

Abstract
We develop and discuss a methodology for batch-level analysis of hyperspectral stimulated Raman scattering (hsSRS) data sets of human meibum in the CH-stretching vibrational range. The analysis consists of two steps. The first step uses a training set (n=19) to determine chemically meaningful reference spectra that jointly constitute a basis set for the sample. This procedure makes use of batch-level vertex component analysis (VCA), followed by unsupervised k-means clustering to express the data set in terms of spectra that represent lipid and protein mixtures in changing proportions. The second step uses a random forest classifier to rapidly classify hsSRS stacks in terms of the pre-determined basis set. The overall procedure allows a rapid quantitative analysis of large hsSRS data sets, enabling a direct comparison among samples using a single set of reference spectra. We apply this procedure to assess 50 specimens of expressed human meibum, rich in both protein and lipid, and show that the batch-level analysis reveals marked variation among samples that potentially correlate with meibum health quality.
Keywords: multi-image analysis, machine learning, hyperspectral stimulated Raman scattering microscopy, human meibum
1 Introduction
Raman spectroscopy probes molecular vibrational modes under benign conditions, allowing label-free identification of important molecular classes in biological samples of unknown composition. This capability qualifies Raman spectroscopy as a noninvasive technique for examining the molecular content of superficial tissues. For instance, dermatology studies have shown the potential of the technique to detect skin cancer1,2. Another example is the use of Raman spectroscopy as a probe for tissue health in accessible body cavities, such as the examination of colonic3, stomach4, and upper gastrointestinal tissues5 in vivo. Beyond superficial tissues, Raman spectroscopy has shown its diagnostic potential for excised tissues, enabling for instance the identification of lesions in human arteries6,7 and breast tissues8. These successes have solidified the potential of Raman spectroscopy as a biomedical tool and continue to drive the clinical translation of the technique2,9–11.
In Raman spectroscopy, the illumination spot on the sample is typically much larger than the dimensions of a cell, implying that individual cells cannot be resolved and the resulting spectra are spatial averages over several structures in the tissue. Although much can be learned from spatially averaged spectra, the ability to spatially resolve important tissue structures adds significant analytical value. Different structures in the tissue can display distinct spectra, and can thus be discriminated in a manner not easily achieved with spatially averaged spectra. Raman microscopy, on the other hand, generates images with Raman contrast at sub-micrometer resolution, thus enabling the identification of individual cells and even intra-cellular structures12–15. On a tissue level, the additional spatial information offered by microscopy introduces another axis along which the spectral analysis can be refined. With the aid of multivariate analysis methods16–18, the spatial separation of meaningful spectral features improves the analytical capabilities of the Raman technique, and enables detailed chemical mapping of healthy and diseased tissue5,19,20.
To speed up data acquisition, nonlinear Raman microscopy methods can be used. Both coherent anti-Stokes Raman scattering (CARS) and stimulated Raman scattering (SRS) microscopy offer a significant gain in image acquisition speed21,22, especially if the spectral region of interest is relatively narrow23. Coherent Raman methods are also much less sensitive to tissue fluorescence, which can plague spontaneous Raman microscopy experiments of tissues and cells24. SRS microscopy, in particular, is a direct analogue of spontaneous Raman microscopy, producing images with contrast based on the same spectral information as probed in linear Raman imaging. When the spectral dimension is limited to a single Raman line, SRS images can be acquired at video rate25. Adding more spectral information generally slows down the acquisition speed, but recent advances in hyperspectral SRS (hsSRS) imaging have pushed the pixel dwell time into the microsecond regime while still collecting a broad range of spectral data points26,27. The combination of spectral information and imaging speed makes it possible to map out larger sections of tissues or assess larger volumes of samples. The analysis of the larger data sets thus generated benefits enormously from advanced multivariate analysis tools. Several strategies have already been implemented to analyze hsSRS data sets, including multivariate curve resolution (MCR)28,29, independent component analysis (ICA)30, vertex component analysis (VCA)31, or k-means clustering analysis (KMCA)32. We have chosen to combine the latter two approaches. An analysis based on VCA allows segmentation with a large dynamic range, producing a reduced dataset that forms the starting point for a refined clustering using simple yet robust algorithms such as KMCA. VCA is a good choice for multivariate analysis of spectral data sets when pure spectra are present or can be easily identified, such as is the case in meibum samples which contain a high concentration of lipids and isolated protein clusters.
Most multivariate approaches used for hsSRS operate on the level of single image stacks, which are three dimensional data sets of position (typically x, y) and frequency (Raman shift). However, the hsSRS image acquisition speed allows for collecting multiple stacks from multiple samples, which emphasizes the need for global multivariate analysis methods. A global analysis of hsSRS data sets would allow a direct comparison between samples, using both space and frequency as discriminators. Because the information content of combined data sets is rich, machine learning tools become important for identifying meaningful features in both spatial and spectral frequency dimensions.
In this work, we discuss a global analysis approach for hsSRS data stacks collected from multiple samples. We implement spectral unmixing and machine learning strategies to extract meaningful spectral information across samples and enable a direct comparison between specimens. To illustrate the clinical utility of this approach, we apply the methodology to analyze lipid samples (meibum) expressed from the eyelids of 50 human subjects. Meibum secretions are lipid and protein amalgams deposited in the tear film lipid layer of the ocular surface. Meibum has been the subject of chromatographic and spectroscopic studies, which point out its different composition in healthy and dry eye patients33,34. Conventional Raman spectroscopy has been used to characterize the lipid content of human meibum, but clear Raman markers for discriminating between meibum from healthy and dry eye patients have remained elusive35. Recent hsSRS microscopy experiments pointed out the utility of adding the spatial dimension, enabling the observation that the lipid-to-protein ratio changes within the Meibomian glands from the acinus to the central duct, before the meibum is finally released (by a blink) into the tear film of the eye36. The proposed framework is capable of extracting detailed spectral information from a relatively narrow but rich portion of the spectra, such as the CH stretching spectral range. We show that the global analysis discussed in the current work provides additional insight in the relation between composition and spatial distribution that may prove useful in addressing meibum quality and its association with ocular surface diseases.
