

Abstract
Protein kinases are able to govern large-scale cellular changes in response to complex arrays of stimuli, and much effort has been directed at uncovering allosteric details of their regulation. Kinases comprise signaling networks whose defects are often hallmarks of multiple forms of cancer and related diseases, making an assay platform amenable to manipulation of upstream regulatory factors and validation of reaction requirements critical in the search for improved therapeutics. Here, we describe a basic kinase assay that can be easily adapted to suit specific experimental questions including but not limited to testing the effects of biochemical and pharmacological agents, genetic manipulations such as mutation and deletion, as well as cell culture conditions and treatments to probe cell signaling mechanisms. This assay utilizes radiolabeled [γ-32P] ATP, which allows for quantitative comparisons and clear visualization of results, and can be modified for use with immunoprecipitated or recombinant kinase, specific or typified substrates, all over a wide range of reaction conditions.
Keywords: Biochemistry, Issue 123, Protein kinase, signal transduction, biochemical assay, radiolabeled assay
Introduction
Protein kinases are sophisticated enzymes critical for the transmission of cellular signals into appropriate responses1. Given their roles in maintaining homeostasis and the prevention or promotion of disease states2, biochemical methods for assessing kinase activity continue to be powerful tools for delineating the particulars of eukaryotic signaling3. Although strategies utilizing phospho-specific antibodies have been highly informative in terms of measuring the effects of different treatment conditions on cell signaling status4, a kinase assay allows for measuring the effects of different treatment conditions directly in terms of the enzymatic activity of a kinase of interest. While there are several options for similar assays that do not use radioactive materials5, we continue to rely on this method for robust quantitation of results. There are two typical applications of this assay, both valuable for different reasons: the immunoprecipitated (IP) kinase assay (Figure 1) and the recombinant protein kinase assay (Figure 2).
The IP kinase assay is tremendously useful for identifying factors capable of activating specific protein kinases as well as gauging inhibitory treatment conditions. Briefly, an epitope-tagged kinase of interest is transfected into cultured eukaryotic cells, subjected to a variety of treatments, immunoprecipitated, and assayed for the ability to incorporate radiolabeled phosphate into a model substrate (e.g. myelin basic protein (MBP)). IP kinase assay can also be performed without resorting to overexpression, either by immunoprecipitation of endogenous protein or any number of genome-editing techniques. Because treatments are administered in culture, this method can detect stimulations transmitted through multiple upstream factors or parallel pathways by in vitro readout. One major advantage of this method is that it does not require prior knowledge of direct upstream or downstream factors or phosphorylation sites therein. Moreover, once specific substrates for a kinase of interest have been identified, the same kinase assay protocol can be used with recombinant components to measure specific activity towards natural substrates and identify specific phosphorylation sites when combined with mass spectrometry analysis. Sites identified in this manner can be further validated with kinase assays utilizing substrate mutants. Lastly, this method can also be used to detect and measure autophosphorylation.
The protocol provided here assumes either an optimized protein purification scheme or transfection method for expressing an affinity-tagged kinase of interest in cultured cells. For more detailed expositions of transfection, lysis, immunoprecipitation, and protein purification protocols, we suggest referring to Cold Spring Harbor Protocols6. For more information regarding assay development and modification, please refer to Protein Phosphorylation: Selected Methods in Enzymology7.
Protocol
1. Kinase Purification Resources and General Immunoprecipitation Pipeline
NOTE: Kinases for use with this assay may be sourced from immunoprecipitates of cultured cells or by recombinant means such as affinity-tagged purification6. Below is a general protocol for flag-tagged immunoprecipitation that may have to be modified depending on the kinase of interest. Everything should be kept on ice when possible.
Add 2 µL of 1 mg/mL antibody to 200 µL of cell lysates (usually ~1 mg/mL total protein concentration) and incubate at 4 °C for 1 h while rocking.
- Wash protein A sepharose beads 2-3 times with lysis buffer (see below) by briefly spinning at 4 °C for 30 s to 1 min at 5,000 x g ("touch spin"), removing supernatant with a pipette, and resuspending beads in buffer.
