

Abstract
The classification “gastrointestinal stromal tumor” (GIST) became commonplace in the 1990s and since that time various advances have characterized the GIST lineage of origin, tyrosine kinase mutations, and mechanisms of response and resistance to targeted therapies. In addition to tyrosine kinase mutations and their constitutive activation of downstream signaling pathways, GISTs acquire a sequence of chromosomal aberrations. These include deletions of chromosomes 14q, 22q, 1p, and 15q, which harbor putative tumor suppressor genes required for stepwise progression from microscopic, preclinical forms of GIST (“microGIST”) to clinically relevant tumors with malignant potential. Recent advances extend our understanding of GIST biology beyond that of the oncogenic KIT/PDGFRA tyrosine kinases and beyond mechanisms of KIT/PDGFRA-inhibitor treatment response and resistance. These advances have characterized ETV1 as an essential interstitial cell of Cajal-GIST transcription factor in oncogenic KIT signaling pathways, and have characterized the biologically distinct subgroup of SDH-deficient GIST, which are particularly common in young adults. Also, recent discoveries of MAX and dystrophin genomic inactivation have expanded our understanding of GIST development and progression, showing that MAX inactivation is an early event fostering cell cycle activity, whereas dystrophin inactivation promotes invasion and metastasis.
Keywords: GIST, receptor tyrosine kinase, tumor suppressor, cell cycle, progression
Introduction
This discussion of recent advances in GIST will be preceded by a brief overview of the substantial achievements made in the past two decades since the discovery that most gastrointestinal stromal tumors (GISTs) arise from oncogenic receptor tyrosine kinase mutations. This backdrop is the framework for our current understandings of GIST biology, pathology, and treatment.
In 1983, Mazur and Clark first recognized GISTs as a unique variety of “stromal tumor”.1 These investigators showed that some gastric tumors diagnosed as leiomyomas or leiomyosarcomas lacked ultrastructural features of smooth muscle or schwannian differentiation, but instead contained interposed cell processes, primitive junctions, and large cytoplasmic vacuoles, suggesting origin from the myenteric nervous system.1 The hypothesis that GISTs may develop from the gastrointestinal (GI) autonomic nervous system gained further support from a study of gastric GISTs arising in the Carney triad syndrome, in which Perez-Atayde et al. noted ultrastructural features of neuroectodermal differentiation and proposed that the cells of origin were the interstitial cells of Cajal.2 Discovery that GISTs feature immunohistochemical expression of CD343 and KIT4 further supported an origin from the interstitial cell of Cajal lineage and differentiated GISTs from leiomyomas and gastric schwannomas.
In 1998, Hirota et al. ushered in the “molecular era” of GIST biology in groundbreaking work showing KIT gain-of-function mutations as oncogenic driver events in GISTs.5 At the time, GISTs were refractory to all conventional systemic therapies, but this study paved the way for the development of GIST targeted therapies. In 2000, imatinib was first used to treat patients with advanced GIST, achieving unprecedented treatment responses in a tumor type that – when metastatic – was previously rapidly lethal.6 Since then, imatinib has been first-line standard of care for both palliative and adjuvant treatments of GIST patients. The broad clinical spectrum of GIST, varying from incidental indolent tumors to highly aggressive tumors with widely metastatic disease, necessitated implementation of risk stratification schemes to predict biologic behavior and determine which patients particularly benefit from adjuvant imatinib treatment. In 2002, the first such risk stratification system was introduced by Fletcher et al.7 and modified by Miettinen and Lasota in 2006.8 It was soon also appreciated that the location of the KIT initiating mutation (particularly exon 9 vs. exon 11) influenced imatinib response in a given GIST.9, 10 Rapid research progress showed that PDGFRA mutations, present in ∼10% of GIST, are mutually exclusive with KIT mutations (Fig. 1).11 Imatinib responses are most dramatic in GISTs with KIT exon 11 mutations, where 400 mg/day is invariably an effective dose, whereas GISTs with KIT exon 9 mutations typically require 800 mg/day of imatinib for optimal clinical response. GISTs with certain kinase mutations – such as PDGFRA D842V – are imatinib-resistant. The dramatically high GIST response rates to imatinib have been surprisingly durable, and a subset of patients have benefitted from daily imatinib for more than 10 years, with effective control of metastatic GIST.12, 13 However, many patients develop clinical progression due to drug-resistant GIST after several years of imatinib therapy, resulting from heterogeneous secondary KIT mutations (on the same allele as the primary KIT mutation) generally affecting the KIT ATP binding-pocket (exons 13-14) or activation loop (exon 17-18) domains.14
Figure 1.

Distribution of primary KIT and PDGFRA tyrosine kinase mutations in GIST.
Since 2010, major discoveries have been achieved that extend our understanding of GIST biology beyond that of the oncogenic tyrosine kinases and related signaling pathways, and beyond mechanisms of KIT/PDGFRA-inhibitor treatment response and resistance. These advances include characterization of ETV1 as an essential transcription factor for the interstitial cell of Cajal-GIST lineage which cooperates with oncogenic KIT mutations in interstitial cell of Cajal lineages to initiate an oncogenic program. Another key advance has been the molecular characterization of SDH subunit deficiency in GISTs, particularly those arising in young adults, supporting that such GISTs are a distinct molecular subgroup (Fig. 2). Other studies have shed light on tumor suppressors responsible for GIST genetic and biologic progression, including inactivation of the 14q tumor suppressor MAX as an early event fostering cell cycle activity, and inactivation of dystrophin on Xp21 as an event promoting invasion and metastasis.
Figure 2.