2 Materials and Methods
2.1 Sample collection and preparation
This study conformed to the tenets of the Declaration of Helsinki and was approved by the institutional review boards of both the University of California at Irvine and Marshall B. Ketchum University, Fullerton. All subjects provided written informed consent following explanation of study procedures prior to initiation of any study procedures.
Pooled meibum samples (obtained from 2 or more orifices) were obtained from the central meibomian glands of the patients lower eyelids, following gentle cleaning with a dry sterile cotton tipped applicator to remove debris, eye makeup, etc. At 10X magnification at the biomicroscope, the Meibomian Gland Evaluator was used to exert the constant force of a hard blink (1.25 g/mm2) for 10–15 seconds37. Meibum was collected from the orifices using a sterile foreign body spud (Miltex Ellis 18–380, Steele Supply Co., Michigan, USA) and smeared on a clean microscope slide labeled with the subject number, eye, and clinical secretion grade. Meibum samples were covered with a thin cover glass, and stored at room temperature until further analysis. Meibum samples were collected from both eyes of 50 human subjects in a primarily clinic-based sample. Included subjects were over the age of 18, but with no upper age limit, and without restriction as to sex or race. Healthy (non-dry eye) subjects were recruited as well as those with aqueous tear deficiency and evaporative (e.g., meibomian gland dysfunction) types of dry eye38.
2.2 Hyperspectral stimulated Raman scattering
Stimulated Raman scattering signals were obtained as described previously31. Briefly, a 76MHz mode-locked Nd:Vanadate laser (PicoTrain, High-Q, Hohenems, Austria) delivered a fundamental beam at 1064nm (Stokes beam) of 7ps pulses, and a frequency-doubled beam at 532nm that pumped an optical parametric oscillator (OPO; Levante, Emerald OPO, Berlin, Germany). The OPO beam was spectrally tuned by adjusting its crystal temperature, Lyot filter, and cavity length with the aid of automated computer code. A portion of the 1064nm beam was used as the Stokes beam in the SRS process, whereas the signal output of the OPO was used as the pump beam. The two beams were overlapped both temporally and spatially, and passed through a laser scanner (Fluoview 300, Olympus, Center Valley, PA), interfaced with an inverted microscope (IX71, Olympus). The combined beams were then focused with a 20x, 0.75 NA objective lens (UplanS Apo, Olympus) onto the sample.
SRS images were obtained by detecting the stimulated Raman loss of the pump beam. For this purpose, the Stokes beam was modulated at 10MHz with an acousto-optic modulator (AOM; Crystal Technology, Palo Alto, CA). The modulation of the pump intensity was detected by a photodiode (FDS1010; Thorlabs, Newton, NJ), and the signal was demodulated with a custom lock-in amplifier. The average combined power of Stokes and pump beams at the specimen was kept under 30mW throughout this study to minimize sample photodamage.
Hyperspectral SRS image stacks were acquired with custom software. The spectral range in the experiments reported in this work spanned the CH-stretching region of the Raman spectrum from ∼ 2800 to 3050 cm−1, with a ∼ 7 cm−1 resolution. The hsSRS stacks consisted of 37 images of 512 by 512 pixels, with a pixel size of 0.46μm.
2.3 Methodology
For clinical purposes, it is critical that the acquisition and analysis method allows comparison of sample composition across specimens and time points. For samples of unknown composition, unsupervised algorithms can help retrieve their main descriptors. Unsupervised multivariate analysis is data-driven and thus dependent on the individual hsSRS image to which it is applied; the extracted basis spectra may vary from image to image, complicating the comparison among specimens. Instead, batch-level analysis, where the basis spectra are obtained considering the data of all the images in the data set, enables a much more meaningful comparison. However, batch-level analysis of complete data sets is inefficient when the number of hsSRS image stacks is high, and impractical when the data set is incomplete (i.e. if more samples want to be added and analyzed in the future for clinical purposes). To systematically analyze and quantify a large number of images and compare samples from different patients, we have developed a two-step, batch-level image analysis framework based on machine learning strategies. First, we characterize the chemical classes that best describe the specimens with unsupervised multivariate analysis on a subset of images that we call training set. Second, we classify the rest of the images into the previously defined chemical classes by means of a supervised classifier. The overall approach is summarized in the diagram of Fig. 1 and explained below.
Figure 1.
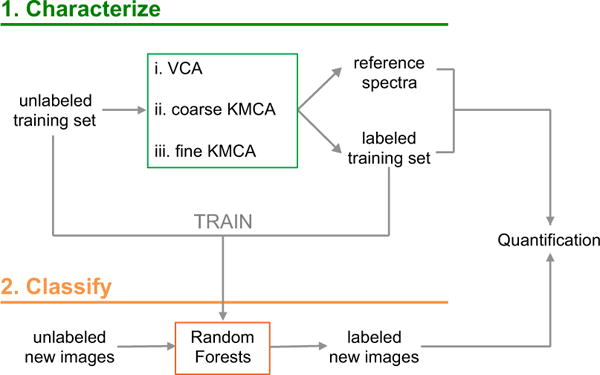
Methodology implemented to quantify hyperspectral SRS images. In a first step, the data are characterized by unsupervised multivariate analysis based on vertex component analysis (VCA) k-means clustering analysis (KMCA). The outcome of the first step is used to train a random forests classifier, which is later used to describe the rest of the data set. The final labeled images are further used for quantification purposes.
2.3.1 Step 1: Generating reference spectra from a training set
Given a set of hyperspectral images of unknown composition, unsupervised multivariate analysis provides a rapid characterization. To describe the samples in terms of their spectral (chemical) content, vertex component analysis (VCA) is used to unmix the spectral components providing an initial, intuitive segmentation. The result can be ultimately mapped into color-coded images with chemical meaning. VCA projects unlabeled data into a simplex geometry by means of singular value decomposition, assuming the different components of the sample are linearly mixed together. The vertices of the defined simplex are occupied by the extremes of the projection, the so-called end members. VCA assumes there is at least one pixel in the data set for each pure component of the mixture (for each end member). A detailed derivation of VCA was given by Nascimento and Dias39.