- Prepare lysis buffer: 50 mM HEPES pH 7.7, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 10% glycerol, 0.2 mM sodium orthovanadate, 100 mM sodium fluoride, 50 mM β-glycerophosphate, 0.1% Triton X 100 or 0.1% NP-40.
Add 30 µL of 50% slurry of protein A sepharose beads in lysis buffer to lysates; incubate at 4 °C for 1 h while rocking.
Touch spin at 4 °C to pellet the beads and remove the supernatant.
Wash 3 times with 1 mL of bead wash buffer (1 M NaCl, 20 mM Tris pH 7.4).
Wash once with 1x kinase reaction buffer (10 mM HEPES pH 8.0, 10 mM MgCl2). Remove as much buffer as possible without removing beads.
2. Initializing Kinase Reactions
NOTE: For IP kinase assays, ~15 µL of unsuspended beads is a typical starting sample. For recombinant protein kinase assays, typical starting amounts range from 0.1-1.0 µg in 1-10 for 25-50 µL final reaction volumes. Adjust the volumes of recombinant protein samples to be equal with the buffer they are stored in prior to initializing the assay. When using high amounts of kinase, include a carrier protein such as BSA to aid in maintaining protein stability for the duration of the assay.
Prepare the 5x kinase reaction buffer given below: 50 mM HEPES pH 8.0 50 mM MgCl2 50 mM Benzamidine (protease inhibitor) 50 mM DTT (reduction agent) 250 µM ATP (unlabeled, or "cold")
Keep kinase samples on ice in 1.5 mL tubes. Prepare the following reaction mixture in separate, chilled 1.5 mL tubes: 21 µL H2O 6 µL 5x kinase reaction buffer 1 µL radiolabeled ("hot") ATP 2 µL substrate (~5 mg/mL) NOTE: For recombinant kinase assays, volume may be adjusted to accommodate purified protein. Calculate such that final reaction volume is 30 µL. Use this recipe to prepare a master mix. Upon receipt of labeled ATP, dilute stock in H2O to a specific activity of 0.01 mCi/µL prior to adding to reaction mixture.
To initialize the assay, add entire reaction mixture to kinase sample and incubate reaction at 30 °C for 5 min to 1 h depending on activity of kinase being assayed. NOTE: When working with a kinase whose activity has not been previously assayed, perform a time course of kinase activity with 5 min intervals between time points.
3. Reaction Termination and SDS-PAGE
Stop reaction by putting on ice and adding 7.5 µL 5x Laemmli sample buffer 6.
Heat at 100 °C for 30 s to 2 min in heat block.
- Touch spin and load 20 µL per well on 10-15% SDS-PAGE gel. Run gel long enough to separate kinase and substrate (typically 1 h at constant power of 5 W) Make sure to keep gel apparatus shielded to limit exposure to 32P.
- Here, use homemade gel apparatuses, but standard commercially available mini-gels are perfectly suitable. Use the dimensions as follows: Gels: 8 cm width, 5.5 cm length (resolving), 1.5 mm thickness. Combs: 15 wells, 1.5 mm thickness, 2.9 mm width, 16 mm depth
4. Gel Staining and Drying
Caution: For all steps be sure to reduce personal exposure to 32P by using radioactive shielding and wearing personal protective equipment. For more information on specific steps some helpful references are included.
Remove the gel from glass/alumina plates and place in 50 mL Coomassie stain (10% glacial acetic acid, 50% methanol, 0.25% R-250 dye) for 1 h on an orbital shaker set to 50 rpm. For this step use a container that is slightly larger than the gel itself.8
Move the gel from stain to fixing solution (10% glacial acetic acid, 20% methanol) in order to de-stain. Rock the gel in 600-700 mL of fixing solution overnight on orbital shaker at 50 rpm. Place pieces of foam or knotted laboratory wipes in the container with the gel to absorb the Coomassie dye 8,9.
Remove gel from fixing solution and soak in 200 mL methanol for 1-2 min with gentle agitation until gel turns milky white. This will help prevent cracking during the drying step.