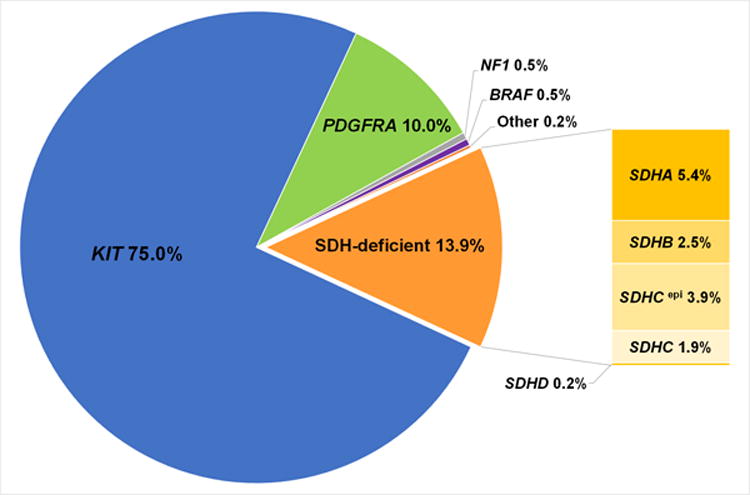
Overview of the frequency of the molecular subtypes of GIST.58
GIST origins
Although GISTs recapitulate interstitial cell of Cajal differentiation, the exact cell(s) of origin for GIST are unknown, i.e., whether precursors of interstitial cells of Cajal or committed interstitial cells of Cajal. GISTs share expression of many biomarkers with interstitial cells of Cajal, and some of these, such as KIT and ANO1 (DOG-1) have proven to be essential diagnostic markers for GIST. Other shared markers in GIST and interstitials cell of Cajal have highlighted crucial biologic mechanisms in GIST development. One such marker is ETV115, which is discussed below. Multifocal interstitial cell of Cajal hyperplasia is a GIST precursor state found in many individuals with GIST syndromes, including those with germline KIT or PDGFRA mutations, Carney triad (SDHC methylation) or neurofibromatosis type 1 (NF1). These polyclonal interstitial cell of Cajal hyperplasias predispose to development of multiple GISTs.16 KIT is not only highly expressed in interstitial cells of Cajal and GIST but also in hematopoietic stem cells, melanocytes, mast cells, and germ cells. Notably, some oncogenic KIT mutations have transforming activity primarily in the interstitial cell of Cajal/GIST context15, whereas others not so. For example, germline oncogenic KIT D816V mutation fosters mastocytosis but not GIST.17 Similarly, it has been shown that human GIST-associated KIT mutations, when expressed in mouse models, almost exclusively foster interstitial cell of Cajal hyperplasia and GIST, but do not engender other types of KIT-positive neoplasia. These observations show that the correct cellular context is needed for a particular KIT mutation to have transforming activity.
So-called microscopic GISTs (microGISTs) are preclinical forms of GIST that measure <1.0 cm and are remarkably frequent in the general population. Autopsy studies have identified microGISTs in the GI tract of ∼30% of unselected individuals, and oncogenic tyrosine kinase mutations can already be detected in these early forms of GIST.18-20 Because <0.1% of these microGISTs progress to clinically relevant tumors, it is clear that KIT/PDGFRA oncogenic mutations are necessary but insufficient to foster most advanced, clinically-evident GISTs. Other genomic alterations, beyond the initiating KIT/PDGFRA mutations, are required to enable biologic tumor progression.
Many GISTs depend on the lineage-specific transcription factor ETV1
ETV1 is a member of the ETS family of transcription factors. Chi et al. have shown that ETV1 is both highly expressed and requisite for development of interstitial cells of Cajal subtypes that depend on KIT signaling.15 These studies demonstrated that ETV1 is a master regulator of the interstitial cell of Cajal and GIST lineage-specific transcription network and is essential for GIST growth.15 Beyond the role in GIST growth, ETV1 is directly responsible for transcriptionally activating many of the known GIST biomarkers. The KIT oncoproteins in GIST signal through the RAS/RAF/MEK pathway to stabilize ETV1 at the protein level, thereby promoting GIST tumorigenesis.15 By contrast, treatment of GIST cells with KIT-inhibitors or MEK/MAPK-inhibitors causes immediate ETV1 downregulation, by proteasomal degradation, leading to GIST growth arrest. These findings highlight therapeutic opportunities for RAS/RAF/MEK pathway inhibitors in GIST.
Various molecular GIST subtypes depend on RAS/RAF/MEK and PI3K/AKT pathways
Most GISTs (∼85%) have KIT or PDGFRA oncogenic mutations (Fig. 1) that constitutively activate downstream RAS/RAF/MEK and PI3K/AKT/mTOR pathways, causing cell proliferation and survival. A smaller subset of GISTs arise from mutational inactivation of the neurofibromatosis 1 protein (NF1), or mutational activation of RAS or BRAF21-25: each of these alternate mutational mechanisms results in constitutive activation of RAS/RAF/MEK pathways. In so doing, the alternate molecular mechanisms supplant the need for upstream KIT/PDGFRA activation and are therefore biologically analogous to KIT- and PDGFRA-mutant GIST. Several reports suggest that dual mutations, one activating RAS/RAF/MEK and the other activating PI3K/AKT/mTOR pathways, are needed to optimally recapitulate upstream KIT/PDGFRA activation.22, 26 In addition, KIT-, PDGFRA-, and NF1-mutant GISTs share mechanisms of genetic progression (discussed below), further credentialing their biologic similarities. These GIST molecular subtypes are distinct from the SDH-deficient GIST subgroup, as will be discussed below. Other alternative kinase mechanisms have been described, in GISTs, involving the FGFR1 and NTRK3 genes27, and it can be assumed (although not yet demonstrated) that these also foment oncogenic signaling via the RAS/RAF/MEK and PI3K/AKT/mTOR pathways.
