

Abstract
The discovery of cell type-specific markers can provide insight into cellular function and the origins of cellular heterogeneity. With a recent push for the improved understanding of neuronal diversity, it is important to identify genes whose expression defines various subpopulations of cells. The retina serves as an excellent model for the study of central nervous system diversity, as it is composed of multiple major cell types. The study of each major class of cells has yielded genetic markers that facilitate the identification of these populations. However, multiple subtypes of cells exist within each of these major retinal cell classes, and few of these subtypes have known genetic markers, although many have been characterized by morphology or function. A knowledge of genetic markers for individual retinal subtypes would allow for the study and mapping of brain targets related to specific visual functions and may also lend insight into the gene networks that maintain cellular diversity. Current avenues used to identify the genetic markers of subtypes possess drawbacks, such as the classification of cell types following sequencing. This presents a challenge for data analysis and requires rigorous validation methods to ensure that clusters contain cells of the same function. We propose a technique for identifying the morphology and functionality of a cell prior to isolation and sequencing, which will allow for the easier identification of subtype-specific markers. This technique may be extended to non-neuronal cell types, as well as to rare populations of cells with minor variations. This protocol yields excellent-quality data, as many of the libraries have provided read depths greater than 20 million reads for single cells. This methodology overcomes many of the hurdles presented by Single-cell RNA-Seq and may be suitable for researchers aiming to profile cell types in a straightforward and highly efficient manner.
Keywords: Neuroscience, Issue 123, Intrinsically photosensitive retinal ganglion cells, electrophysiology, RNA sequencing, single-cell isolation, low input RNA sequencing, neurons
Introduction
Neuronal diversity is observed throughout the central nervous system, particularly in the vertebrate retina, a highly specialized tissue consisting of 1 glial and 6 neuronal cell types that arise from one population of retinal progenitor cells1,2,3. Many subtypes of cells can be classified functionally, morphologically, and genetically. The goal of this protocol is to tie the genetic variability of cell types to their identifiable functional and/or morphological characteristics. A number of genes have been identified for the classification of cells, but many subtypes continue to go uncharacterized, as they represent a small fraction of the overall population. The identification of genes within these specific subtypes will allow for a greater understanding of neuronal diversity within the retina and may also shed light on the diversification of neural cells elsewhere. Furthermore, single-cell studies allow for the uncovering of new cell types, which may have been overlooked due to their low representation among the overall population4,5,6,7.
One of the benefits of single-cell transcriptomics is that unique markers or combinations of markers that define a particular cellular subtype can be discovered. These can then be used to gain genetic access to that cell type for different manipulations. For example, we are using this protocol to characterize the cell type-specific genes of a subset of retinal ganglion cells that express the photopigment melanopsin. The use of a fluorescent marker in melanopsin-expressing retinal ganglion cells enables the study of these cells, as they are clustered together due to their expression of a known gene. Interestingly, there are five known subtypes of this cell population in the mouse retina8. Thus, in order to isolate RNA from cells of each type, we have used established morphological classifications within the transgenic model to identify each subtype prior to cell isolation. This technique allows for the characterization of cells as well as for their isolation directly from the retina, without the need for tissue dissociation, which may cause a stress response within cells and contamination due to severed dendrites9.
A multitude of new techniques have come to light in the past few years as the RNA-Seq method continues to develop. These tools allow for maximized cell acquisition and greater cost efficiency while approaching the question at hand4,7,10,11,12,13. However, while these techniques have been excellent stepping stones, there are a number of hurdles still encountered that this protocol is able to address. First, many of the current procedures isolate cells from dissociated tissue and attempt to use either principal component analysis or hierarchical clustering post-hoc to determine cell classification. Relying on these tools to classify subtypes may not produce reliable results and may force one to find new ways to validate this data for the correlation of a genetic marker to a functional cell type. The requirement for dissociation in other protocols can sometimes result in tissue damage and can cause neuronal processes to be severed, resulting in a potential loss of mRNA. Furthermore, in dissociated cell preparations, the stress responses may begin to affect the transcriptomes of these cells14. This protocol overcomes these challenges by determining the functional cell type prior to isolation, and it better maintains the health of the cells by keeping the retinal tissue intact.
One technique was introduced in 2014 and consisted of the in vivo analysis of the transcriptome of live cells15. While this technique allows for the examination of the transcriptome with minimal mechanical disruption to the tissue, it lacks the ability to classify specific cell types within the tissue before examining their transcriptomes without using a very specific reporter mouse. Our protocol does not require a specific reporter, as we utilize cell filling and electrophysiology to characterize cells before their isolation. Another limitation of this previous protocol is that it requires a specific wavelength to excite the photoactivatable element, whereas our protocol allows for the use of a fluorescent reporter and fluorescent dye, which are readily available or can be chosen by each lab individually. Still, other laboratories have married the two methods of electrophysiology and transcriptomics for the study of cellular diversity. The use of patch-clamp recordings to characterize the function of a cell prior to its isolation has been performed on dissociated neurons16 and, in some cases, it has preceded the use of microarray analysis17 for these studies. The same complications are encountered by those approaches, as they require tissue dissociation or the use of microarray technology, which relies on the hybridization of samples to available probes. One of the most recent advances has been the development of Patch-Seq, a technique that combines the use of patch-clamp recordings and RNA-Seq technology to understand cells from whole-brain slices18. While this technique has its similarities to the protocol presented here, it is again important to note that our approach allows the tissue to remain intact for the health of the cells. Here, we present a protocol for the optimization of this alliance, which generates high-quality, single-cell libraries for the use of RNA-Seq to obtain a high read depth and mapping coverage.
Protocol
All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Northwestern University.
1. Preparation of Solutions for Electrophysiology (4 h)
Make 0.1% DEPC-treated H2O by adding 1 mL of diethyl pyrocarbonate (DEPC) to 999 mL of reverse osmosis-purified H2O. Mix thoroughly and let the mixture incubate for 1 h at room temperature (RT). Then, autoclave the DEPC-mixed H2O for 15 min on a liquid cycle. Let the DEPC-treated H2O cool at RT.