The present model (see Eq.1e) defines each spectrum of the data set (S; size p × q) as a linear combination of the pure end member spectra (M; size l × q), weighted by an abundance matrix (a; size p × l). The model also assumes there is a noise level (n; size p × q) associated with each measurement. For the study of the meibum samples the total number of pixels, and thus of spectra, in the training set is p = x × y × n, where x, y are the number of spatial pixels and n is the number of samples in the training set. Using x = y = 512 and n = 19, the total number of data points is 4,980,736. Each point in this data set has a spectral dimension, which is spanned by q = 37 spectral points. The chosen number of end members is l = 3, based on prior knowledge of the samples, which contain three main spectral classes: lipids, proteins, and the background introduced by the microscope slide glass.
The initial spectral data set for a given pixel p is written as:
| (1a) |
| (1b) (1c) |
For the vertex component analysis, we define the j component of the spectrum of pixel p as follows:
| (1d) |
In its generalized form, this can be written as:
| (1e) |
| (1f) |
| (1g) |
| (1h) |
VCA reduces the dimension of the spectral space from q to l features, with q ≫ l. In our case, from 37 spectral data points to 3 basis spectra. Each spectrum of the data set is now defined by three components that represent the weight of each end member spectrum. The end member spectra correspond to the vertices of a triangle, as schematically depicted in Fig. 2a. Each end member spectrum is assigned to a color, which permits the reconstruction of color-coded images with chemical meaning. For convenience, we have chosen the RGB color space, with red attributed to spectra rich in lipids, green for spectra rich in proteins, and blue for background, as depicted in Fig. 2d. All other spectra in the training set are defined as linear combinations of the three end members, with [αr, αg, αb] the color coefficients of each hyperpixel as determined by the abundance matrix. These hyperpixels reconstruct the spectra of each pixel in the image as:
| (2) |
Figure 2.
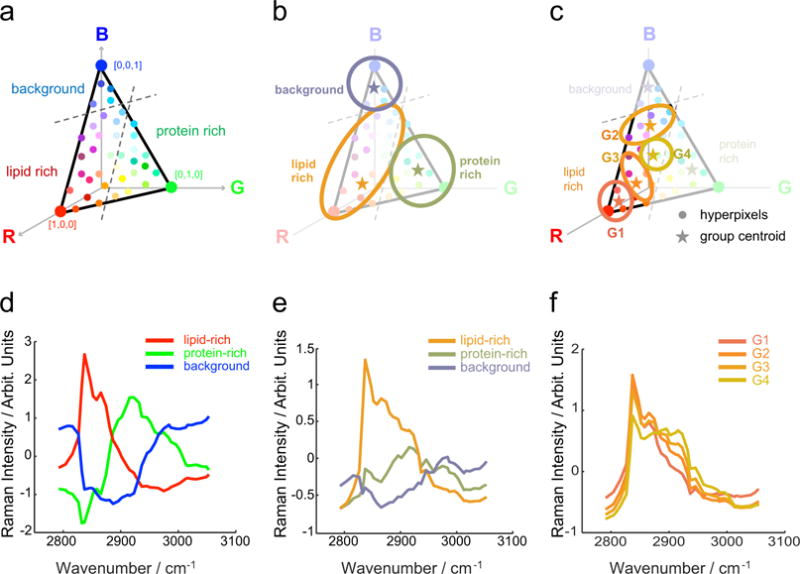
Multivariate analysis characterized the chemical composition of meibum samples. a,d) Vertex component analysis (VCA) is used to describe the spectra in terms of three end members, each one assigned to an RGB base color. b,e) K-means clustering analysis (KMCA) groups the VCA output by color similarities and provides biochemical meaningful reference spectra. c,f) A nested KMCA within the lipid-rich group of the previous KMCA unveils extra grouping relevant to the meibum samples.
It is important to note that the data applied to this first step is pretreated and z-scored (to have zero mean and a standard deviation of one), in order to avoid intensity variation effects. We note that additional data preprocessing steps, such as signal-to-noise ratio enhancement strategies, can be implemented prior to VCA, which may lead to improved results.
Color-coded VCA generates images that display a rich variation of spectral and morphological features (see Fig. 3a). Despite being informative and detailed, these images are difficult to quantify. In order to select statistically meaningful thresholds for grouping the spectra in representative classes, we implement a k-means clustering analysis (KMCA) within the 3D color space spanned by the VCA end members. KMCA is another unsupervised algorithm that groups data into a predefined number of clusters k. KMCA assigns each spectrum within the data set, now defined by 3 color coefficients instead of 37 frequency features, to a group by minimizing the sum of distances between original spectra to the mean spectrum of the assigned cluster (ck), the so-called centroids40:
| (3a) |
| (3b) |
Figure 3.
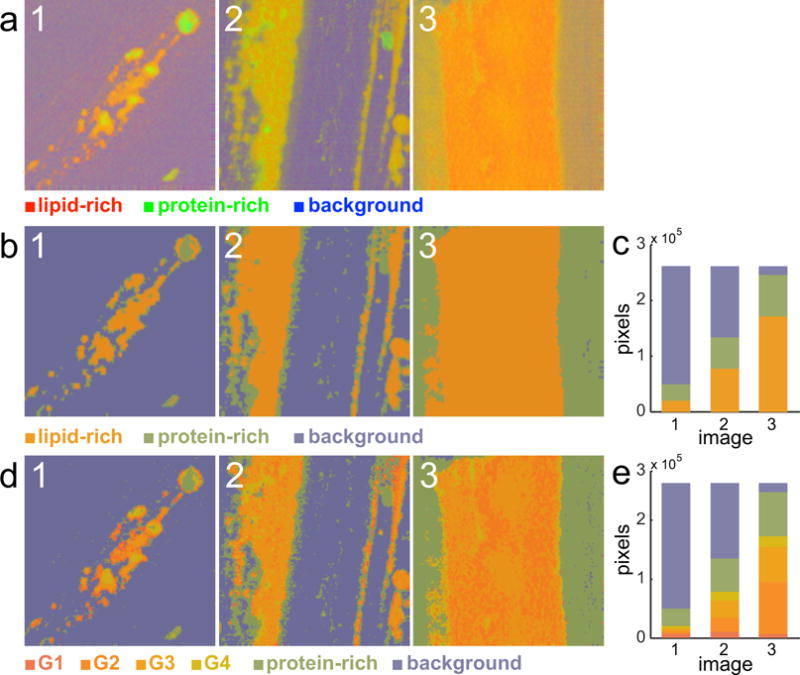
Characterization of the training set. a) Result of applying vertex component analysis (VCA) on three representative images in the training set. b) Result of applying the coarse K-means clustering analysis (KMCA) on the previous images and c) corresponding histogram quantification. d) Final result after applying the fine KMCA on the images and e) corresponding histogram quantification. Image size is 235.5 × 235.5 μm.