Histopathologic findings and risk assessment
GISTs usually present as sharply demarcated subserosal or submucosal tumors in the GI tract and display either a spindled (70%), epithelioid (20%) or mixed (10%) cytomorphology. Expression of KIT is present in 95% of cases, DOG-1 in 98%, PDGFRA in 80%, CD34 in 70-80%, and – with the exception of SDH-deficient GISTs – expression of SDHA and SDHB is retained (Fig. 3).
Figure 3.

Typical histologic features in GIST. A KIT-mutant GIST with sheet-like, solid growth (A) showing expression of DOG-1 (A, inset), SDHB (B), and SDHA (C). In contrast, SDH-deficient GISTs exhibit characteristic multinodular growth pattern at low power (D), are positive for DOG-1 (D, inset) and lack SDHB expression (E). In this case, SDHA expression (F) is retained, indicating the GIST arises from mutation of SDHB, SDHC or SDHD, rather than SDHA. Another example of an SDH-deficient GIST showing the characteristic epithelioid morphology (G) and expression of DOG-1 (G, inset) with SDHB (H) and SDHA (I) loss of expression indicating an underlying SDHA mutation; vessels (bottom left) serve as positive internal control.
Approximately 30% of GISTs are malignant, and prediction of malignant potential based on histopathologic criteria is crucial in identifying patients with high likelihood of local recurrence or distant metastases. The first risk stratification system was introduced by Fletcher et al., predicting GIST malignant behavior by classification into very low, low, intermediate, and high risk categories based on tumor size and mitotic rate.7 A modified classification system developed by Miettinen and Lasota8 introduced tumor site as a third independent factor. This classification system led to the Armed Forces Institute of Pathology (AFIP) criteria and the National Comprehensive Cancer Network (NCCN) risk criteria28, reliably predicting risk of progression in GIST (with the exception of SDH-deficient GIST, see below) (Table 1).
Table 1.
Summary of the Armed Forces Institute of Pathology (AFIP)8 (A) and the National Comprehensive Cancer Network (NCCN)28 (B) criteria for risk assessment in GIST.
| A | ||||||
|---|---|---|---|---|---|---|
| AFIP Criteria: | ||||||
| Tumor parameters | Patients with progressive disease during long-term follow up and characterization of risk for metastasis | |||||
|
| ||||||
| Group | Tumor size (cm) | Mitotic rate (per 50 HPFs) | Gastric GISTs | Jejunal and Ileal GISTs | Duodenal GISTs | Rectal GISTs |
| 1 | ≤2 | ≤5 | 0% None | 0% None | 0% None | 0% None |
| 2 | >2 ≤5 | ≤5 | 1.9% Very low | 4.3% Low | 8.3% Low | 8.5% Low |
| 3a | >5 ≤10 | ≤5 | 3.6% Low | 25% Moderate | 34% High** | 57%* High** |
| 3b | >10 | ≤5 | 12% Moderate | 52% High | ||
| 4 | ≤2 | >5 | 0%* | 50%* | ∫ | 54% High |
| 5 | >2 ≤5 | >5 | 16% Moderate | 73% High | 50% High | 52% High |
| 6a | >5 ≤10 | >5 | 55% High | 85% High | 86% High** | 71% High** |
| 6b | >10 | >5 | 86% High | 90% High | ||
| B | |||||
|---|---|---|---|---|---|
| NCCN Criteria: | |||||
| Tumor parameters | Risk of Progressive Disease | ||||
|
| |||||
| Mitotic Index (per 50 HPFs) | Size (cm) | Gastric | Duodenum | Jejunum/Ileum | Rectum |
| ≤5 | ≤2 | 0% None | 0% None | 0% None | 0% None |
| ≤5 | >2 ≤5 | 1.9% Very low | 4.3% Low | 8.3% Low | 8.5% Low |
| ≤5 | >5 ≤10 | 3.6% Low | 24% Moderate | Insufficient data | Insufficient data |
| ≤5 | >10 | 10% Moderate | 52% High | 34% High | 57% High |
| >5 | ≤2 | None* | High* | Insufficient data | 54% High |
| >5 | >2 ≤5 | 16% Moderate | 73% High | 50% High | 52% High |
| >5 | >5 ≤10 | 55% High | 85% High | Insufficient data | Insufficient data |
| >5 | >10 | 86% High | 90% High | 86% High | 71% High |
Small number of cases
Combined groups because of small number of cases
No tumors of such category included
HPF: High power field
Extragastrointestinal GISTs and GISTs arising at visceral locations
The concept of so-called “extragastrointestinal GISTs” (or “E-GISTs”) was introduced when it was recognized that GISTs occasionally appear to arise outside the GI tract, such as the omentum, mesentery, retroperitoneum, or pleura.29-33 E-GISTs generally share canonical morphologic, immunohistochemical, and molecular features of conventional GISTs, and E-GISTs primarily arising in the mesentery or retroperitoneum seem to follow a more aggressive clinical course31, 33, compared to cases arising in the omentum.30 Some E-GISTs arise in the tubular GI tract but have become substantially detached from the gastric or intestinal primary site, which can therefore be clinically occult. Explanations for the genesis of true E-GISTs might include origin from ectopic interstitial cells of Cajal or from pluripotential mesenchymal progenitor cells. Origin from developmental abnormalities (such as enteric duplication) has also been proposed.33 GISTs arising primarily in visceral organs are exceedingly rare, and have not been systematically studied.34, 35 GISTs manifesting primarily in visceral organs such as the liver or spleen most likely represent distant metastases from an occult GI tract primary GIST.