Make the extracellular solution by mixing one bottle of Ames' medium and 1.9 g (23 mM) of sodium bicarbonate into 1 L of H2O. Bubble the extracellular solution with 95% O2/5% CO2 and maintain it at a pH of 7.3-7.4.
Generate the intracellular solution by combining 125 mM K-gluconate, 2 mM MgCl2,10 mM EGTA, 10 mM HEPES, and 0.1% DEPC-treated H2O. Store it in 1 mL aliquots at -20 °C. Add 10 µM of fluorescent tracer at the beginning of each experiment.
Make enzyme solution by adding 10,000 units of collagenase and 83 mg of hyaluronidase to 4.15 mL of extracellular solution. The enzyme solution should be stored in 50 µL aliquots at -20 °C.
2. Preparation of Retinal Tissue (2 h)
NOTE: All procedures in this section should be performed under dim red illumination
Dark-adapt animals for at least 1 h prior to dissection. Perform all procedures under dim red illumination.
Euthanize the animals by CO2 asphyxiation and enucleate the eyeballs into a Petri dish with previously oxygenated extracellular solution.
Poke the cornea with a needle and cut it away by cutting with ophthalmic scissors at the border of the cornea and sclera19.
Remove the lens using #5 forceps. Gently make a tear in the sclera with the forceps and sever the optic nerve where the retina and sclera meet. Carefully finish removing the sclera from the retina.
Remove the transparent vitreous using #5 forceps; once removed, the vitreous appears as a gelatinous substance stuck to the forceps. Slice the retinas in half (so that there are 4 pieces/animal) and store them in oxygenated extracellular solution at RT until use.
- When ready to mount the tissue in the recording chamber, place a piece of retina to incubate in enzyme solution diluted in 500 µL of oxygenated extracellular solution. Incubate in a Petri dish for 2 min at RT on a shaker.
- Wash the piece of retina in oxygenated extracellular solution and place the tissue in a glass-bottom recording chamber; use a plastic transfer pipette with the tip cut off to allow for the retina to be transferred without causing damage to the tissue.
Use forceps to carefully flatten the tissue with the photoreceptor layer facing down. Remove excess fluid using a pipette. Anchor the tissue using a platinum ring with nylon mesh. NOTE: This method could also be used to prepare the tissue for the isolation of RNA from labeled amacrine and bipolar cells.
Fill the chamber with oxygenated extracellular solution and mount it onto a microscope stage. Perfuse tissue with oxygenated extracellular solution at 2-4 mL/min.
3. Visualization and Targeting of GFP+ Retinal Ganglion Cells (10 min)
NOTE: All procedures in this section should be performed under dim red illumination
Before beginning, pull glass micropipettes (OD: 1.2 mm, ID: 0.69 mm) for electrophysiological recordings using a micropipette puller. Use the following protocol for the electrodes (please note that the parameters should be adjusted accordingly to achieve the desired resistance and will vary across pullers and with different glass): Heat: Ramp +10; Pull: 0; Vel: 23; Delay: 1; Pressure: 500; Program loop: 5 times. Ensure that the tips are ~1 µm in diameter, with resistances of 2-4 MΩ for targeting large cells and of 5-7 MΩ for targeting smaller cells.
Observe the ganglion cell layer using Infrared Differential Interference Contrast (IR-DIC) optics (Figure 1A). Identify GFP+ Retinal Ganglion Cells (RGCs) using epifluorescence (~480 nm) (Figure 1B).
Locate the pipette filled with intracellular solution in DIC. Apply slight positive pressure and zero any voltage offsets on the amplifier.
Press the glass micropipette against a GFP+ cell and apply negative pressure to form a GΩ seal between the pipette and the cell membrane. Apply test voltage command steps (e.g., 5 mV) to monitor the seal resistance. After forming a stable seal, rupture the membrane by applying brief pulses of negative pressure to gain whole-cell access.
Wait 1-2 min for the dendrites of the cell to fill with fluorescent tracer. NOTE: The cell can be morphologically typed by examining the morphology in epifluorescence (Figure 1C). In the case of melanopsin-expressing RGCs, dendritic stratification in the inner plexiform layer is visualized by examining the dendrites filled with fluorescent tracer under epifluorescent illumination and determining whether they stratify far from the soma in the OFF sublamina (M1 ipRGCs), near the ganglion cell layer in the ON sublamina (M2 & M4 ipRGCs), or both (M3 ipRGCs). This observation, combined with soma size (M4s have distinctly large somas compared to all other ipRGC subtypes), allow for the identification of cell type20,21,22. Thus, this technique allows for the identification of cell type in vitro prior to RNA isolation. This method could be modified for other cell type identification protocols involving either dendritic morphology or cellular physiology.
4. Cell Isolation (2 min)
Before beginning, set the tabletop microcentrifuge to 2,000 x g. Prepare a sample-expelling apparatus by connecting tubing (OD: 3/32 in, ID: 1/32 in) with a 1 cc syringe.
Place 0.2 mL PCR tubes containing 10 µL of lysis buffer and 1% β-mercaptoethanol on ice. Prepare a 1 cc syringe containing DEPC-treated H2O to rinse the pipette tips. Prepare a container of dry ice to freeze the lysis buffer after sample collection.
- Carefully extract the cytoplasmic content of the cell pipette by applying negative pressure using a 10 mL syringe; all cytoplasmic content, including organelles, should be extracted if possible.
- Monitor the extraction in DIC by visualizing the cell body decreasing in size. After extracting the cytoplasmic contents, lift the pipette carefully off of the tissue and quickly remove the pipette from the solution.
Quickly remove the pipette from the head-stage holder and rinse the pipette tip briefly with DEPC-treated H2O using a 1 mL syringe. Connect the pipette to a 1 mL syringe via tight-fitting tubing to expel the sample.
- Immediately expel the cells into 10 µL of lysis buffer 1 containing 1% β-mercaptoethanol in 0.2 mL PCR tubes. NOTE: The entire aspirate with the cells should be expelled gently so as to not introduce bubbles.
- Briefly centrifuge the tube in a tabletop mini centrifuge at 2,000 x g for 10 s. Immediately flash-freeze the samples for 5 min on dry ice. After freezing, store them at -80 °C for up to two weeks for best results; the samples may last longer, but it is recommended that they are processed as quickly as possible.