Wet a 14 cm x 14 cm piece of qualitative filter paper with methanol and place onto slab gel vacuum dryer. Lay the gel down onto the filter paper front side facing up.
Cover the gel with plastic wrap carefully to avoid wrinkles and air bubbles. Run the dryer for 1.5 h at 80 °C. After the vacuum has been attained open the lid and roll out any air bubbles if needed with a soft rubber brayer.
5. Autoradiogram and Scintillation Counts
Remove dried gel and attach an autorad marker, phosphorescent ruler or dots to the filter paper on the side of the gel. If using dots make sure they are in an asymmetric pattern. This will align the gel with the film after exposure and development.
Use a Geiger counter to check the intensity of the signal. For weaker signals exposure at -70 °C to -80 °C with an intensifying screen will increase the film band density.
In a dark room put dried gel in a film cassette with film and an intensifying screen in the following order from top to bottom: gel, film, intensifying screen.
- Attach the intensifying screen to the lid of the cassette and tape the gel in place to make this step easier. The intensifying screen emits light when irradiated by the beta particles from the 32P. These light emissions penetrate the film more efficiently than the beta particles.
- Make sure the wavelength emitted from the intensifying screen corresponds to a wavelength the film is most sensitive to.
- Use a Geiger counter to determine how long to leave exposure for: if cpm count is ~100 or below, try overnight at -70 °C. If count is ~10,000, start with a 1 h exposure and optimize from there.
At the conclusion of the exposure remove the film and develop in a medical/X-ray film processor. If the exposure was performed at -70 to -80 °C, either allow the cassette to warm to room temperature to minimize condensation on the film or remove the film immediately before condensation forms10.
Lay the film over the dried gel/filter paper and align the image of the marker/dots with the markers/dots on the dried gel. Mark on the film the bands corresponding to the protein standards. Label the protein standards and which lanes the reactions are for future reference.
Excise the bands from the gel that correspond to bands of interest on the film and place them in 7 mL scintillation vials. Add 4 mL of scintillation fluid and count with liquid scintillation counter. Confirm the counter is set to monitor the correct energy spectrum window for 32P. NOTE: Be sure to also excise the band corresponding to substrate from the no-kinase control lane. This will be used to calculate background radiation that will be subtracted from all sample scintillation counts.
6. Analysis of Results
NOTE: The autoradiogram provides a qualitative visualization of results. For accurate quantitation, 32P incorporation can be measured with a scintillation counter. Data are usually expressed in terms of relative activity, as shown in Figure 1B. As long as uniform conditions are maintained for all samples, relative measurements of specific activity are sufficient to compare treatments.
When calculating absolute specific activity values, perform a rigorous optimization of all reaction conditions, including kinase, substrate, and cold ATP concentrations, in order to ensure a linear range of activity over a time course (e.g. 2 x [kinase] = 2 x specific activity). For kinases with high specific activity, perform assays with increased substrate concentration in case kinase activity is limited by amount of substrate.
- Calculate the specific activity of kinase of interest in units of nanomoles phosphate transferred per min per mg kinase.
- Subtract blanks from the cpm in the counted bands.
- Calculate the decay of hot ATP: [(1/2)^(t/t1/2)] x 0.95 where t is the number of days since the reference date (should be provided by vendor), t1/2is the half-life of 32P, which is 14.3 days, and 0.95 reflects the efficiency of the scintillation counter. Example: if using hot ATP that is 28 days old, the decay value should be 0.245.
- Calculate the picomoles (pmol) of total ATP in the assay (usually this is equal to the amount of cold ATP in the reaction): assay volume (µL) x [cold ATP] (µM) = total ATP (pmols). Example: 30 µL x 50 µM = 1500 pmol
- Calculate cpm/pmol ATP: [µL of hot ATP x (2.2 x 107) x decay factor]/ pmol of cold ATP. NOTE: The value of 2.2 x 107 converts 0.01 mCi/µL of ATP to cpm.
- Calculate the amount of kinase in the assay in mg. For both recombinant kinase and immunoprecipitated kinases, standard methods such as BCA assays are suitable to determine starting concentrations. Example: wtERK2 0.0002 µg/µL in a 50 µL kinase reaction is 0.01 µg, or 1 x 10-5 mg.
