please click here to view a larger version of this figure.
Discussion
Live single-cell imaging of fluorescent probes, such as CatchER+, is an effective technique to analyze intricate ER/SR Ca2+ signaling processes in each cell in response to receptor agonists or antagonists. This technique is also useful for imaging using multiple wavelengths concurrently, such as needed for Fura-2 or to image CatchER+ and Rhod-2 together to monitor both ER and cytosolic calcium changes, respectively. There are several critical steps in this protocol; cell transfection can have a great effect on the viability of imaging. Low transfection rates can result in insufficient expression of the GECI for monitoring calcium responses. On the other hand, overexpression of CatchER+ will not have a buffering effect on ER/ER Ca2+, a problem associated with synthetic calcium dyes. Buffering effects of Ca2+ probes are influenced by the [Ca2+] in the organelle, the Kd of the probe and the concentration of the probe required for measurement. Cytosolic [Ca2+] is typically in the low nanomolar range, however, the required probe concentration is in a comparable range. In this case, the amount of dye concentration and its capability in buffering cytosolic calcium was, and is, a major concern for cellular imaging. In contrast, ER/SR [Ca2+] is in the 100s of micromolar to millimolar range31 as we determined for different mammalian cell lines25. Since 0.1-1 µM expression of CatchER+ is sufficient to enable detection of ER calcium, the buffering effect of CatchER is negligible. Thus, CatchER+ can monitor Ca2+ flux in high [Ca2+] environments without any buffering effect on luminal ER/SR Ca2+32.
Several factors can affect transfection efficiency of the GECI such as the incubation time, the transfection reagent, the incubation temperature, and the cell confluency. The cell confluency, when seeding the cells on the slide, can significantly affect the imaging quality. Low confluency can result in a low number of cells (n) and less statistical accuracy, while high levels of confluency lead to overlapping layers of cells producing large variables in imaging. The time and temperature for transfection should be optimized based on the cell type. Additionally, adding DMEM with reduced serum media is optimal for longer transfection times to prevent apoptosis. The original GECI CatchER fluorescence in mammalian cells, when cultured and transfected, was only 30 °C. The fluorescence of CatchER was successfully optimized and improved for expression at 37 °C resulting in the new, improved variant CatchER+. A western blot using an antibody against EGFP can be performed to confirm the expression of Ca2+ probe.
Moreover, the method and consistency of reagent delivery is critical. Reagents can be added to the chamber by perfusion, small volume diffusion, or by mechanical pumps, all of which can elicit differing responses. It is imperative that the solution chamber is properly sealed by applying a layer of sealant grease on the bottom of the chamber that will be placed onto the slide. The correct wavelengths must be selected for the probe. Excitation and emission wavelengths for CatchER+ are 395/488 nm and 510 nm, respectively. For cell imaging, only 488 nm is used for excitation to avoid the detrimental effect of UV light on cells. Therefore, using any optical filters which excite at 488 nm and collect the emission at 510 nm would be acceptable to use to image with CatchER+. To prevent photobleaching, efforts may be focused to optimize light intensity and light exposure such as frames/s33.
While this protocol is ideal to analyze the effect of ER/SR changes using CatchER+ Ca2+ ER/SR fluorescent probe, there are limitations as expected in any experiment. Since single-cell live imaging only analyzes a small frame of cells, the number of cells (n) can vary from 1-100 cells, on average, but can be lower for larger cell lines. This leads to lower cell numbers and statistically varied results without multiple trials. An n>6 is required for statistical accuracy. To get a larger n, many dishes must be imaged or other techniques must be used, concurrently. Additionally, CatchER+ is a single wavelength ER/SR Ca2+ probe, this leads to the issues of other single wavelength probes and dyes with not being able to quantify the signal, not knowing if signal is true as opposed to disruption of the cells artifact, and having larger noise as compared to ratiometric systems34.
This work shows that these optimized Ca2+ probes can be applied in different cell types or tissue types to monitor receptor-mediated ER/SR Ca2+ release. CatchER+ is a single wavelength ER/SR Ca2+ probe. Therefore, it is important that the experimental parameters and settings are consistent for quantitative measurement. A detailed calibration of calcium concentration and Kd of calcium sensor in the cell lines are additional important measurements for quantitative analysis.