First, J spectra are randomly assigned to each cluster k, and the mean of every cluster (ck) is computed. The k-means algorithm examines the distance between each spectrum and the computed cluster centroids. If the spectrum of interest is not assigned to the group with the closest mean, it is re-assigned to the one that minimizes such distance. As an iterative process, this step is repeated until all spectra are located in the cluster with the nearest centroid, or until the overall sum is minimized18.
As depicted in Fig. 2b, KMCA groups the spectra out of the VCA analysis by color similarity, providing as a result three spectra that better describe the sample’s content (Fig. 2e). This step facilitates further quantification of the samples, as now each group is composed of a finite number of pixels. For our particular data set, a detailed analysis of the spectral content in each group unveiled a wide variation in the spectra in the lipid-rich group. Therefore, we applied a nested KMCA only to those pixels that constitute the lipid-rich group. This latter step allowed us to obtain a subset of groups hidden in the first KMCA, which are relevant to the case of study (Fig. 2c and Fig. 2f).
Overall, the nested clustering approach defines a set of reference spectra that simplify the description of the samples: in the first (coarse) KMCA the background and the protein spectra are determined (Fig. 2b and 2e), and in the second (fine) KMCA various levels of lipid-protein mixtures are discerned (Fig. 2c and 2f).
2.3.2 Step 2: Classifying images according to the reference spectra
The characterization step is slow and computationally intense, impractical to apply to hundreds (or even dozens) of hsSRS image stacks. Therefore, only a reduced number of hsSRS images (training set) are used to characterize the set of samples, while the rest of the image stacks are directly classified into predefined reference spectra, speeding up the process significantly. This approach also allows for an increase in the number of images that can be added to the analysis a posteriori.
A random forests classifier was implemented to label hsSRS images with the 6 predefined groups established with the training set. Random forests classifiers are supervised algorithms that generate multiple classification outputs for a given entry and the most popular one is chosen as the final classification decision41. Once the algorithm is trained against a specific classification, it assigns spectra from new data sets in the context of the predefined groups. To setup the classifier we implemented a 5-fold cross-validation, with 20 trees and a minimum leaf size of 5, and obtained an accuracy of 87%. Training the classifier required 4 hours (for the 19 images of the training set), but once set the posterior classification of new images was very fast, 50 images were classified in less than 10 minutes. For the entire analysis, customized code was written in Matlab on an Intel(R) Xeon(R) PC running Windows 7 with 16.0 GB of RAM.
3 Results
We have chosen to limit the Raman spectral range to 2800 – 3050 cm−1, which captures the CH-stretching vibrational modes. Although additional information can be obtained in the finger print region, the CH-stretching has previously been shown to provide useful spectral features that can be used for spectral unmixing42,43. In addition, this range provides the strongest SRS signals, which facilitates signal acquisition. In the following, we describe the implementation of the batch-level analysis to extract meaningful information from hsSRS images of meibum samples obtained from 50 patients. First, we will describe the success of the procedure to identify spectra in the training set that are representative of lipid and protein components in the meibum sample. Second, we will use the obtained spectral groups to rapidly classify meaningful features in the complete data set.
3.1 Step 1: Characterization of meibum spectral content
The first step consists of characterizing the meibum content in terms of its biochemical composition, provided by the Raman spectra reconstructed from the hsSRS images. A random set of 19 hsSRS images was selected to act as training set. The main spectra that define the training set are extracted by spectral unmixing and nesting 2 clustering methods: VCA, coarse KMCA, and fine KMCA, as described in Section 2.3. The results are summarized in Fig. 3. The first row (Fig. 3a) contains the VCA reconstructed images of three meibum samples. The color scheme is defined by the basis spectra in Fig. 2d. Red denotes lipid-rich areas, green is assigned to protein-rich parts, and blue is left for background. The second row shown in Fig. 3b depicts the same images after the implementation of the coarse KMCA on the VCA output. Notice how only three colors compose the images, which match the average group spectra depicted in Fig. 2e. The lipid-rich pixels are orange, the protein pixels green, and the background indigo. Importantly, the KMCA facilitates the quantification of the image in terms of chemically meaningful reference spectra, as demonstrated by the histogram in Fig. 3c. Application of the fine KMCA enables a partitioning of the lipid fraction into four groups (Fig. 3c). Although the result qualitatively resembles the images of the VCA result shown in Fig. 3a, the fine KMCA produces reference spectra that make it easier to quantitatively analyze the images in terms of biochemical content. The histogram shown in Fig. 3e identifies groups that relate directly to the images in Fig. 3d. The color coding of the lipid-rich spectra is given in Fig. 2f.
The result of the second KMCA provides a graded variation of the contribution of the ∼ 2940 cm−1 spectral component in the samples. This contribution coincides with the methyl stretch vibrational mode, which is indicative of protein. Spatially, this spectral contribution shows up most prominently in areas that are devoid of lipids, further suggesting that this spectral component can be ascribed to protein. Together with the protein-rich spectra from the coarse KMCA, the 5 reference spectra define a scale of relative protein contribution in each area of the sample, from lipid-rich (dark orange, G1) to protein-rich (green). These reference spectra are normalized and depicted in Fig. 4a, and, in combination with the background spectrum (for total of 6 reference spectra), will be used for classification of the spectral features in the remainder of the data set.
Figure 4.