5. RNA Purification (30 min)
Before beginning, set up a magnetic separator device by taping the top part of an inverted P20 or P200 tip holder to the 96-well magnetic stand23.
Prepare fresh 70% ethanol (EtOH) – approximately 1 mL per sample will suffice. Remove the RNA magnetic beads from 4 °C storage and thaw them at RT for at least 30 min. NOTE: No more than 8 samples should be processed at one time, as many steps in this protocol rely on efficiency and quick handling.
Once the magnetic beads are at RT, vortex for 30 s to ensure that the solution is well mixed. NOTE: The beads use an RNA-specific buffer to selectively bind RNA, and they allow for the removal of other cellular waste when employed with a magnetic plate stand.
Thaw cells at RT for 1 min, and then add 5 µL of RNase-free H2O to each sample; pipette up and down. Add 22 µL of RNA beads to each tube and pipette thoroughly to mix. Incubate the samples at RT for 5 min to allow the RNA to interact and bind with the magnetic beads.
Place the tubes on a magnetic separator device and let sit for 8 min; before proceeding, ensure that the supernatant is clear. Observe the beads from one pellet and be sure to not detach it during pipetting.
Remove the supernatant from the samples and add 150 µL of 70% EtOH. Remove the EtOH and repeat the wash twice more.
Allow the samples to air dry for 6 min. Check intermittently to see if more EtOH has collected at the bottom of the tube. Remove it accordingly.
While the samples are drying, prepare 10X reaction buffer by combining 19 µL of lysis buffer 2 and 1 µL of RNase Inhibitor (40 U/µL). Briefly spin it down and keep it on ice.
Once the samples are dry and the bead pellets no longer appear glossy, remove the tubes from the magnetic separator and add 9.5 µL of RNase-free H2O to rehydrate the samples. Place the samples on ice and add 1 µL of 10x reaction buffer to each sample.
6. Reverse Transcription (10 min)
NOTE: Before beginning, thaw the necessary reagents for reverse transcription (RT; except for the enzyme) on ice. These include: primer II, buffer 1, oligonucleotide, and RNase inhibitor.
To each tube, add 2 µL of primer II (AAGCAGTGGTATCAACGCAGAGTACT(30)N-1N, for which N-1 can be A, C, or G and N can be A, C, G, or T; 12 µM). Place the tubes in a thermocycler that has been preheated at 72 °C for 3 min.
During incubation, prepare the RT master mix. For each reaction, add 4 µL of buffer 1 (250 mM Tris-HCl, pH 8.3; 375 mM KCl; and 30 mM MgCl2), 1 µL of oligonucleotide (48 µM), and 0.5 µL of RNase inhibitor (40 U/µL).
Immediately following the incubation, place the tubes on ice for 2 min.
Add 2 µL per reaction of the reverse transcriptase (100 U/µL) to the master mix and pipette thoroughly. Add 7.5 µL of master mix to each tube and mix by gently pipetting. Briefly spin the tubes to collect the contents at the bottom and place them into a preheated thermocycler with the following program: 42 °C for 90 min, 70 °C for 10 min, and a hold at 4 °C.
Store the tubes at -20 °C O/N before proceeding, although it is recommended that the samples be carried through the amplification step before being stored for long periods of time; other sources suggest O/N storage at 4 °C would also be acceptable at this step24.
7. cDNA Amplification (2.5 h)
NOTE: Before beginning, thaw PCR buffer and PCR primer on ice and spin the tubes down in a tabletop mini centrifuge before making the PCR master mix.
For each reaction, prepare a PCR master mix containing 25 µL of PCR buffer, 1 µL of PCR primer (12 µM), 1 µL of DNA polymerase, and 3 µL of nuclease-free H2O. Add the DNA polymerase last, just prior to the addition of master mix to the samples.
Add 30 µL of master mix to each tube and spin at 2,000 x g for 10 s to collect the contents at the bottom of the tubes.
Place the tubes in a preheated thermocycler with the following program: 95 °C for 1 min; 34 cycles of 98 °C for 10 s, 65 °C for 30 s, and 68 °C for 3 min; 72 °C for 10 min; and a hold at 4 °C. NOTE: The number of cycles for this PCR has been increased to 34, differing from the suggested manufacturer's instructions. After repeat testing, this cycle number was found to produce consistently reliable results.
Store the tubes at -20 °C for up to one year before proceeding.
8. Purification of Amplified cDNA (30 min)
Before beginning purification, bring the DNA beads and elution buffer to RT for at least 30 min. Prepare fresh 80% EtOH; 1 mL per sample should suffice. Add 1 µL of 10X lysis buffer to each sample.
Vortex the DNA beads for 30 s and add 50 µL of DNA beads to each sample. Mix thoroughly by pipetting, and then briefly spin them down at 2,000 x g for 10 s. Incubate the tubes at RT for 8 min.
Place the tubes on the magnetic separation device for 5 min. Gently pipette the supernatant up and down twice and allow the samples to sit for 2 min. While the samples are on the magnetic device, remove the supernatant and discard.
Add 150 µL of freshly made 80% EtOH to each sample and allow them to sit at RT for 30 s. Remove the EtOH and repeat the EtOH wash once.
Briefly spin the samples and place them back on the magnetic separator for 1 min. Remove any remaining EtOH and allow the samples to air dry for 5 min.
Check the samples intermittently to see if any EtOH has collected and remove it with a pipette.
Once the bead pellet no longer appears glossy, but before cracks begin to appear, remove the tube from the magnetic separator and add 17 µL of elution buffer. Gently pipette up and down to resuspend the beads completely.
Incubate the resuspended samples at RT for 2 min.
Briefly spin the samples to collect all liquid at the bottom and place them on the magnetic separator for 2 min. Transfer 15 µL of clear supernatant to a 1.5 mL RNase-free tube and store it at -20 °C.
Examine quality and size of the cDNA library with a high-sensitivity bioanalyzer chip using 1 µL of cDNA. The ideal size of a cDNA library is 0.3-2 Kb, with a concentration of at least 10 ng/µL (Figure 2). NOTE: You may choose to employ PCR as a way to screen samples to ensure the expression of known markers before proceeding with sample preparation.