These developed calcium sensors can also be targeted to specific locations within the ER/SR to monitor the vastly different Ca2+ transients that exist compared to global Ca2+ changes in various biological and pathological conditions. These findings will continue to push the fields of Ca2+ imaging and probe design forward to provide future tools for diagnosing Ca2+-related diseases. The reported protocols and developed sensor can also be adapted for drug discoveries against diseases associated with calcium signaling.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was funded by NIH GM62999, NIH EB007268, NIH AG15820, B&B Seed Grant, and a NIH Supplemental Grant to FR, BB fellowship to CM, CDT fellowship to RG.
References
- Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000;1(1):11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Calcium signalling remodelling and disease. Biochem. Soc. Trans. 2012;40:297–309. doi: 10.1042/BST20110766. [DOI] [PubMed] [Google Scholar]
- Tang S, Reddish F, Zhuo Y, Yang JJ. Fast kinetics of calcium signaling and sensor. Curr. Opin. Chem. Biol. 2015;27:90–97. doi: 10.1016/j.cbpa.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: Molecular determinants and functional consequences. Physiol. Rev. 2006;86(1):369–408. doi: 10.1152/physrev.00004.2005. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium. 2002;32(5-6):235–249. doi: 10.1016/s0143416002001823. [DOI] [PubMed] [Google Scholar]
- Clapham DE. Calcium Signaling. Cell. 2007;131(6):1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- HerrmannFrank A, Richter M, Sarkozi S, Mohr U, LehmannHorn F. 4-chloro-m-cresol, a potent and specific activator of the skeletal muscle ryanodine receptor. Biochimica Et Biophysica Acta-General Subjects. 1996;1289(1):31–40. doi: 10.1016/0304-4165(95)00131-x. [DOI] [PubMed] [Google Scholar]
- Ferris CD, Huganir RL, Bredt DS, Cameron AM, Snyder SH. Inositol trisphosphate receptor: phosphorylation by protein kinase C and calcium calmodulin-dependent protein kinases in reconstituted lipid vesicles. Proc. Natl. Acad. Sci. 1991;88(6):2232–2235. doi: 10.1073/pnas.88.6.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubyak GR, el-Moatassim C. Signal transduction via P2-purinergic receptors for extracellular ATP and other nucleotides. Am. J. Physiol., Cell Physiol. 1993;265(3 Pt 1):C577–C606. doi: 10.1152/ajpcell.1993.265.3.C577. [DOI] [PubMed] [Google Scholar]
- Song Z, Vijayaraghavan S, Sladek CD. ATP increases intracellular calcium in supraoptic neurons by activation of both P2X and P2Y purinergic receptors. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007;292(1):R423–R431. doi: 10.1152/ajpregu.00495.2006. [DOI] [PubMed] [Google Scholar]
- Brini M, Carafoli E. Calcium Pumps in Health and Disease. Physiol. Rev. 2009;89(4):1341–1378. doi: 10.1152/physrev.00032.2008. [DOI] [PubMed] [Google Scholar]
- Michelangeli F, East JM. A diversity of SERCA Ca2+ pump inhibitors. Biochem. Soc. Trans. 2011;39(3):789–797. doi: 10.1042/BST0390789. [DOI] [PubMed] [Google Scholar]
- Hofer AM, Landolfi B, Debellis L, Pozzan T, Free Curci S. Ca2+ dynamics measured in agonist-sensitive stores of single living intact cells: a new look at the refilling process. Embo J. 1998;17(7):1986–1995. doi: 10.1093/emboj/17.7.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer AM, Machen TE. Technique For InSitu Measurement of Calcium in Intracellular Inositol 1,4,5-Trisphosphate-Sensitive Stores Using The Fluorescent Indicator Mag-Fura-2. Proc. Natl. Acad. Sci. U. S. A. 1993;90(7):2598–2602. doi: 10.1073/pnas.90.7.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju B, Murphy E, Levy LA, Hall RD, London RE. A fluorescent indicator for measuring cytosolic free magnesium. Am J Physiol. 1989;256 doi: 10.1152/ajpcell.1989.256.3.C540. [DOI] [PubMed] [Google Scholar]