Treatment of localized and advanced disease
The mainstay of treatment for localized GIST remains surgery. Low and intermediate risk GISTs do not require adjuvant treatment, whereas high risk GISTs with mutations sensitive to imatinib are usually treated with three years of imatinib (i.e., KIT exon 9 and 11, PDGFRA exon 12, 14, and 18 with the exception of the exon 18 D842V mutation).36-39 Adjuvant imatinib recurrence-free survival benefit, for patients with KIT exon 11 mutant GISTs, depends on the location of the mutations, with those involving KIT exon 11 codons 557 and/or 558 seeming to benefit most.37, 40 Imatinib has revolutionized GIST treatment by extending the median overall survival for patients with metastatic disease from 19 months to 5 years, and ∼80% of patients show initial treatment response.41 However, most patients with initial response develop secondary drug resistance which is mainly caused by selection for subclones harboring KIT mutations in the ATP-binding pocket (exons 13 and 14) or activation loop (exons 17 and 18). Sunitinib has been approved as second-line treatment for imatinib-resistant GIST and shows efficacy in GIST with imatinib-resistance mutations in the ATP-binding pocket42, whereas third-line regorafenib therapy shows predominantly complementary activity by best inhibiting GISTs with imatinib-resistance due to activation loop mutations.43 However, treatment of metastatic GISTs containing multiple resistant subclones and heterogeneous genomic subpopulations remains a therapeutic challenge.14
Despite the expression of activated KIT in NF1-mutant and SDH-deficient GIST, therapeutic KIT inhibition is not clinically effective in these GIST subtypes.44, 45 Alternative molecular targets need to be developed to effectively treat these molecular subsets of GIST.
Genomic progression in GIST
Although most GISTs arise from KIT, PDGFRA or NF1 mutations, additional chromosomal aberrations are required to foster GIST progression. Most microGISTs already contain KIT, PDGFRA or NF1 mutations and yet have exceedingly low potential for malignant progression. Therefore, GISTs provide an ideal model by which to study constraints to tumorigenic progression. Further, even advanced GISTs have non-complex genomic landscapes compared to most other cancers, and therefore serve as tractable models by which to elucidate mechanisms of genomic and biologic progression in cancer. Another notable difference between GISTs and most other sarcomas is the fact that the benign precursor state has been well-characterized in GIST. The opportunity to study precursor lesions such as interstitial cell of Cajal hyperplasia and microGIST enables evaluations of the sequence of mutations accounting for oncogenic progression. Such studies are more challenging in sarcomas and other cancers where a benign precursor state cannot be identified and where the cancer progenitor cell is unknown.
GIST cytogenetic progression has been studied extensively46-50, and it seems that most GISTs develop by means of a stepwise accumulation of chromosomal aberrations. Loss of 14q is observed in 60-70% of cases as the earliest and most frequent aberration, followed by loss of 22q (∼50%), 1p (∼50%), and 15q (∼40%) in intermediate and higher risk tumors.46-50 Stepwise genomic inactivation of putative tumor suppressor genes located on these specific chromosomes is likely responsible for genomic progression, following the initiating tyrosine kinase or NF1-pathway mutations (Fig. 4).
Figure 4.

Model of GIST genomic progression. Primary KIT, PDGFRA or NF1 mutations represent the initiating oncogenic driver events in most GISTs and are followed by stepwise accumulation of chromosomal aberrations, harboring putative tumor suppressor genes, and cell cycle dysregulating events. Metastatic GISTs develop treatment resistance through evolving TKI-resistant subclones with additional secondary KIT or PDGFRA mutations.
The first 14q GIST tumor suppressor was recently shown to be the MYC-associated factor X (MAX), a basic helix-loop-helix leucine zipper (bHLHZ) transcription factor and the essential binding partner of MYC.51, 52 Homozygous MAX inactivation by intragenic deletion or mononucleotide mutations was found in ∼20% of KIT-, PDGFRA-, and NF1-mutant GISTs, and ∼50% of GISTs had extinction of MAX protein expression (Fig. 5).51 These studies demonstrated MAX inactivation in microGISTs and low risk GISTs and showed identical MAX inactivating events in all metastases from individual patients, indicating that MAX tumor suppressor inactivation is an early step in GIST progression.51 MAX inactivation resulted in p16 inactivation and cell cycle perturbations in GIST, suggesting that these tumor suppressor events serve to increase proliferation in the early GIST.51
Figure 5.

A metastatic GIST (A) without MAX or p16/INK4A coding region deletion with retained expression of MAX (B) and p16 (C); Another metastatic GIST (D) with homozygous MAX deletion and without p16/INK4A coding region deletion shows loss of MAX (E) and p16 (F) expression; vessels and inflammatory cells serve as positive internal controls. Dystrophin immunohistochemistry (using the DYS-A antibody) shows predominantly membranous expression in normal skeletal muscle (G). A GIST with retained expression of dystrophin (H) and another GIST showing loss of dystrophin expression (I); infiltrated smooth muscle cells (bottom left) serve as positive internal control.
Further cell cycle dysregulation occurs in higher risk GISTs, resulting in transition to a high-grade cancer, and generally results from inactivating mutations in the p16, p53, or RB1 tumor suppressors.53, 54 These mutations rarely occur in low risk GISTs and therefore might be useful as prognostic biomarkers, or as predictive markers to identify patients that stand to benefit most from adjuvant imatinib therapy.
Inactivation of dystrophin, encoded by the DMD gene on Xp21.1, has been shown to occur as a late event in GIST progression and is present in more than 90% of metastatic GISTs.55 DMD inhibits cell migration, invasion, anchorage independence, and invadopodia formation, and its inactivation facilitates metastatic spread in GIST. Ongoing studies aim at validating dystrophin expression in GIST as a prognostic and predictive biomarker: patients with intermediate/high risk GISTs that show a loss of dystrophin expression by immunohistochemistry (Fig. 5). One can hypothesize that primary GISTs with dystrophin inactivation might particularly benefit from long-term adjuvant/palliative tyrosine kinase inhibitor (TKI) treatment as these tumors might have high likelihood of occult metastases – in contrast, patients whose GISTs retain dystrophin expression might be at low risk for metastases and therefore could be spared the side effects and costs of long-term imatinib therapy. Targeted gene therapies which have been developed in patients with muscular dystrophies (type Duchenne or Becker) provide compelling opportunities to develop targeted therapies to restore or replace dystrophin function in myogenic cancers with DMD inactivation.