9. Determine Concentrations and Tagment cDNA (20 min)
Before beginning, bring the assay reagent, dilution buffer, and standards to RT for 30 min. Prepare a master mix containing 1 µL of assay reagent and 199 µL of dilution buffer per reaction. Aliquot 199 µL of this mixture into 500 µL assay tubes. Add 1 µL of sample and mix thoroughly.
Aliquot 190 µL of master mix to tube 1 & 2. Add 10 µL of standard 1 to tube 1 and 10 µL of standard 2 to tube 2. Vortex all sample tubes and standard tubes for 5 s; incubate at RT for 3 min.
Determine concentration of samples with a high-sensitivity fluorometer. Ensure the correct sample amount has been input by selecting the "calculate stock solution" option and highlighting "1 µL". Dilute each sample in a separate 1.5 mL tube to a final concentration of 0.2 ng/µL in RNase-free H2O. NOTE: Whereas the manufacturer's instructions suggest that one should begin with 1 ng/µL total of DNA, we have used a smaller starting amount and have also adjusted the protocol to account for this amendment.
In new 0.2 mL PCR tubes, pipette 2.5 µL of buffer 2. Add 1.25 µL of cDNA to the appropriate tube for a total of 250 pg. Finally, add 1.25 µL of tagmentation mix to each tube and pipette thoroughly to mix; the transposase within the tagmentation mix will fragment the DNA into short strands and anneal the adapters to either end of each strand, for later use by the sequencing instrument. NOTE: The volumes used for tagmentation and index coupling are a fraction of the amount suggested by the manufacturer's instructions. This not only allows for the conservation of reagents, but it also has consistently produced tagmented samples in our experience.
Centrifuge the tubes on a countertop microcentrifuge for 1 min.
Place the tubes in a preheated thermocycler with the following program: 55 °C for 10 min and a hold at 10 °C. NOTE: The length of this incubation is 10 min, as opposed to the proposed 5 min from the manufacturer's instructions.
Immediately after this program is complete, remove the tubes and add 1.25 µL of tagmentation neutralizing buffer to each. Pipette well to mix and incubate at RT for 5 min. Complete this step promptly, as the transposase remains active until the buffer neutralizes the enzyme and stops the reaction.
10. Index Coupling and Purification (1 h)
NOTE: Before beginning, bring the DNA beads and resuspension buffer to RT for at least 30 min. Decide which indices to use for each of the samples.
NOTE: These indices will be attached to the respective 5' and 3' ends of the fragmented DNA for the identification of samples following sequencing. Ensure that no two pairings are the same for samples that may be sequenced together. For example, if sample 1 will use indices white 1 and orange 1, sample two should use white 1 and orange 2 or white 2 and orange 2, but never the same combination of indices. This kit contains 4 distinct white and 6 distinct orange indices. All of the different possible combinations allow up to 24 samples to be pooled in one sequencing lane. Although we typically only pool 10 samples in a lane, one could also use the kit containing 24 indices, which would allow for the pooling of 96 samples in a single lane of sequencing, if desired.
To each tube, add 1.25 µL of the left index and 1.25 µL of the right index for that specific sample. Add 3.75 µL of PCR master mix and pipette well to mix.
Centrifuge in a countertop microcentrifuge for 1 min.
Place the tubes in a preheated thermocycler with the following program: 72 °C for 3 min; 95 °C for 30 s; 12 cycles of 95 °C for 10 s, 50 °C for 30 s, and 72 °C for 1 min; 72 °C for 5 min; and a hold at 4 °C. NOTE: The first 95 °C step was changed from 98 °C, which was suggested by manufacturer's instructions. Also, the cycle details were adjusted so that the temperatures and time lengths allowed for the optimal tagmentation of our samples. The final 72 °C incubation was also added to our protocol and was not included in the manufacturer's instructions.
Briefly spin the tubes to collect the contents at the bottom. Vortex the DNA beads for 30 s, and then add 30 µL to each tube and mix thoroughly by pipetting.
Incubate the samples at RT for 5 min.
Place the samples on a magnetic separator for 2 min. Pipette the supernatant up and down twice and incubate samples for 1 min.
Remove and discard the clear supernatant. Add 150 µL of 80% EtOH to each sample and remove. Repeat the EtOH wash once.
Allow the samples to air dry for 10 min. Check intermittently to see if any EtOH has collected at the bottom of the tubes and remove as necessary.
Once a crack begins to appear in the DNA bead pellet, remove the sample from the magnetic separator and add 27.5 µL of resuspension buffer to rehydrate the pellet. Ensure that the pellet is completely resuspended, and then incubate at RT for 2 min. NOTE: The volume used for the resuspension buffer was modified to account for the smaller amount of starting material in our protocol and differs from the suggested volume in the manufacturer's instructions.
Place the tubes on the magnetic separator for 2 min. Transfer 25 µL of clear supernatant into an RNase-free tube and store at -20 °C. NOTE: Do not proceed with the manufacturer's instructions to complete library normalization. The samples are successfully prepared following this step and are prepared for sequencing as is.
Analyze the tagmentation of the samples using a bioanalyzer chip and 1 µL of each sample. NOTE: The analysis of smears will be conducted as earlier; however, cDNA should now be detected at a size range of 0.2-1 Kb (Figure 3). Smears below this point likely represent the degradation of samples, while larger smears suggest incomplete tagmentation.
11. Pooling of Samples (10 min)
Obtain concentrations of tagmentation samples with the high-sensitivity fluorometer from earlier and determine the concentration in nM. NOTE: This calculation can be computed with the use of an internet conversion tool and relies on the average fragment length from the bioanalyzer trace. To determine the average fragment length, observe the trace for each sample individually and determine the size of the average DNA fragment. This may also be calculated when examining the bioanalyzer trace by highlighting the range of the smear and examining the molarity calculated by the program.
Combine samples such that the pool contains the same concentration of each sample. Do not dilute samples; the pool should be only as dilute as the lowest-concentrated sample and would ideally have a total concentration of at least 15 nM.
Store the pooled samples at -20 °C prior to sequencing; it is suggested that samples undergo sequencing within 1 week of pooling. Perform paired-end, 100 bp sequencing with a HiSeq platform.