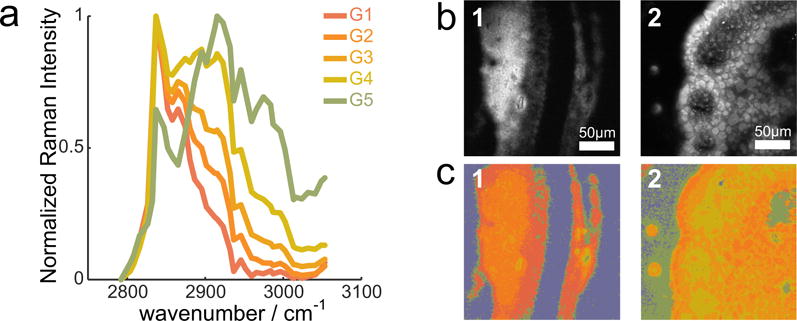
Classification results. a) Reference spectra used to train the random forests classifier. b) Maximum intensity projection hsSRS images of expressed meibum and c) corresponding classification. Image size is 235.5 × 235.5 μm.
3.2 Step 2: Classification of meibum spectral content
Once the main spectra that characterize the biochemical composition of the meibum secretions are established, we apply a random forests classifier that sorts the spectra in the remaining hsSRS images into the six reference spectral classes (Fig. 4a). Once trained, the classifier allows for a rapid analysis of each hsSRS image stack. The only pre-processing performed on the raw data is the z-score normalization.
50 images are classified according to this reference in less than 10 minutes, and room is still left for optimizing the classification process. Fig. 4 shows two examples of hsSRS images of meibum samples, in the form of a maximum intensity projection of the spectral stack (Fig. 4b) and its corresponding classified image (Fig. 4c). The classified images show that the extracted meibum consists of mixtures of protein and lipid with various degrees of protein to lipid ratios. The classification procedure makes it possible to directly compare the images obtained from sample 1 and 2. In sample 1, the streak contains a high proportion of the lipid class G1, a spectrum that is indicative of wax esters. In sample 2, the overall protein content is relatively higher. In addition, signatures of phase separation are observed, with patches of higher protein content (greener patches) surrounded by lipid-rich pools, which are not evident in sample 1. This example illustrates that the combined spectral and spatial features provide useful metrics that enable categorization of the samples.
3.3 Global analysis and findings
The importance of batch-level analysis of SRS data is shown in Fig. 5, which displays the difference between VCA implemented on individual images and the corresponding batch-level analysis. VCA yields different end member spectra for each image when these are analyzed separately, precluding direct comparison among them. In this case, the generated colors have different meaning in each image on Fig. 5a, as illustrated by the different spectra in Fig. 5b. The differences are small, but relevant, as red pixels in image 1 have a different biochemical composition than red pixels in image 2. For example, end member one, depicted in red, has nonexistent contributions of the 2940cm−1 band for images 1 and 3, whereas for image 2 these methyl stretches are contributing to the spectra of end member one. Further quantification of these samples is complicated due to the lack of reference. For images 1 and 3, red areas evoke a purer wax ester contribution, whereas for image 2, red areas have a significant CH3 stretches contribution, a difference that is relevant to the meibum quality. On the other hand, the batch-level analysis yields a set of end members common to all the images; the colors across samples refer to the same basis spectra allowing their direct comparison, and facilitating further quantification. In this case, red pixels translate to pure wax esters, and mixtures with CH3 stretches are depicted in oranges shifting to green as these become dominant. Additionally, batch-level analysis also reduces spectral noise on the resulting end members and subsequent group spectra, and yields a more accurate representation of the most prominent spectral features in the samples.
Figure 5.
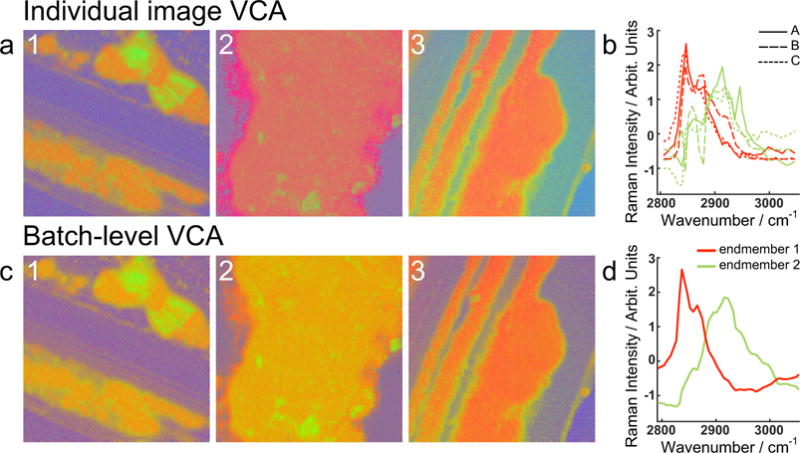
Vertex component analysis (VCA) implemented a) on single images, with b) the corresponding end member spectra 1 (red) and 2 (green), and c) at the batch-level, with d) the corresponding end member spectra 1 (red) and 2 (green). Image size is 235.5 × 235.5 μm.
The chemical maps thus obtained unveil complex meibum composition only made visible by the global analysis of spatial and spectral distributions. Inspection of the classified hyperspectral images reveals that different structures in the tissue display distinct spectra. Some patterns can be recognized upon examining these spatio-spectral correlations. Common structures found within the data set are displayed in Fig. 6. The majority of the meibum secretions are composed of lipid-rich areas that are mixtures of different lipid types, and appear as homogeneous lipid-protein mixtures on the micrometer scale (sample 1). A subset of samples displays a structure of lipid blobs that are characterized by a wax ester Raman signature (the darker red hyper-pixels), embedded in a surrounding protein-rich matrix (sample 2). In some occasions, protein aggregates appear as crystalline structures of variable size, enveloped by lipid-rich areas or isolated (sample 3). Fig. 6a shows the maximum intensity projection of the hsSRS, rich in spatial information but poor in spectral content. Fig. 6b displays the corresponding VCA images, which keep the spatial distribution information complemented with detailed composition evaluation. Thresholds that delineate the extent of a bio-molecular group facilitate the quantification of these VCA images in terms of reference spectra, which is accomplished in the corresponding KMCA images shown in Fig. 6c.
Figure 6.