Based on these recent discoveries, homozygous deletions are emerging as a major mechanism of tumor suppressor inactivation in GIST. This may, in part, explain why these events may have been missed by previous screening approaches, as the size of these intragenic deletions is often too large to be detected by conventional targeted sequencing algorithms and at the same time too small to be revealed by SNP arrays.
SDH-deficient GIST
Most GISTs lacking KIT, PDGFRA, or NF1 mutations arise from loss-of-function alterations of the succinate dehydrogenase complex (SDH), an enzymatic complex involved in the citric acid cycle and the electron transport chain (Fig. 3). The SDH complex is comprised of proteins encoded by SDHA, SDHB, SDHC, and SDHD, and loss of function of any of these four components translates into a loss of SDHB expression (Fig. 3).56 Subunit inactivation is achieved by mutations of subunit encoding genes (accounting for ∼80% of cases) or SDHC promoter methylation resulting in epigenetic inactivation of the SDHC gene (SDHC-“epimutated”, accounting for ∼20% of cases) (Fig. 2).57, 58
SDH-deficiency is found in GISTs arising as part of the non-hereditary Carney triad59 (including paraganglioma and pulmonary chondroma) and the autosomal-dominant Carney-Stratakis syndrome (together with paragangliomas) with predisposing germline SDH subunit mutations.60, 61 SDH-deficient GISTs are characterized by several features distinct from KIT/PDGFRA-mutant GISTs: they occur nearly exclusively in the stomach and show either epithelioid or mixed morphology but virtually never pure spindle cell morphology.62 Their growth pattern is characterized by a unique multilobular or plexiform architecture, with nests of tumor cells separated by septae of smooth muscle which facilitates their recognition on conventional HE-stained slides (Fig. 3). Immunohistochemical staining with SDHB reveals a loss of expression in tumor cells and additional SDHA loss points towards an underlying SDHA mutation, whereas retained SDHA expression is observed in SDHB-, SDHC-, and SDHD-mutated/epimutated GISTs (Fig. 3). SDH-deficient GISTs express activated KIT56, but the mechanism of KIT activation is unclear. SDH-deficient GISTs feature a hypermethylation program58, 63 that differs from the more common GISTs with KIT, PDGFRA or NF1 mutations. Further, SDH-deficient GISTs lack the canonical series of cytogenetic deletions that invariably accumulate during neoplastic progression in GISTs with KIT, PDGFRA or NF1 mutations.64 Although SDH-deficient GISTs generally lack large-scale genomic aberrations, one exception is occasional 1q deletion, which can apparently target the SDHC gene.59, 61 Despite propensity for lymphatic spread and multifocality, SDH-deficient GIST usually follow an indolent clinical course.65 The aforementioned morphologic and genomic features underscore that SDH-deficient GISTs are a truly distinct biologic subgroup, arising from mechanisms different from those in KIT/PDGFRA/NF1-mutant GISTs, and virtually always restricted to the stomach as the primary site.
As shown recently in a study of 76 SDH-deficient GISTs, conventional risk stratification approaches do not apply to this particular subgroup as they fail to predict disease progression.65 Specifically, 60-80% of patients with SDH-deficient GIST were shown to develop distant metastases regardless of risk category, and models that more accurately predict SDH-deficient GIST progression and patient survival remain to be established.65
Conclusions
Recent research progress has substantially advanced our understanding of GIST biology and has shown that GISTs arising by virtue of different genetic alterations also have different clinicopathological features. One hopes this progress will lead to new therapies, augmenting the dramatic therapeutic progress and survival benefits already accomplished with the approved KIT/PDGFRA-inhibitors imatinib, sunitinib, and regorafenib. GIST translational research spans from early (and even initiating) kinase or SDH mutations, as driver oncogenic events, on to dystrophin inactivation as a late event in GIST progression leading to metastatic spread. Future studies defining the patient subsets that benefit most from long-term adjuvant TKI therapy are expected to further improve individualized treatment options. Likewise, creative new approaches are needed to suppress the heterogeneous TKI-resistant GIST subclones in individual patients. Investigating the steps of genomic progression in the distinct subset of SDH-deficient GISTs and developing specific treatment options for SDH-deficient and NF1-mutant tumors also remain urgent challenges to be addressed in translational research efforts.
Acknowledgments
The authors thank Dr. Jason L. Hornick, Department of Pathology, Brigham and Women's Hospital, Harvard Medical School, for immunohistochemical stains.
Footnotes
Conflicts of Interest and Source of Funding: The authors have no conflicts of interest to disclose. This work was supported by US National Institutes of Health grants 1P50CA127003 (JAF) and 1P50CA168512 (JAF, AME), and by the GIST Cancer Research Fund (JAF).