Representative Results
Cell types are easily classified following the dye injection
Figure 1 shows an example of a GFP+ RGC before and after fluorescent tracer filling. This cell was identified based on its expression of GFP in the transgenic line (Figure 1A). A tight seal was formed with a fine-tip, pulled-glass electrode onto the soma of this cell. In order to characterize the subtype, the fluorescent dye was injected into the soma and allowed to fill all associated processes (Figure 1B). The classification as an M4 ipRGC was enabled through the observation that this cell has a very large soma and that its processes terminate within the ON sublamina of the inner plexiform layer (Figure 1C). As an illustration of differences in stratification that can be discerned in an isolated retinal preparation, we filled M1 (OFF-stratifying), M3 (bistratified), and M4 (ON-stratifying) ipRGCs with a fluorescent tracer, fixed them, and performed confocal imaging of the ON and OFF sublaminae (Figure 1D). This visualization of the dendrites via confocal imaging is very similar to how the dendrites look in unfixed in vitro tissue when cells are filled with a fluorescent tracer (Schmidt and Kofuji 2009, 2010, & 2011).
cDNA libraries from single cells
The use of an Oligo d(T) primer allows for the selective reverse transcription and amplification of mRNA. After generating and amplifying the cDNA libraries from each cell, bioanalyzer chips were used to assess the quality and size of the libraries. The results of this analysis show that the ideal cDNA smears are 0.5-2 Kb in a good sample (Figure 2A). Some samples show a few bands or smears below the 300 bp mark, suggesting that these libraries are of poor quality (see Table 1 for troubleshooting). An example of a good-quality bioanalyzer trace shows few or no DNA bands below 300bp and a robust smear between 500bp and 2Kb (Figure 2B). Empty control lanes should display lower and upper markers at 35 & 10380 bp, respectively, as well as a steady baseline that does not fluctuate. Various libraries can produce quality data, as can be seen by both Figure 2B and 2C, which produced excellent read depths and high mapping rates. Only those samples with strong cDNA libraries of the expected size should be carried through to tagmentation (see Table 1 for troubleshooting).
Low-input tagmentation prepares high-quality samples for sequencing
This protocol is optimized for tagmentation with a minor amount of starting material. Just 250 pg works best for a successful tagmentation, as lower amounts will not amplify properly and higher amounts will not fragment completely. After completing the tagmentation and PCR amplification/sample clean-up, the samples were again analyzed on a bioanalyzer chip. The ideal cDNA smears at this point should be 0.2-1 Kb (Figure 3A). The trace should appear smooth and evenly distributed between those two sizes (Figure 3B); this trace corresponds to the sample in lane 1. Figure 3C shows an incomplete tagmentation, which can be resolved easily (see Table 1 for troubleshooting). We have performed data analysis on the samples and found that this protocol produced samples with excellent read depths, often surpassing 12 million reads per sample; averages of 69.1% mapping; and more than 5,000 expressed genes.
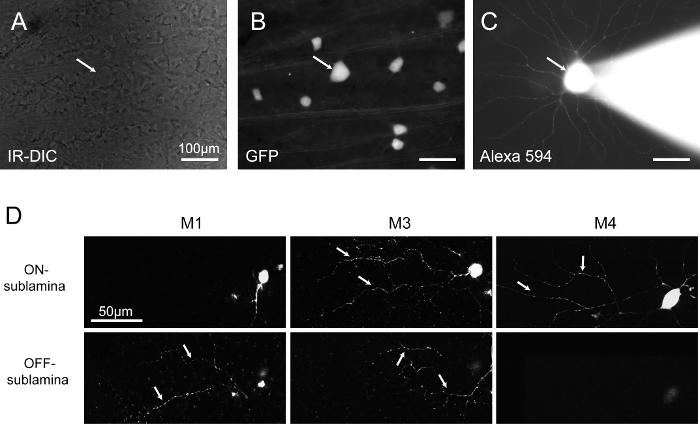 Figure 1: Identification of RGCs Types based on GFP Expression in Melanopsin-expressing ipRGCs and on Morphological Properties. (A) The ganglion-cell layer of the retina, visualized using IR-DIC in the whole-mount retina preparation. (B)The same preparation visualized in epifluorescence (~480 nm) to identify GFP-expressing ganglion cells. (C) A GFP-expressing cell targeted for patch-clamp recording and filled with a fluorescent tracer. The cell can be identified as an M4 ipRGC based on its very large soma size and ON stratifying dendrites25,26. (D) Confocal image of ipRGC dendrites imaged in the ON and/or OFF sublamina of the IPL (M1), in the ON sublamina of the IPL (M4), and in both the ON and OFF sublaminae of the IPL (M3). IR-DIC: infrared differential interference contrast. Please click here to view a larger version of this figure.
Figure 1: Identification of RGCs Types based on GFP Expression in Melanopsin-expressing ipRGCs and on Morphological Properties. (A) The ganglion-cell layer of the retina, visualized using IR-DIC in the whole-mount retina preparation. (B)The same preparation visualized in epifluorescence (~480 nm) to identify GFP-expressing ganglion cells. (C) A GFP-expressing cell targeted for patch-clamp recording and filled with a fluorescent tracer. The cell can be identified as an M4 ipRGC based on its very large soma size and ON stratifying dendrites25,26. (D) Confocal image of ipRGC dendrites imaged in the ON and/or OFF sublamina of the IPL (M1), in the ON sublamina of the IPL (M4), and in both the ON and OFF sublaminae of the IPL (M3). IR-DIC: infrared differential interference contrast. Please click here to view a larger version of this figure.