- Calculate total cpm in assay. Assemble 30 µL reactions and run 20 µL from each reaction on a gel. Multiply the cpm counted (after blank subtraction) by a factor of total assay volume/ volume loaded. Continuing the above example, multiply all counts by 1.5, or 30/20.
- Calculate the specific activity of the assayed kinase: Total cpm in assay)/(cpm/pmol ATP)/(assay time in minutes)/(amount of kinase in mg) NOTE: Dividing the above value by 1,000 yields the specific activity in nanomoles/minute/mg
Calculate how many mols of phosphate are incorporated into a substrate (as long as its molecular weight is known). This is used to determine the number of phosphosites on a particular protein once the reaction has gone to completion. Total cpm in assay)/(cpm/pmol ATP)]/(amount of substrate in pmols)
Representative Results
WNK1 IP kinase assays
Myc-tagged WNK1 was transfected into HEK293 cells and immunoprecipitated with an anti-Myc antibody11. The immunoprecipitate displayed kinase activity towards the model substrate MBP as well as toward itself (Figure 1A). WNK1 mutants were then tested for kinase activity towards MBP by the same method, this time employing GST-tagged constructs (Figure 1B). Analysis of mutant kinase behavior relative to wildtype revealed residues critical for optimal WNK1 activity.
HEK293 cells were exposed to a panel of pharmacological and biochemical treatments. Using an antibody raised against a WNK1 N-terminal peptide, immunoprecipitated endogenous WNK1 kinase activity was assayed using MBP as substrate. Epidermal growth factor (EGF), a known activator of ERK1/2 signaling, nocodazole, a drug that targets microtubule dynamics, anisomycin, an inhibitor of eukaryotic translation, and lysophosphatidic acid (LPA), a potent mitogen, were all unable to elicit a robust increase in phosphorylated MBP. In contrast, NaCl was demonstrated to be a regulator of WNK1 kinase activity.
ERK2 recombinant protein kinase assays
Rat ERK2 bearing an N-terminal 6His tag was expressed in bacterial cells and affinity purified with nickel resin12. Protein was further purified by ion exchange chromatography on a MonoQ column, wherein several elution fractions were tested for kinase activity (Figure 2A). Because ERK2 requires dual phosphorylation by MEK to reach maximal kinase activity, ERK2 purified from bacteria in the absence of MEK is largely inactive. Therefore, in order to test fractions of recombinant ERK2 for activity towards MBP, a constitutively active form of MEK1 called MEK1R4F was included in kinase reactions. Notably, MEKR4F does not harbor high kinase activity on MBP (Figure 2).
Using an assay similar to the one shown in Figure 2A, an MBP kinase activity was used to measure activity of recombinant ERK2 mutants purified from bacterial cultures relative to wildtype protein (Figure 2B). Although ERK2 is able to phosphorylate MBP when stimulated by MEKR4F, mutation of either the catalytic lysine (K52R) or a threonine proximal to the canonical sites of dual phosphorylation (T188D and T188E) dramatically abrogates ERK2 kinase activity. ERK2-T188D and ERK2-T188E display marginal kinase activity toward a small, flexible peptide (Figure 2C), however, they are unable to robustly phosphorylate the known ERK2 substrates Nup153 and PDX1 (Figure 2D).