References
- 1.Mazur MT, Clark HB. Gastric stromal tumors. Reappraisal of histogenesis. Am J Surg Pathol. 1983;7:507–519. doi: 10.1097/00000478-198309000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Perez-Atayde AR, Shamberger RC, Kozakewich HW. Neuroectodermal differentiation of the gastrointestinal tumors in the Carney triad. An ultrastructural and immunohistochemical study. Am J Surg Pathol. 1993;17:706–714. doi: 10.1097/00000478-199307000-00008. [DOI] [PubMed] [Google Scholar]
- 3.Miettinen M, Virolainen M, Maarit Sarlomo R. Gastrointestinal stromal tumors--value of CD34 antigen in their identification and separation from true leiomyomas and schwannomas. Am J Surg Pathol. 1995;19:207–216. doi: 10.1097/00000478-199502000-00009. [DOI] [PubMed] [Google Scholar]
- 4.Sarlomo-Rikala M, Kovatich AJ, Barusevicius A, et al. CD117: a sensitive marker for gastrointestinal stromal tumors that is more specific than CD34. Mod Pathol. 1998;11:728–734. [PubMed] [Google Scholar]
- 5.Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–580. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- 6.Joensuu H, Roberts PJ, Sarlomo-Rikala M, et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med. 2001;344:1052–1056. doi: 10.1056/NEJM200104053441404. [DOI] [PubMed] [Google Scholar]
- 7.Fletcher CD, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Int J Surg Pathol. 2002;10:81–89. doi: 10.1177/106689690201000201. [DOI] [PubMed] [Google Scholar]
- 8.Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol. 2006;23:70–83. doi: 10.1053/j.semdp.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 9.Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342–4349. doi: 10.1200/JCO.2003.04.190. [DOI] [PubMed] [Google Scholar]
- 10.Rubin BP, Singer S, Tsao C, et al. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res. 2001;61:8118–8121. [PubMed] [Google Scholar]
- 11.Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708–710. doi: 10.1126/science.1079666. [DOI] [PubMed] [Google Scholar]
- 12.Heinrich M, Rankin C, Blanke CD, et al. Correlation of Long-term Results of Imatinib in Advanced Gastrointestinal Stromal Tumors With Next-Generation Sequencing Results: Analysis of Phase 3 SWOG Intergroup Trial S0033. JAMA Oncol. 2017 doi: 10.1001/jamaoncol.2016.6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cameron S, Schaefer IM, Schwoerer H, et al. Ten Years of Treatment with 400 mg Imatinib per Day in a Case of Advanced Gastrointestinal Stromal Tumor. Case Rep Oncol. 2011;4:505–511. doi: 10.1159/000333471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liegl B, Kepten I, Le C, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol. 2008;216:64–74. doi: 10.1002/path. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chi P, Chen Y, Zhang L, et al. ETV1 is a lineage survival factor that cooperates with KIT in gastrointestinal stromal tumours. Nature. 2010;467:849–853. doi: 10.1038/nature09409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen H, Hirota S, Isozaki K, et al. Polyclonal nature of diffuse proliferation of interstitial cells of Cajal in patients with familial and multiple gastrointestinal stromal tumours. Gut. 2002;51:793–796. doi: 10.1136/gut.51.6.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Longley BJ, Jr, Metcalfe DD, Tharp M, et al. Activating and dominant inactivating c-KIT catalytic domain mutations in distinct clinical forms of human mastocytosis. Proc Natl Acad Sci U S A. 1999;96:1609–1614. doi: 10.1073/pnas.96.4.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agaimy A, Wunsch PH, Hofstaedter F, et al. Minute gastric sclerosing stromal tumors (GIST tumorlets) are common in adults and frequently show c-KIT mutations. Am J Surg Pathol. 2007;31:113–120. doi: 10.1097/01.pas.0000213307.05811.f0. [DOI] [PubMed] [Google Scholar]
- 19.Agaimy A, Wunsch PH, Dirnhofer S, et al. Microscopic gastrointestinal stromal tumors in esophageal and intestinal surgical resection specimens: a clinicopathologic, immunohistochemical, and molecular study of 19 lesions. Am J Surg Pathol. 2008;32:867–873. doi: 10.1097/PAS.0b013e31815c0417. [DOI] [PubMed] [Google Scholar]
- 20.Kawanowa K, Sakuma Y, Sakurai S, et al. High incidence of microscopic gastrointestinal stromal tumors in the stomach. Hum Pathol. 2006;37:1527–1535. doi: 10.1016/j.humpath.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 21.Miettinen M, Fetsch JF, Sobin LH, et al. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: a clinicopathologic and molecular genetic study of 45 cases. Am J Surg Pathol. 2006;30:90–96. doi: 10.1097/01.pas.0000176433.81079.bd. [DOI] [PubMed] [Google Scholar]
- 22.Falchook GS, Trent JC, Heinrich MC, et al. BRAF mutant gastrointestinal stromal tumor: first report of regression with BRAF inhibitor dabrafenib (GSK2118436) and whole exomic sequencing for analysis of acquired resistance. Oncotarget. 2013;4:310–315. doi: 10.18632/oncotarget.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miranda C, Nucifora M, Molinari F, et al. KRAS and BRAF mutations predict primary resistance to imatinib in gastrointestinal stromal tumors. Clin Cancer Res. 2012;18:1769–1776. doi: 10.1158/1078-0432.CCR-11-2230. [DOI] [PubMed] [Google Scholar]
- 24.Gasparotto D, Rossi S, Polano M, et al. Quadruple-Negative GIST Is a Sentinel for Unrecognized Neurofibromatosis Type 1 Syndrome. Clin Cancer Res. 2017;23:273–282. doi: 10.1158/1078-0432.CCR-16-0152. [DOI] [PubMed] [Google Scholar]