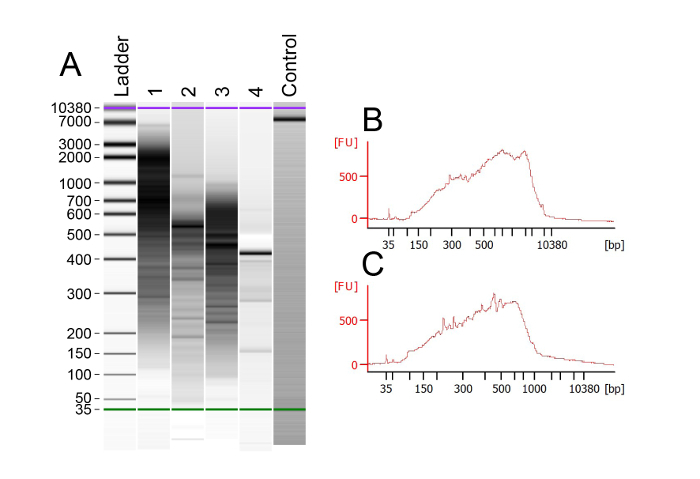 Figure 2: Analysis of cDNA Library Quality with a Bioanalyzer Trace. (A) Bioanalyzer output example for multiple samples that have been reverse transcribed, amplified, and purified. Lanes 1-3 show the ideal DNA smears, with the majority of DNA larger than 300 bp. Libraries with smears in this range have consistently provided excellent sequencing data, showing an average of 5,683 genes expressed per cell. Lane 4 represents a sample that was unsuccessfully processed and thus produced no cDNA. The successful control lane has a constant baseline and two clean peaks at 35 and10,380 bp. (B) Example of a successful bioanalyzer trace with a high intensity of cDNA around 2 Kb. This smear corresponds to the sample in lane 1. (C) Example of a successful bioanalyzer trace with the cDNA centered around 500 bp. This trace corresponds to the sample in lane 3. Please click here to view a larger version of this figure.
Figure 2: Analysis of cDNA Library Quality with a Bioanalyzer Trace. (A) Bioanalyzer output example for multiple samples that have been reverse transcribed, amplified, and purified. Lanes 1-3 show the ideal DNA smears, with the majority of DNA larger than 300 bp. Libraries with smears in this range have consistently provided excellent sequencing data, showing an average of 5,683 genes expressed per cell. Lane 4 represents a sample that was unsuccessfully processed and thus produced no cDNA. The successful control lane has a constant baseline and two clean peaks at 35 and10,380 bp. (B) Example of a successful bioanalyzer trace with a high intensity of cDNA around 2 Kb. This smear corresponds to the sample in lane 1. (C) Example of a successful bioanalyzer trace with the cDNA centered around 500 bp. This trace corresponds to the sample in lane 3. Please click here to view a larger version of this figure.
 Figure 3: Tagmented Samples Show Robust Smears between 0.2 and 2 Kb. (A) A representative example bioanalyzer output following tagmentation, amplification, and PCR clean-up. (B) Trace of a successfully tagmented sample corresponding to lane 1 in (A). (C) Example of the trace for a sample with incomplete tagmentation, clear by the peak intensity around 1 Kb. Please click here to view a larger version of this figure.
Figure 3: Tagmented Samples Show Robust Smears between 0.2 and 2 Kb. (A) A representative example bioanalyzer output following tagmentation, amplification, and PCR clean-up. (B) Trace of a successfully tagmented sample corresponding to lane 1 in (A). (C) Example of the trace for a sample with incomplete tagmentation, clear by the peak intensity around 1 Kb. Please click here to view a larger version of this figure.
| Problem | Possible Cause | Solution |
| Step 3.3) Cannot form GΩ seal | Surface of cell is not clean enough | Clean further using positive pressure |
| Step 3.3) Cell appears to deflate/die | Prepare new pipette and target a new cell | |
| Step 3.4) Cannot determine terminal location of dendrites | Alexafluor has not had enough time to diffuse throughout cell or concentration of Alexafluor is not high enough | Check to make sure gain and exposure time on camera are high enough. Wait an additional 5 min before visualizing dendrites. If dendrites still are not visible, prepare solution with higher Alexafluor concentration. |
| Step 4.1) Unable to aspirate all of cytoplasm | ||
| Step 5.5) Supernatant is not clear after 8 minutes | Gently pipette entire solution twice, while still on magnetic separator, and let sit for another 5 min | |
| Step 5.5) Pellet disperses during pipetting | Sample is too far from magnet | Keep tubes on magnetic separating device during all pipetting. Expel solution and allow beads to re-pellet for 5 min |
| Step 5.7) After 5 min, samples still appear glossy | Maximum amount of EtOH has not been removed | Continue to monitor samples during drying. Every 2 min, use a P10 pipette and remove all EtOH at bottom of tube |
| Step 8.9) Pellet had cracks before rehydration | Allow sample to rehydrate for a total of 4 min, rather than 2 (Step 8.10) | |
| Step 8.11) Small amount of bead was brought up with supernatant | Eject entire sample back into tube and place on magnetic separator for 1 min; Pipet up gently and make sure to avoid pellet | |
| Step 8.12) DNA smears are inconsistent and fluorescence scale continuously changes | Too high DNA concentration for a HS chip | Check concentration of sample and dilute between 1 - 10 ng/µL. Rerun bioanalyzer |
| Step 8.12) Low molecular weight smear | RNA Degradation | Make sure to flash freeze cell immediately after collection. Discard this cDNA library |
| Step 8.12) Fluctuating marker baseline in control lane | Contamination or old reagent | Discard DNA marker and employ new tube for Bioanalyzer run |
| Step 8.12) No DNA smear | Failure to expel cell | Pull new needles with a slightly larger tip |
| RNA Bead Failure | Make sure beads have been fully resuspended before use and allow samples to incubate before placing on magnetic separator | |
| Step 8.12) Weak DNA smear | Not enough amplification | Employ more PCR cycles |
| Step 8.12) DNA smear outside of 500bp-2Kb range | DNA smears below 0.5 and above 2 Kb are likely contamination. Discard sample | |
| Step 8.12) Regularly spaced spikes on DNA trace | Contamination of sample | Always wear fresh gloves and make new ethanol for rinses; filter tips should be used at all times |
| Step 9.3) Unable to detect concentration of sample on fluorometer | Samples below detection level have too little DNA for tagmentation and cannot be used | |
| Step 10.10) Samples are not dry after 10 min | Continue to remove excess EtOH | Allow samples to airdry longer, check on them every minutes |
| Step 10.13) Smear skewed toward 2Kb | Incomplete fragmentation | Re-dilute sample to a concentration of 0.15 ng/µL, rather than 0.2 ng/µL; rerun tagmentation |
| Step 10.13) Smear skewed toward 200bp | Too little DNA input | Dilute sample less, try a concentration of 0.4 ng/µL; rerun tagmentation |
| Tagmentation reaction too long | Reduce reaction time of tagmentation to 8 min | |
| Step 10.13) Weak smears | Improper Amplification of DNA | Dilute sample to concentration of 0.4 ng/µL and rerun tagmentation |
| Step 11.2) Maximum obtainable pool concentration is below 5 nM | Identify which sample(s) have significantly low concentrations and rerun tagmentation with new dilution |
Table 1: Solutions and Suggestions for Potential Hindrances in the Protocol. This table lists potential difficulties that may occur during this protocol and the steps at which one may encounter them. This table lists possible causes for many of these problems, as well as solutions that may help to solve any issues.