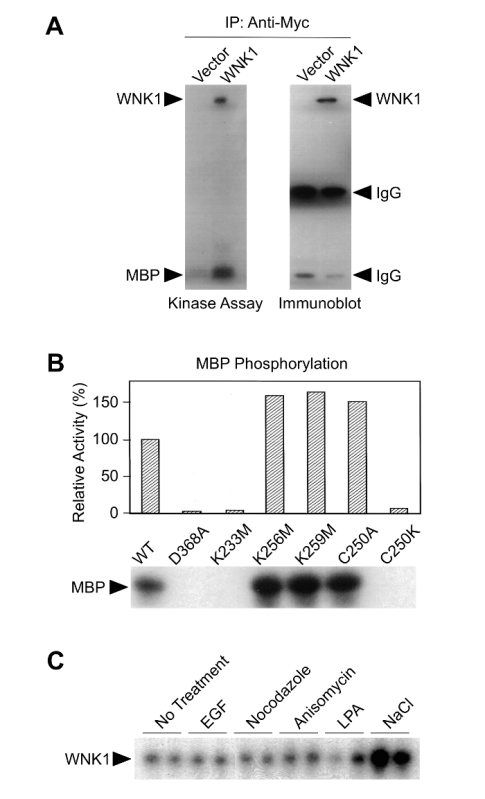 Figure 1: Immunoprecipitation kinase assays of WNK1. (A) HEK293 cells were transfected with either pCMV5-Myc without an insert or pCMV5-Myc-WNK1, tagged proteins were immunoprecipitated with the anti-Myc antibody followed by kinase assays using MBP as substrate. Autoradiography is shown on the left, and an immunoblot of the immunoprecipitates on the right. (B) Various GST-WNK1 mutant proteins were used in kinase assays with MBP as substrate; MBP phosphorylation is expressed as activity relative to wildtype WNK1. (C) Endogenous WNK1 was immunoprecipitated from HEK293 cells treated with various stimuli and assayed for autophosphorylation. This figure was modified from Xu et al., 200011. Please click here to view a larger version of this figure.
Figure 1: Immunoprecipitation kinase assays of WNK1. (A) HEK293 cells were transfected with either pCMV5-Myc without an insert or pCMV5-Myc-WNK1, tagged proteins were immunoprecipitated with the anti-Myc antibody followed by kinase assays using MBP as substrate. Autoradiography is shown on the left, and an immunoblot of the immunoprecipitates on the right. (B) Various GST-WNK1 mutant proteins were used in kinase assays with MBP as substrate; MBP phosphorylation is expressed as activity relative to wildtype WNK1. (C) Endogenous WNK1 was immunoprecipitated from HEK293 cells treated with various stimuli and assayed for autophosphorylation. This figure was modified from Xu et al., 200011. Please click here to view a larger version of this figure.
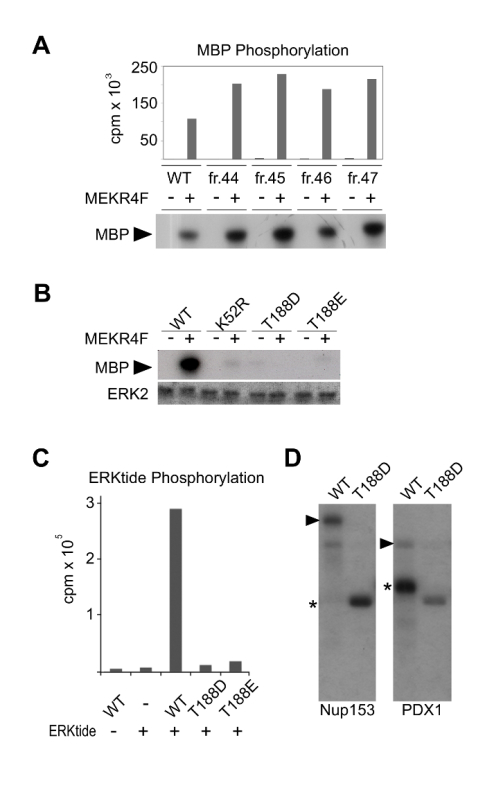 Figure 2: Recombinant protein kinase assays of ERK2. (A) Purified ERK2 fractions from a MonoQ anion exchange column were used in a kinase assay with MBP in the presence or absence of MEK1R4F, a constitutively active stimulator of ERK2 kinase activity. (B) ERK2 mutants were tested for kinase activity on MBP. (C) Activation loop mutants ERK2-T188D and ERK2-T188E display marginal kinase activity toward a small typified peptide substrate. (D) Comparison of ERK2 or T188D kinase activities following activation by MEK1R4F with known ERK2 substrates nucleoporin-153 (Nup153) and pancreatic and duodenal homeobox 1 (PDX1). Phosphorylated substrates are denoted by wedges and phosphorylated ERK2 and T188 by asterisks. Reprinted (and modified) with permission from McReynolds et al., 201612 (Copyright 2016 American Chemical Society). Please click here to view a larger version of this figure.