Characteristic features of expressed maibum. a) Maximum intensity projection hsSRS. b) VCA reconstructed images. c) KMCA reconstructed images. Image size is 235.5 × 235.5 μm.
The usefulness of reducing the classification to 5 interpretable reference spectra is evident when analyzing larger batches. In Fig. 7, we show the classification of the data set comprising of 50 human subjects, of varying dry eye diagnosis. The quantification is normalized to only account for the pixels that contain meibum (excluding the background - in indigo on the maps). The KMCA on the VCA output permits a data-driven quantification to generate the presented histograms, where every color represents a biochemical class. The procedure followed here avoids setting arbitrary thresholds assigned by human bias. Importantly, all images within the set can be quantitatively compared, as all the classified images are expressed within the same basis set of reference spectra. Comparison of the histograms allows a rapid inspection of pure lipid (red), mixtures of protein and lipid (orange shades) to pure protein (green). We see that the biochemical composition among the samples shown here varies markedly. In some specimens, such as sample #48, the protein-rich material dominates. In other samples, such as sample #29, the protein-to-lipid ratio is low with protein only present in microscopically homogeneous mixtures. Sample #26 exemplifies a segregated area, with protein-rich material on the top pf the image, and lower lipid-to-protein ratio small areas surrounding it, and larger ones below it. The global analysis and representation in terms of SRS reference spectra facilitates comparison among specimens, at sample quantities that are commensurate with clinical studies.
Figure 7.
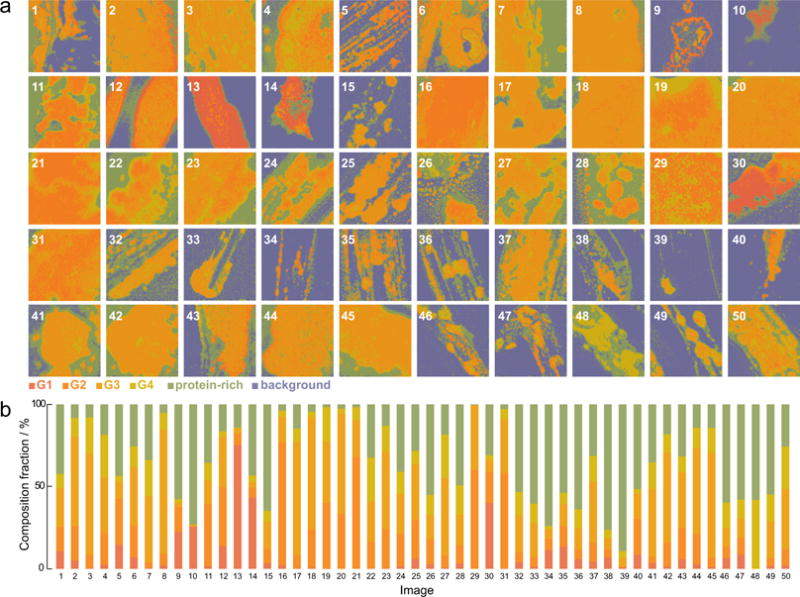
High throughput analysis and quantification of human expressed meibum. a) Biochemical maps, and b) corresponding composition fraction histograms. Image size is 235.5 × 235.5 μm.
Further examination of these biochemical metrics and correlation with clinical data may spur insights into the biochemical underpinnings of dry eye disease, its diagnostic observables, and its response to treatment.
4 Discussion and Conclusion
In Raman spectroscopy, the use of batch-level multivariate analysis is a common approach to compare measured spectra from different samples in terms of a joint basis set. In Raman microscopy, which enjoys both spatial and spectral information, multivariate approaches are growing more and more important for the analysis of spectroscopic images. Most of these approaches have been carried out on the single cell level, where multivariate analysis has proven indispensable for the identification of intracellular structures and organelles44. Similar strategies have also found their way in the analysis of coherent Raman hyperspectral images of cells and tissues23,45,46. There are a few examples of studies that have implemented multivariate analysis and machine learning approaches in Raman microscopy on a batch level47,48. In these studies, multiple Raman images of single cells were jointly analyzed with the aid of a single set of reference spectra. The work presented here employs a similar strategy for the batch-analysis of hsSRS images. Since hsSRS images can be acquired at faster rates compared to spontaneous Raman images, the optimization of batch-level analysis for SRS is an important step towards making vibrational microscopic methods suitable for clinical purposes.
In this study, we combined hsSRS microscopy with machine learning analysis tools to identify the biochemcial content of human expressed meibum and map out their spatial distribution. The methodology consists of a two step process. First, a subset of samples are examined with unsupervised spectral unmixing via vertex component analysis, and clustered with k-means cluster analysis. This step provides insight in the nature of the samples, it identifies their spectral content and groups those spectra to facilitate quantification. Second, a classifier is trained to group all other samples into the identified spectral clusters. A Random Forests classifier was selected for the task as it provided accurate and fast classifications. Further fine-tuning of the classifier, or even other classifiers, can further improve the degree of accuracy. The relevance of the two-step process is also two-fold. First, it permits a direct quantitative comparison across samples, as they are all analyzed in terms of the same reference spectra. Second, it allows to add new images/samples to the analysis in an efficient and rapid manner.
We have implemented the batch-level analysis by using a limited range of the Raman spectrum, namely the CH stretching spectral band. Even though the spectral features are broad, this range is rich in spectral information. In particular, spectral unmixing of the lipid and protein molecular classes can be achieved in this spectral range, which is very relevant for the application chosen here. We have applied the batch-level multivariate analysis and classification to assess a data set comprised of meibum preparations from 50 patients. All samples were analyzed in terms of 5 reference spectra, which represent mixtures of proteins and lipids in various ratios. Even though all samples are composed of lipids and proteins, the batch-level analysis shows that the variability among samples is high. The method identified samples with regions where proteins and lipids are almost completely segregated, whereas other samples display regions where the protein and lipid components are homogeneously mixed on the micro-scale. Chemical separation and phase segregation of mixtures are potentially important observables that can be used to assess meibum health quality. Such information cannot be obtained by spatially averaging the spectra over the sample, underlining the usefulness of the spatial dimensions to extract meaningful demarcation criteria from the hyperspectral data set.