Discussion
Our protocol demonstrates, through a quick and easy-to-use guide, a method to prepare single cells of identified morphological classes for high-quality sequencing, with little injury to the sample. In the present manuscript, intrinsically photosensitive retinal ganglion cells are morphologically characterized, isolated, and prepared for RNA-Seq. Cellular stresses may occur during retinal handling; for this reason, we replace each piece of tissue after no more than 4 h of use. We can assess the state of the cells by using the electrophysiology rig to record from these cells and to monitor their responses, which allows us to ensure that the cells are of good health. Our protocol allowed for the generation of successful cDNA libraries from 15 out of 23 samples processed, giving a success rate of 65%. Furthermore, the quality of our sequencing data was excellent, as the average number of genes expressed by our cells ranged from 2,316-10,353, with an average of 5,683 genes registering with a non-zero FPKM value. These numbers are similar to expressed gene counts found by other studies of neurons5, showing that our data is also of good quality.
While we have had success with this protocol for these specific neurons, it should be noted that minor amendments to this technique can be applied for a number of different applications. Using Fluorescence-activated Cell Sorter (FACS) within a separate mouse model, we have isolated small populations of cells (1,000 -25,000) labeled with a GFP marker from the central nervous system. The RNA from these populations was isolated, and 1 µL (at or slightly below 12 ng/µL) of this RNA was used to begin the reverse transcription at step 6.1. The only adjustment that should be made to the protocol after this step occurs at step 7.4, at which point one may choose to employ fewer PCR cycles. We have used 19 cycles in cases when the initial sample contained greater than 10,000 cells and have found that this produces good-quality cDNA libraries. For example, cell samples were prepared from a FACSorted population, and the number of genes expressed ranged from 12,340-14,052, with an average of 13,265 genes with a non-zero FPKM value. This technique can be applied to various mouse models for which a fluorescent reporter is present. Due to this amendment, researchers could potentially study any cell population for which a fluorescent reporter mouse is available, extending studies past the field of neuronal diversity.
A second adjustment can be made to profile cells based on functionality rather than just morphology. This project relies on a transgenic model, but this technique can be applied to neurons with no known molecular identifiers. For example, there are over 30 functional subtypes of retinal ganglion cells currently identified27, very few of which have unique markers. As this technique already relies on an electrophysiology rig for the cell filling, one could employ patch clamping to identify the functional response profile of each cell based on its spiking pattern. Using light-evoked spike recordings, the classification of retinal neurons could be determined prior to cell isolation. This technique works on retinal ganglion cells, but it may be extended to examine other retinal neurons. It should be noted, however, that if this protocol is used to type retinal bipolar or amacrine cells in the inner nuclear layer, for example, one must be careful to provide constant positive pressure at the tip of the electrode to avoid contacting cells as the electrode passes through the ganglion cell layer and inner plexiform layer. For retinal neurons within the inner nuclear layer, the preparation of retinal sections prior to cellular recordings would likely work best to consistently avoid contamination. Each cell would be isolated and prepared as described here following the classification step. Furthermore, we propose the use of this protocol for the study of neurons, but this technique may be applied to any cell type that can be morphologically characterized or isolated through the use of a transgenic model.
This technique can also be modified to work with previously prepared cDNA libraries, as we have generated in the past for microarray hybridization28. With a separately prepared library, one begins with cDNA quantities in the range of 50-150 ng and starts the protocol at step 8.4; higher and lower concentrations have not been attempted, although they may work as well. Purification of the cDNA using DNA beads would occur first, followed by tagmentation and PCR amplification. We have tested this adjustment with cDNA from library preparations, such as those for microarray hybridization29, and have successfully produced tagmented libraries for RNA-Seq.
Finally, this protocol can be used in the assessment of the success of a particular induction of cell populations from induced Pluripotent Stem Cells (iPSCs). The driving force behind differentiation of these re-programmed pluripotent cells relies on an understanding of the transcription factors that lead cell types to develop separately from one another. Driving iPSCs toward particular cell fates is a new method for studying neuronal diversity, as it can be used to selectively generate cell types. This protocol can be adapted to assess the quality of diversity among differentiated cells from this model. As previously mentioned, there are more than 30 functional subtypes of retinal ganglion cells, and many efforts have been made to generate functional RGCs from iPSCs. The identification of markers for subtypes of RGCs – in our case, ipRGCs – would allow the researcher to evaluate the success of their protocol in producing various subtypes of RGCs. The evaluation of cell types among these neurons would benefit from this protocol and may also serve as a tool for the continued investigation of subtype-specific markers as this model system becomes readily available.
In the present manuscript, we describe a simple technique for the isolation and preparation of single cells for transcriptomic analysis. Moreover, we suggest ways to edit the protocol so that this technique can be used for a multitude of experiments with a range of goals in mind. The protocol described here was adapted from a protocol described by Trombetta et al.24 as an adjustment of the recommended kit instructions for low-input RNA-Seq. The major differences lie within cell isolation and various volumetric changes that we have chosen to best suit the needs of this project. It is important to highlight that the most important step in this protocol is the isolation of cells from the retinal tissue. This is the point in the protocol during which most errors can occur, and it should be carried out with the most careful attention. Any amount of contamination can result in unusable samples, while the loss of cytoplasm may cause a severe depletion of mRNA molecules. This step should be carried out with care and by the same individual from experiment to experiment to ensure that the samples have been handled precisely.