- 25.Huss S, Pasternack H, Ihle MA, et al. Clinicopathological and molecular features of a large cohort of gastrointestinal stromal tumors (GISTs) and review of the literature: BRAF mutations in KIT/PDGFRA wildtype GISTs are rare events. Hum Pathol. 2017 doi: 10.1016/j.humpath.2017.01.005. [DOI] [PubMed] [Google Scholar]
- 26.Serrano C, Wang Y, Marino-Enriquez A, et al. KRAS and KIT Gatekeeper Mutations Confer Polyclonal Primary Imatinib Resistance in GI Stromal Tumors: Relevance of Concomitant Phosphatidylinositol 3-Kinase/AKT Dysregulation. J Clin Oncol. 2015;33:e93–96. doi: 10.1200/JCO.2013.48.7488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi E, Chmielecki J, Tang CM, et al. FGFR1 and NTRK3 actionable alterations in “Wild-Type” gastrointestinal stromal tumors. J Transl Med. 2016;14:339. doi: 10.1186/s12967-016-1075-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Demetri GD, Benjamin RS, Blanke CD, et al. NCCN Task Force report: management of patients with gastrointestinal stromal tumor (GIST)--update of the NCCN clinical practice guidelines. J Natl Compr Canc Netw. 2007;5(Suppl 2):S1–29. quiz S30. [PubMed] [Google Scholar]
- 29.Long KB, Butrynski JE, Blank SD, et al. Primary extragastrointestinal stromal tumor of the pleura: report of a unique case with genetic confirmation. Am J Surg Pathol. 2010;34:907–912. doi: 10.1097/PAS.0b013e3181d9f18f. [DOI] [PubMed] [Google Scholar]
- 30.Miettinen M, Sobin LH, Lasota J. Gastrointestinal stromal tumors presenting as omental masses--a clinicopathologic analysis of 95 cases. Am J Surg Pathol. 2009;33:1267–1275. doi: 10.1097/PAS.0b013e3181a13e99. [DOI] [PubMed] [Google Scholar]
- 31.Reith JD, Goldblum JR, Lyles RH, et al. Extragastrointestinal (soft tissue) stromal tumors: an analysis of 48 cases with emphasis on histologic predictors of outcome. Mod Pathol. 2000;13:577–585. doi: 10.1038/modpathol.3880099. [DOI] [PubMed] [Google Scholar]
- 32.Miettinen M, Monihan JM, Sarlomo-Rikala M, et al. Gastrointestinal stromal tumors/smooth muscle tumors (GISTs) primary in the omentum and mesentery: clinicopathologic and immunohistochemical study of 26 cases. Am J Surg Pathol. 1999;23:1109–1118. doi: 10.1097/00000478-199909000-00015. [DOI] [PubMed] [Google Scholar]
- 33.Miettinen M, Felisiak-Golabek A, Wang Z, et al. GIST Manifesting as a Retroperitoneal Tumor: Clinicopathologic Immunohistochemical, and Molecular Genetic Study of 112 Cases. Am J Surg Pathol. 2017;41:577–585. doi: 10.1097/PAS.0000000000000807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barros A, Linhares E, Valadao M, et al. Extragastrointestinal stromal tumors (EGIST): a series of case reports. Hepatogastroenterology. 2011;58:865–868. [PubMed] [Google Scholar]
- 35.Mao L, Chen J, Liu Z, et al. Extracorporeal hepatic resection and autotransplantation for primary gastrointestinal stromal tumor of the liver. Transplant Proc. 2015;47:174–178. doi: 10.1016/j.transproceed.2014.09.111. [DOI] [PubMed] [Google Scholar]
- 36.Joensuu H, Eriksson M, Sundby Hall K, et al. Adjuvant Imatinib for High-Risk GI Stromal Tumor: Analysis of a Randomized Trial. J Clin Oncol. 2016;34:244–250. doi: 10.1200/JCO.2015.62.9170. [DOI] [PubMed] [Google Scholar]
- 37.Joensuu H, Wardelmann E, Sihto H, et al. Effect of KIT and PDGFRA Mutations on Survival in Patients With Gastrointestinal Stromal Tumors Treated With Adjuvant Imatinib: An Exploratory Analysis of a Randomized Clinical Trial. JAMA Oncol. 2017 doi: 10.1001/jamaoncol.2016.5751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Le Cesne A, Blay JY, Reichardt P, et al. Optimizing tyrosine kinase inhibitor therapy in gastrointestinal stromal tumors: exploring the benefits of continuous kinase suppression. Oncologist. 2013;18:1192–1199. doi: 10.1634/theoncologist.2012-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marrari A, Wagner AJ, Hornick JL. Predictors of response to targeted therapies for gastrointestinal stromal tumors. Arch Pathol Lab Med. 2012;136:483–489. doi: 10.5858/arpa.2011-0082-RA. [DOI] [PubMed] [Google Scholar]
- 40.Heinrich MC, Corless CL, Demetri GD. Defining the Impact of Adjuvant Therapy in Molecularly Defined Subsets of Gastrointestinal Stromal Tumor : From Lumping to Splitting. JAMA Oncol. 2017 doi: 10.1001/jamaoncol.2016.5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blanke CD, Demetri GD, von Mehren M, et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26:620–625. doi: 10.1200/JCO.2007.13.4403. [DOI] [PubMed] [Google Scholar]
- 42.Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26:5352–5359. doi: 10.1200/JCO.2007.15.7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ben-Ami E, Barysauskas CM, von Mehren M, et al. Long-term follow-up results of the multicenter phase II trial of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of standard tyrosine kinase inhibitor therapy. Ann Oncol. 2016;27:1794–1799. doi: 10.1093/annonc/mdw228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mussi C, Schildhaus HU, Gronchi A, et al. Therapeutic consequences from molecular biology for gastrointestinal stromal tumor patients affected by neurofibromatosis type 1. Clin Cancer Res. 2008;14:4550–4555. doi: 10.1158/1078-0432.CCR-08-0086. [DOI] [PubMed] [Google Scholar]