- Tsien RY. The green fluorescent protein. Annu. Rev. Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- Miyawaki A, et al. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 1997;388(6645):882–887. doi: 10.1038/42264. [DOI] [PubMed] [Google Scholar]
- Palmer AE, Jin C, Reed JC, Tsien RY. Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc. Natl. Acad. Sci. U. S. A. 2004;101(50):17404–17409. doi: 10.1073/pnas.0408030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim N, Griesbeck O. Genetically Encoded Indicators of Cellular Calcium Dynamics Based on Troponin C and Green Fluorescent Protein. J. Biol. Chem. 2004;279(14):14280–14286. doi: 10.1074/jbc.M312751200. [DOI] [PubMed] [Google Scholar]
- Mank M, et al. A FRET-based calcium biosensor with fast signal kinetics and high fluorescence change. Biophys. J. 2006;90(5):1790–1796. doi: 10.1529/biophysj.105.073536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garaschuk O, Griesbeck O, Konnerth A. Troponin C-based biosensors: A new family of genetically encoded indicators for in vivo calcium imaging in the nervous system. Cell Calcium. 2007;42(4-5):351–361. doi: 10.1016/j.ceca.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Carlson HJ, Campbell RE. Mutational Analysis of a Red Fluorescent Protein-Based Calcium Ion Indicator. Sensors. 2013;13(9):11507–11521. doi: 10.3390/s130911507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Shui B, Kotlikoff MI, Sondermann H. Structural Basis for Calcium Sensing by GCaMP2. Structure. 2008;16(12):1817–1827. doi: 10.1016/j.str.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koldenkova VP, Nagai T. Genetically encoded Ca2+ indicators: Properties and evaluation. Biochim. Biophys. Acta-Mol. Cell Res. 2013;1833(7):1787–1797. doi: 10.1016/j.bbamcr.2013.01.011. [DOI] [PubMed] [Google Scholar]
- Tang S, et al. Design and application of a class of sensors to monitor Ca2+ dynamics in high Ca2+ concentration cellular compartments. Proc. Natl. Acad. Sci. U. S. A. 2011;108(39):16265–16270. doi: 10.1073/pnas.1103015108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, et al. Developing sensors for real-time measurement of high Ca2+ concentrations. Biochemistry. 2007;46(43):12275–12288. doi: 10.1021/bi7007307. [DOI] [PubMed] [Google Scholar]
- Wang Z-M, Tang S, Messi ML, Yang JJ, Delbono O. Residual sarcoplasmic reticulum Ca2+ concentration after Ca2+ release in skeletal myofibers from young adult and old mice. Pflugers Arch., EJP. 2012;463(4):615–624. doi: 10.1007/s00424-012-1073-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 2006;24(1):79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- Siemering KR, Golbik R, Sever R, Haseloff J. Mutations that suppress the thermosensitivity of green fluorescent protein. Current Biology. 1996;6(12):1653–1663. doi: 10.1016/s0960-9822(02)70789-6. [DOI] [PubMed] [Google Scholar]
- Reddish FN. Design Of Genetically-Encoded Ca2+ Probes With Rapid Kinetics for Sub-Cellular Application [dissertation] Georgia State University; 2016. [Google Scholar]
- Seo MD, Enomoto M, Ishiyama N, Stathopulos PB, Ikura M. Structural insights into endoplasmic reticulum stored calcium regulation by inositol 1,4,5-trisphosphate and ryanodine receptors. Biochim. Biophys. Acta-Mol. Cell Res. 2015;1853(9):1980–1991. doi: 10.1016/j.bbamcr.2014.11.023. [DOI] [PubMed] [Google Scholar]
- Lambert DG. Calcium signaling protocols. Humana Press; 1999. [Google Scholar]
- Stephens DJ, Allan VJ. Light microscopy techniques for live cell imaging. Science. 2003;300(5616):82–86. doi: 10.1126/science.1082160. [DOI] [PubMed] [Google Scholar]
- Bootman MD, Rietdorf K, Collins T, Walker S, Sanderson M. Cold Spring Harb. 2. Vol. 2013. Cold Spring; 2013. Ca2+-sensitive fluorescent dyes and intracellular Ca2+ imaging; pp. 83–99. [DOI] [PubMed] [Google Scholar]