In summary, this protocol describes a technique for the classification, isolation, and preparation of cells for high-quality RNA-Seq. This is an efficient, relatively low-cost way to optimize the quality of data from single cells. This technique is versatile and may be minimally modified for application to various studies.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We would like to acknowledge Jennifer Bair and Einat Snir, as well as the University of Iowa Institute for Human Genetics, for their assistance in preparing and handling samples.
References
- Austin C, Cepko CL. Specification of Cell Fate in the Vertebrate Retina. Neural Cell Specif. 1995;3:139–143. [Google Scholar]
- Masland RH. The Neuronal Organization of the Retina. Neuron. 2012;76(2):266–280. doi: 10.1016/j.neuron.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodieck R. The First Steps in Seeing. Sunderland, MA: Sinauer Associates, Inc; 1998. [Google Scholar]
- Chiu IM, et al. Transcriptional profiling at whole population and single cell levels reveals somatosensory neuron molecular diversity. Elife. 2014;3:e04660. doi: 10.7554/eLife.04660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B, et al. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat. Neurosci. 2016;19(2):335–346. doi: 10.1038/nn.4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C. Defining cell types and states with single-cell genomics. Genome Res. 2015;25(10):1491–1498. doi: 10.1101/gr.190595.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, et al. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science. 2015;347(6226):1138–1142. doi: 10.1126/science.aaa1934. [DOI] [PubMed] [Google Scholar]
- Schmidt TM, Do MTH, Dacey D, Lucas R, Hattar S, Matynia A. Melanopsin-Positive Intrinsically Photosensitive Retinal Ganglion Cells: From Form to Function. J. Neurosci. 2011;31(45):16094–16101. doi: 10.1523/JNEUROSCI.4132-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberwine J, Miyashiro K, Kacharmina JE, Job C. Local translation of classes of mRNAs that are targeted to neuronal dendrites. Proc. Natl. Acad. Sci. 2001;98(13):7080–7085. doi: 10.1073/pnas.121146698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmanis S, et al. A survey of human brain transcriptome diversity at the single cell level. Proc. Natl. Acad. Sci. 2015;112(23):7285–7290. doi: 10.1073/pnas.1507125112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko EZ, et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell. 2015;161(5):1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usoskin D, et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 2015;18(1):145–153. doi: 10.1038/nn.3881. [DOI] [PubMed] [Google Scholar]
- Poulin JF, Zou J, Drouin-Ouellet J, Kim KYA, Cicchetti F, Awatramani RB. Defining midbrain dopaminergic neuron diversity by single-cell gene expression profiling. Cell. Rep. 2014;9(3):930–943. doi: 10.1016/j.celrep.2014.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross A, Schoendube J, Zimmermann S, Steeb M, Zengerle R, Koltay P. Technologies for Single-Cell Isolation. Int. J. Mol. Sci. 2015;16:16897–16919. doi: 10.3390/ijms160816897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovatt D, et al. Transcriptome in vivo analysis (TIVA) of spatially defined single cells in live tissue. Nat Methods. 2014;11(2):190–196. doi: 10.1038/nmeth.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu S, et al. Single-neuron RNA-Seq: Technical feasibility and reproducibility. Front. Genet. 2012;3(124):1–8. doi: 10.3389/fgene.2012.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subkhankulova T, Yano K, Robinson HPC, Livesey FJ. Grouping and classifying electrophysiologically-defined classes of neocortical neurons by single cell, whole-genome expression profiling. Front. Mol. Neurosci. 2010;3(10):1–11. doi: 10.3389/fnmol.2010.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuzik J, et al. Integration of electrophysiological recordings with single-cell RNA-seq data identifies neuronal subtypes. Nat. Biotechnol. 2016;34(2):175–183. doi: 10.1038/nbt.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt TM, Kofuji P. An isolated retinal preparation to record light response from genetically labeled retinal ganglion cells. J. Vis. Exp. 2011. p. e2367. [DOI] [PMC free article] [PubMed]
- Schmidt TM, Kofuji P. Functional and morphological differences among intrinsically photosensitive retinal ganglion cells. J Neurosci. 2009;29(2):476–482. doi: 10.1523/JNEUROSCI.4117-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt TM, Kofuji P. Structure and Function of Bistratified Intrinsically Photosensitive Retinal Ganglion Cells in the Mouse. J Comp Neurol. 2011;519(8):1492–1504. doi: 10.1002/cne.22579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt TM, Kofuji P. Differential cone pathway influence on intrinsically photosensitive retinal ganglion cell subtypes. J Neurosci. 2010;30(48):16262–16271. doi: 10.1523/JNEUROSCI.3656-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clontech Laboratories I. SMARTerTM Ultra Low RNA Kit for Illumina® Sequencing. 2013.
- Trombetta JJ, Gennert D, Lu D, Satija R, Shalek AK, Regev A. Preparation of single-cell RNA-Seq libraries for next generation sequencing. Curr. Protoc. Mol. Biol. 2014;4(22):1–17. doi: 10.1002/0471142727.mb0422s107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecker JL, et al. Melanopsin-expressing retinal ganglion-cell photoreceptors: Cellular diversity and role in pattern vision. Neuron. 2010;67(1):49–60. doi: 10.1016/j.neuron.2010.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt TM, et al. A Role for Melanopsin in Alpha Retinal Ganglion Cells and Contrast Detection. Neuron. 2014;82(4):781–788. doi: 10.1016/j.neuron.2014.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, Masland RH. The Types of Retinal Ganglion Cells: Current Status and Implications for Neuronal Classification. Annu. Rev. Neurosci. 2014;38:221–246. doi: 10.1146/annurev-neuro-071714-034120. [DOI] [PubMed] [Google Scholar]
- Goetz JJ, Trimarchi JM. Single-cell Profiling of Developing and Mature Retinal Neurons. J. Vis. Exp. 2012. p. e3824. [DOI] [PMC free article] [PubMed]
- Trimarchi JM, et al. Molecular Heterogeneity of Developing Retinal Ganglion and Amacrine Cells Revealed through Single Cell Gene Expression Profiling. J. Comp. Neurol. 2007;502:1047–1065. doi: 10.1002/cne.21368. [DOI] [PubMed] [Google Scholar]