Figure 2: Recombinant protein kinase assays of ERK2. (A) Purified ERK2 fractions from a MonoQ anion exchange column were used in a kinase assay with MBP in the presence or absence of MEK1R4F, a constitutively active stimulator of ERK2 kinase activity. (B) ERK2 mutants were tested for kinase activity on MBP. (C) Activation loop mutants ERK2-T188D and ERK2-T188E display marginal kinase activity toward a small typified peptide substrate. (D) Comparison of ERK2 or T188D kinase activities following activation by MEK1R4F with known ERK2 substrates nucleoporin-153 (Nup153) and pancreatic and duodenal homeobox 1 (PDX1). Phosphorylated substrates are denoted by wedges and phosphorylated ERK2 and T188 by asterisks. Reprinted (and modified) with permission from McReynolds et al., 201612 (Copyright 2016 American Chemical Society). Please click here to view a larger version of this figure.
Discussion
Kinases are a diverse family of proteins that have evolved extensive functionality in numerous contexts, and kinase assays have been incredibly useful in studying several signaling proteins and have greatly contributed to our current understanding of cellular communication. Notably, the same basic assay was used in characterizing two disparate kinases despite major differences in structure and activity. WNK1 kinase contains an atypical catalytic pocket where the critical lysine has shifted to a unique position and is known to play roles in regulation of cation-chloride cotransporters through both kinase and scaffolding functions. The kinase activity of WNK1 is known to have a low turnover number, even on its best-characterized substrates, the protein kinases oxidative stress responsive 1 (OSR1) and SPS/STE20-related proline alanine-rich kinase (SPAK). In contrast, ERK2, along with the closely related kinase ERK1, displays robust kinase activity when activated in vitro and is known to phosphorylate over a hundred substrates in cells13,14.
The most critical aspect of the assay is reaction assembly. In order to make accurate time point measurements, special care must be given to initializing kinase reactions. There are four basic requirements for a kinase assay to proceed: kinase, substrate, ATP, and a metal ion. For this protocol, which is primarily used for eukaryotic kinases, MgCl2 is used as a source of magnesium. Both recombinant protein kinase and IP kinase preparations typically contain magnesium, rendering MgCl2 insufficient as a reaction starter. Likewise, addition of unlabeled ("cold") ATP prior to the addition of labeled ("hot") ATP can initiate a reaction inappropriately early. We recommend preparing reactions using a concentrated kinase reaction buffer that facilitates simultaneous addition of MgCl2, hot and cold ATP, and substrate. In the case of measuring autophosphorylation activity, combined addition of magnesium and ATP can be used to initiate reactions. In all cases, samples should be prepared on ice and immediately transferred to a 30 °C water bath once reactions are assembled. It is also important to include a no-kinase control reaction to ascertain whether background signal will interfere with results. It may be necessary to troubleshoot various parameters of the assay, including time, temperature, and buffer content. The number of samples that can be processed at once is limited to the number of wells SDS-PAGE gels, which is high enough to effectively screen different conditions. As mentioned earlier in the protocol, we recommend beginning pilot experiments with a time course before moving on to testing other reaction conditions.
Although there is some distinction between IP kinase assays and recombinant protein kinase assays, the experimental paradigm common to both demonstrates how versatile this assay can be. Indeed, there are instances when features of both kinase assay types may be blended, as with studies on the mitogen-activated protein kinase/ extracellular signal-regulated kinase (MAPK/ERK) signaling pathway. In this pathway, ERK1/2 are activated by dual-phosphorylation by the upstream factor MAPK/ERK kinase 1 (MEK1). Previous studies have shown that while MEK1, as well as a constitutively active mutant termed MEK1R4F, is able to activate ERK1/2, it harbors very low activity towards MBP. Consequently, ERK1/2 purified from bacterial cells display limited kinase activity towards MBP unless treated with MEK1R4F, creating a robust platform for comparing the kinase activity of wildtype ERK1/2 to mutant constructs, as shown in Figure 2. Inclusion of immunoprecipitated components can add even more nuance, highlighting the kinase assay as a highly adaptable method to probe the multifaceted nature of these important signaling molecules.