- 45.Miettinen M, Wang ZF, Sarlomo-Rikala M, et al. Succinate dehydrogenase-deficient GISTs: a clinicopathologic, immunohistochemical, and molecular genetic study of 66 gastric GISTs with predilection to young age. Am J Surg Pathol. 2011;35:1712–1721. doi: 10.1097/PAS.0b013e3182260752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wozniak A, Sciot R, Guillou L, et al. Array CGH analysis in primary gastrointestinal stromal tumors: cytogenetic profile correlates with anatomic site and tumor aggressiveness, irrespective of mutational status. Genes Chromosomes Cancer. 2007;46:261–276. doi: 10.1002/gcc.20408. [DOI] [PubMed] [Google Scholar]
- 47.El-Rifai W, Sarlomo-Rikala M, Andersson LC, et al. DNA sequence copy number changes in gastrointestinal stromal tumors: tumor progression and prognostic significance. Cancer Res. 2000;60:3899–3903. [PubMed] [Google Scholar]
- 48.Gunawan B, Bergmann F, Hoer J, et al. Biological and clinical significance of cytogenetic abnormalities in low-risk and high-risk gastrointestinal stromal tumors. Hum Pathol. 2002;33:316–321. doi: 10.1053/hupa.2002.32216. [DOI] [PubMed] [Google Scholar]
- 49.Gunawan B, von Heydebreck A, Sander B, et al. An oncogenetic tree model in gastrointestinal stromal tumours (GISTs) identifies different pathways of cytogenetic evolution with prognostic implications. J Pathol. 2007;211:463–470. doi: 10.1002/path.2128. [DOI] [PubMed] [Google Scholar]
- 50.Schaefer IM, Delfs C, Cameron S, et al. Chromosomal aberrations in primary PDGFRA-mutated gastrointestinal stromal tumors. Hum Pathol. 2014;45:85–97. doi: 10.1016/j.humpath.2013.05.027. [DOI] [PubMed] [Google Scholar]
- 51.Schaefer IM, Wang Y, Liang CW, et al. MAX inactivation is an early event in GIST development that regulates p16 and cell proliferation. Nat Commun. 2017;8:14674. doi: 10.1038/ncomms14674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Belinsky MG, Rink L, Cai KQ, et al. Somatic loss of function mutations in neurofibromin 1 and MYC associated factor X genes identified by exome-wide sequencing in a wild-type GIST case. BMC Cancer. 2015;15:887. doi: 10.1186/s12885-015-1872-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schneider-Stock R, Boltze C, Lasota J, et al. High prognostic value of p16INK4 alterations in gastrointestinal stromal tumors. J Clin Oncol. 2003;21:1688–1697. doi: 10.1200/JCO.2003.08.101. [DOI] [PubMed] [Google Scholar]
- 54.Romeo S, Debiec-Rychter M, Van Glabbeke M, et al. Cell cycle/apoptosis molecule expression correlates with imatinib response in patients with advanced gastrointestinal stromal tumors. Clin Cancer Res. 2009;15:4191–4198. doi: 10.1158/1078-0432.CCR-08-3297. [DOI] [PubMed] [Google Scholar]
- 55.Wang Y, Marino-Enriquez A, Bennett RR, et al. Dystrophin is a tumor suppressor in human cancers with myogenic programs. Nat Genet. 2014;46:601–606. doi: 10.1038/ng.2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Janeway KA, Kim SY, Lodish M, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci U S A. 2011;108:314–318. doi: 10.1073/pnas.1009199108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haller F, Moskalev EA, Faucz FR, et al. Aberrant DNA hypermethylation of SDHC: a novel mechanism of tumor development in Carney triad. Endocr Relat Cancer. 2014;21:567–577. doi: 10.1530/ERC-14-0254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boikos SA, Pappo AS, Killian JK, et al. Molecular Subtypes of KIT/PDGFRA Wild-Type Gastrointestinal Stromal Tumors: A Report From the National Institutes of Health Gastrointestinal Stromal Tumor Clinic. JAMA Oncol. 2016;2:922–928. doi: 10.1001/jamaoncol.2016.0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matyakhina L, Bei TA, McWhinney SR, et al. Genetics of carney triad: recurrent losses at chromosome 1 but lack of germline mutations in genes associated with paragangliomas and gastrointestinal stromal tumors. J Clin Endocrinol Metab. 2007;92:2938–2943. doi: 10.1210/jc.2007-0797. [DOI] [PubMed] [Google Scholar]
- 60.Pasini B, McWhinney SR, Bei T, et al. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet. 2008;16:79–88. doi: 10.1038/sj.ejhg.5201904. [DOI] [PubMed] [Google Scholar]
- 61.Stratakis CA, Carney JA. The triad of paragangliomas, gastric stromal tumours and pulmonary chondromas (Carney triad), and the dyad of paragangliomas and gastric stromal sarcomas (Carney-Stratakis syndrome): molecular genetics and clinical implications. J Intern Med. 2009;266:43–52. doi: 10.1111/j.1365-2796.2009.02110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Doyle LA, Nelson D, Heinrich MC, et al. Loss of succinate dehydrogenase subunit B (SDHB) expression is limited to a distinctive subset of gastric wild-type gastrointestinal stromal tumours: a comprehensive genotype-phenotype correlation study. Histopathology. 2012;61:801–809. doi: 10.1111/j.1365-2559.2012.04300.x. [DOI] [PubMed] [Google Scholar]
- 63.Killian JK, Miettinen M, Walker RL, et al. Recurrent epimutation of SDHC in gastrointestinal stromal tumors. Sci Transl Med. 2014;6:268ra177. doi: 10.1126/scitranslmed.3009961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Janeway KA, Liegl B, Harlow A, et al. Pediatric KIT wild-type and platelet-derived growth factor receptor alpha-wild-type gastrointestinal stromal tumors share KIT activation but not mechanisms of genetic progression with adult gastrointestinal stromal tumors. Cancer Res. 2007;67:9084–9088. doi: 10.1158/0008-5472.CAN-07-1938. [DOI] [PubMed] [Google Scholar]
- 65.Mason EF, Hornick JL. Conventional Risk Stratification Fails to Predict Progression of Succinate Dehydrogenase-deficient Gastrointestinal Stromal Tumors: A Clinicopathologic Study of 76 Cases. Am J Surg Pathol. 2016;40:1616–1621. doi: 10.1097/PAS.0000000000000685. [DOI] [PubMed] [Google Scholar]