The composition of meibum is known, and contains a mixture of wax esters, sterol esters, and phospholipids49. The spatially averaged Raman spectrum of purified meibum reflects the presence of these ingredients35, but does not necessarily reveal the spatial distribution of its constituents. Small changes in chemical composition can produce large changes in the degree of mixing and segregation of the lipid components, sample characteristics that can only be identified when assessing the spatial distribution of the compounds. In addition, the presence of protein may constitute an important factor that affects meibum fluidity and cloudiness36,45. Information about the distribution of both lipids and protein in meibum expressed from the gland can thus provide important clues toward the link between meibum quality, its biochemical composition and its micro-rheology. Although a full clinical analysis of the meibum specimens is beyond the scope of the present work, the batch-level multivariate hsSRS study presented in here is ideally suited for establishing such links from clinical data sets.
Further improvements of the methodology discussed here include expansion of the training set, thereby improving the quality of the reference spectra. In addition, it would be useful to cross-validate the reference spectra with other analytical tools such as mass (micro-)spectroscopy34. The latter may provide a deeper assessment of the chemical components that underlie the measured (stimulated) Raman spectra.
As the imaging speed and capabilities of hsSRS continue to grow, we expect that batch-level analysis and machine learning will gain in prominence. The method presented here is an important first step that emphasizes the power of analyzing multiple samples in terms of a single set of reference spectra. With further improvements in speed and efficiency, the batch-level analysis will be an important contribution to the translation of coherent Raman scattering techniques for clinical studies.
Acknowledgments
The project was supported in part by NEI EY021510, an unrestricted grant from Research to Prevent Blindness, Inc., and NIH grant P41-RR01192 (Laser Microbeam and Medical Program, LAMMP). Additional support was provided by Alcon Research Ltd.
References
- 1.Gniadecka M, Philipsen PA, Wessel S, Gniadecki R, Wulf HC, Sigurdsson S, Nielsen OF, Christensen DH, Hercogova J, Rossen K, Thomsen HK, Hansen LK. Journal of Investigative Dermatology. 2004;122:443–449. doi: 10.1046/j.0022-202X.2004.22208.x. [DOI] [PubMed] [Google Scholar]
- 2.Lui H, Zhao J, McLean D, Zeng H. Cancer Research. 2012;72:2491–2500. doi: 10.1158/0008-5472.CAN-11-4061. [DOI] [PubMed] [Google Scholar]
- 3.Molckovsky A, Song L-MWK, Shim MG, Marcon NE, Wilson BC. Gastrointestinal Endoscopy. 2003;57:396–402. doi: 10.1067/mge.2003.105. [DOI] [PubMed] [Google Scholar]
- 4.Teh SK, Zheng W, Ho KY, Teh M, Yeoh KG, Huang Z. British Journal of Surgery. 2010;97:550–557. doi: 10.1002/bjs.6913. [DOI] [PubMed] [Google Scholar]
- 5.Duraipandian S. Journal of Biomedical Optics. 2012;17:081418. doi: 10.1117/1.JBO.17.8.081418. [DOI] [PubMed] [Google Scholar]
- 6.Marzec KM, Wrobel TP, Rygula A, Maslak E, Jasztal A, Fedorowicz A, Chlopicki S, Baranska M. Journal of Biophotonics. 2014;7:744–756. doi: 10.1002/jbio.201400014. [DOI] [PubMed] [Google Scholar]
- 7.Dochow S, Fatakdawala H, Phipps JE, Ma D, Bocklitz T, Schmitt M, Bishop JW, Margulies KB, Marcu L, Popp J. Journal of Biophotonics. 2016:1–9. doi: 10.1002/jbio.201500341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haka AS, Shafer-Peltier KE, Fitzmaurice M, Crowe J, Dasari RR, Feld MS. Proceedings of the National Academy of Sciences. 2005;102:12371–12376. doi: 10.1073/pnas.0501390102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choo-Smith LP, Edwards HGM, Endtz HP, Kros JM, Heule F, Barr H, Robinson JS, Bruining HA, Puppels GJ. Biopolymers. 2002;67:1–9. doi: 10.1002/bip.10064. [DOI] [PubMed] [Google Scholar]
- 10.Kallaway C, Almond LM, Barr H, Wood J, Hutchings J, Kendall C, Stone N. Photodiagnosis and Photodynamic Therapy. 2013;10:207–219. doi: 10.1016/j.pdpdt.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 11.Diem M. Modern Vibrational Spectroscopy and Micro-Spectroscopy. John Wiley & Sons, Ltd; 2015. pp. 103–122. [Google Scholar]
- 12.Turrell G, Corset J, editors. Raman Microscopy. Elsevier Academic Press; San Diego: 1996. [Google Scholar]
- 13.Uzunbajakava N, Lenferink A, Kraan Y, Willekens B, Vrensen G, Greve J, Otto C. Biopolymers. 2003;72:1–9. doi: 10.1002/bip.10246. [DOI] [PubMed] [Google Scholar]
- 14.Matthäus C, Bird B, Miljković M, Chernenko T, Romeo M, Diem M. Biophysical Tools for Biologists, Volume Two: In Vivo Techniques. In: Wilson L, Matsudaira P, editors. Methods in Cell Biology. Vol. 89. Academic Press; 2008. pp. 275–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klein K, Gigler AM, Aschenbrenner T, Monetti R, Bunk W, Jamitzky F, Morfill G, Stark RW, Schlegel J. Biophysical Journal. 2012;102:360–368. doi: 10.1016/j.bpj.2011.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adams MJ. Chemometrics in Analytical Spectroscopy. The Royal Society of Chemistry; Cambridge: 2004. [Google Scholar]