While some may find working with radioactive materials cumbersome, the quantitative tractability of the radiolabeled kinase assay is one of its major advantages.However, recent advancements in the fields of natural product chemistry, genomics, and mass spectrometry have created a demand for modified kinase assays with readouts more amenable to high throughput applications15. Because these assays take advantage of different labeling materials, compromises are made in terms of accuracy, but these can be overcome by using a radiolabeled assay to validate screen results. As our understanding of protein kinase signaling increases, it remains clear that techniques able to tease out the particulars of kinase regulatory functions will continue to aid in the development of tools and therapies.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors thank all current and former members of the Cobb laboratory for valuable work and discussions, and Dionne Ware for administrative assistance. These studies were supported by National Institutes of Health Grant R37 DK34128 and Welch Foundation Grant I1243 to M.H.C.
References
- Raman M, Chen W, Cobb MH. Differential regulation and properties of MAPKs. Oncogene. 2007;26(22):3100–3112. doi: 10.1038/sj.onc.1210392. [DOI] [PubMed] [Google Scholar]
- Wellbrock C, Arozarena I. The Complexity of the ERK/MAP-Kinase Pathway and the Treatment of Melanoma Skin Cancer. Front Cell Dev Biol. 2016;4:33. doi: 10.3389/fcell.2016.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Terman JR. Characterizing PKA-Mediated Phosphorylation of Plexin Using Purified Proteins. Methods Mol Biol. 2017;1493:147–159. doi: 10.1007/978-1-4939-6448-2_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross H, Armstrong CG, Cohen P. A non-radioactive method for the assay of many serine/threonine-specific protein kinases. Biochem J. 2002;366(Pt 3):977–981. doi: 10.1042/BJ20020786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akita S, Umezawa N, Kato N, Higuchi T. Array-based fluorescence assay for serine/threonine kinases using specific chemical reaction. Bioorg Med Chem. 2008;16(16):7788–7794. doi: 10.1016/j.bmc.2008.07.007. [DOI] [PubMed] [Google Scholar]
- Cold Spring Harbor Protocols. 2016. Available from: http://cshprotocols.cshlp.org/
- Dalby KN. Protein Phosphorylation: Selected Methods in Enzymology. J Am Chem Soc. 1999;121(51):12215. [Google Scholar]
- Roland J. Coomassie Staining and Destaining. 2007. Available from: http://www.cytographica.com/lab/protocols/gel_destain.html.
- Autoradiography. 2011. Available from: https://www.nationaldiagnostics.com/electrophoresis/article/autoradiography.
- Walker JM. The Protein Protocols Handbook. 3 edn. Humana Press; 2009. [Google Scholar]
- Xu B. WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J Biol Chem. 2000;275(22):16795–16801. doi: 10.1074/jbc.275.22.16795. [DOI] [PubMed] [Google Scholar]
- McReynolds AC. Phosphorylation or Mutation of the ERK2 Activation Loop Alters Oligonucleotide Binding. Biochemistry. 2016;55(12):1909–1917. doi: 10.1021/acs.biochem.6b00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson SM. Large-scale discovery of ERK2 substrates identifies ERK-mediated transcriptional regulation by ETV3. Sci Signal. 2011;4(196):rs11. doi: 10.1126/scisignal.2002010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courcelles M. Phosphoproteome dynamics reveal novel ERK1/2 MAP kinase substrates with broad spectrum of functions. Mol Syst Biol. 2013;9:669. doi: 10.1038/msb.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers SM. High-Throughput Screening and Hit Validation of Extracellular-Related Kinase 5 (ERK5) Inhibitors. ACS Comb Sci. 2016;18(8):444–455. doi: 10.1021/acscombsci.5b00155. [DOI] [PubMed] [Google Scholar]