- 17.Diem M, Matthäus C, Chernenko T, Romeo MJ, Miljković M, Bird B, Schubert J, Papamarkakis K, Laver N. Infrared and Raman Spectroscopic Imaging. Wiley-VCH Verlag GmbH & Co. KGaA; 2009. pp. 173–201. [Google Scholar]
- 18.Miljković M, Chernenko T, Romeo MJ, Bird B, Matthäus C, Diem M. The Analyst. 2010;135:2002–2013. doi: 10.1039/c0an00042f. [DOI] [PubMed] [Google Scholar]
- 19.Diem M, Mazur A, Lenau K, Schubert J, Bird B, Miljković M, Krafft C, Popp J. Journal of Biophotonics. 2013;6:855–886. doi: 10.1002/jbio.201300131. [DOI] [PubMed] [Google Scholar]
- 20.Majzner K, Kochan K, Kachamakova-Trojanowska N, Maslak E, Chlopicki S, Baranska M. Analytical Chemistry. 2014;86:6666–6674. doi: 10.1021/ac501395g. [DOI] [PubMed] [Google Scholar]
- 21.Cheng J, Xie XS. Coherent Raman Scattering Microscopy. CRC Press; 2013. (Series in Cellular and Clinical Imaging). [Google Scholar]
- 22.Alfonso Garcia A, Mittal R, Lee ES, Potma EO. Journal of Biomedical Optics. 2014;19:071407. doi: 10.1117/1.JBO.19.7.071407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suhalim JL, Boik JC, Tromberg BJ, Potma EO. Journal of Biophotonics. 2012;5:387–395. doi: 10.1002/jbio.201200002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krafft C. Journal of Biomedical Optics. 2012;17:040801. doi: 10.1117/1.JBO.17.4.040801. [DOI] [PubMed] [Google Scholar]
- 25.Saar BG, Freudiger CW, Reichman J, Stanley CM, Holtom GR, Xie XS. Science. 2010;330:1368–1370. doi: 10.1126/science.1197236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liao CS, Slipchenko MN, Wang P, Li J, Lee SY, Oglesbee RA, Cheng JX. Light: Science & Applications. 2015;4:e265. doi: 10.1038/lsa.2015.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liao CS, Wang P, Wang P, Li J, Lee HJ, Eakins G, Cheng JX. Science Advances. 2015;1 doi: 10.1126/sciadv.1500738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang D, Wang P, Slipchenko MN, Ben-Amotz D, Weiner AM, Cheng J-X. Analytical chemistry. 2013;85:98–106. doi: 10.1021/ac3019119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang X, de Juan A, Tauler R. Applied spectroscopy. 2015;69:993–1003. doi: 10.1366/14-07819. [DOI] [PubMed] [Google Scholar]
- 30.Ozeki Y, Umemura W, Otsuka Y, Satoh S, Hashimoto H, Sumimura K, Nishizawa N, Fukui K, Itoh K. Nature photonics. 2012;6:845–851. [Google Scholar]
- 31.Alfonso Garcia A, Pfisterer SG, Riezman H, Ikonen E, Potma EO. Journal of Biomedical Optics. 2016;21:061003. doi: 10.1117/1.JBO.21.6.061003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fu D, Xie XS. Analytical chemistry. 2014;86:4115–4119. doi: 10.1021/ac500014b. [DOI] [PubMed] [Google Scholar]
- 33.Foulks GN, Borchman D, Yappert M, Kim S-H, McKay JW. Cornea. 2010;29:1. doi: 10.1097/ICO.0b013e3181cda38f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen J, Green KB, Nichols KK. Investigative Opthalmology & Visual Science. 2013;54:5730. doi: 10.1167/iovs.12-10317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oshima Y, Sato H, Zaghloul A, Foulks GN, Yappert MC, Borchman D. Current Eye Research. 2009;34:824–835. doi: 10.3109/02713680903122029. [DOI] [PubMed] [Google Scholar]
- 36.Suhalim JL, Parfitt GJ, Xie Y, De Pavia CS, Pflugfelder SC, Shah TN, Potma EO, Brown DJ, Jester JV. The Ocular Surface. 2014;12:59–68. doi: 10.1016/j.jtos.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Korb DR, Blackie CA. Cornea. 2008;27:1142–1147. doi: 10.1097/ICO.0b013e3181814cff. [DOI] [PubMed] [Google Scholar]
- 38.Lemp MA, Foulks GN. The Ocular Surface. 2007 [Google Scholar]
- 39.Nascimento JMP, Dias JMB. IEEE Transactions on Geoscience and Remote Sensing. 43:898–910. [Google Scholar]
- 40.Jain Anil K. Pattern Recognition Letters. 2010;31:651–666. [Google Scholar]
- 41.Breiman L. Machine learning. 2001:5–32. [Google Scholar]
- 42.Fu D, Lu F-K, Zhang X, Freudiger C, Pernik DR, Holtom G, Xie XS. Journal of the American Chemical Society. 2012;134:3623–3626. doi: 10.1021/ja210081h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu F-K, Basu S, Igras V, Hoang MP, Ji M, Fu D, Holtom GR, Neel VA, Freudiger CW, Fisher DE, Xie XS. Proceedings of the National Academy of Sciences. 2015;112:11624–11629. doi: 10.1073/pnas.1515121112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hedegaard M, Matthäus C, Hassing S, Krafft C, Diem M, Popp J. Theoretical Chemistry Accounts. 2011;130:1249–1260. [Google Scholar]
- 45.Lin C-Y, Suhalim JL, Nien CL, Miljković MD, Diem M, Jester JV, Potma EO. Journal of Biomedical Optics. 2011;16:021104. doi: 10.1117/1.3533716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.El-Mashtoly SF, Niedieker D, Petersen D, Krauss SD, Freier E, Maghnouj A, Mosig A, Hahn S, Kötting C, Gerwert K. Biophysical Journal. 2014;106:1910–1920. doi: 10.1016/j.bpj.2014.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matthäus C, Krafft C, Dietzek B, Brehm BR, Lorkowski S, Popp J. Analytical Chemistry. 2012;84:8549–8556. doi: 10.1021/ac3012347. [DOI] [PubMed] [Google Scholar]
- 48.Hedegaard MA, Bergholt MS, Stevens MM. Journal of biophotonics. 2016 doi: 10.1002/jbio.201500238. [DOI] [PubMed] [Google Scholar]
- 49.McCulley JP, Shine WE. The Ocular Surface. 2003;1:97–106. doi: 10.1016/s1542-0124(12)70138-6. [DOI] [PubMed] [Google Scholar]