

Abstract
Blood vessels supply oxygen and nutrients throughout the body, and the formation of the vascular network is under tight developmental control. The efficient in vivo visualization of blood vessels and the reliable quantification of their complexity are key to understanding the biology and disease of the vascular network. Here, we provide a detailed method to visualize blood vessels with a commercially available fluorescent dye, human plasma acetylated low density lipoprotein DiI complex (DiI-AcLDL), and to quantify their complexity in Xenopus tropicalis. Blood vessels can be labeled by a simple injection of DiI-AcLDL into the beating heart of an embryo, and blood vessels in the entire embryo can be imaged in live or fixed embryos. Combined with gene perturbation by the targeted microinjection of nucleic acids and/or the bath application of pharmacological reagents, the roles of a gene or of a signaling pathway on vascular development can be investigated within one week without resorting to sophisticated genetically engineered animals. Because of the well-defined venous system of Xenopus and its stereotypic angiogenesis, the sprouting of pre-existing vessels, vessel complexity can be quantified efficiently after perturbation experiments. This relatively simple protocol should serve as an easily accessible tool in diverse fields of cardiovascular research.
Keywords: Developmental Biology, Issue 123, Vasculogenesis, angiogenesis, cardiovascular development, Xenopus tropicalis, microinjection, DiI-AcLDL, imaging, vein complexity index
Introduction
Vasculogenesis, the formation of new blood vessels from newly born endothelial cells, and angiogenesis, the formation of new vessels from pre-existing vessels, are two distinct processes that shape embryonic vasculature1. Any dysregulation in these processes results in various heart diseases and structural abnormalities of vessels. Furthermore, tumor growth is associated with uncontrolled vessel growth. As such, molecular mechanisms underlying vasculogenesis and angiogenesis are the subject of intense investigation2.
Xenopus and zebrafish are attractive vertebrate models for vasculogenesis and angiogenesis studies, for several reasons. First, their embryos are small; therefore, it is relatively easy to image the entire vasculature. Second, embryonic development is rapid; it only takes a couple of days for the entire vasculature to develop, during which time the developing vasculature can be imaged. Third, genetic and pharmacological interventions before and during vessel formation are easy to perform, such as through the microinjection of antisense morpholino nucleotides (MOs) into the developing embryo or through the bath application of drugs3,4,5.
The unique advantage of Xenopus over zebrafish is that embryological manipulations can be performed because Xenopus follows stereotypical holoblastic cleavages and the embryonic fate map is well defined6. For example, it is possible to generate an embryo in which only one lateral side is genetically manipulated by injecting an antisense MO to one cell at the two-cell stage. It is also possible to transplant the heart primordium from one embryo to another to determine whether the gene exerts its function by a cell-intrinsic or -extrinsic mechanism7. Although these techniques have mostly been developed in Xenopus laevis, which is allotetraploid and is therefore not ideal for genetic studies, they can be directly applied to Xenopus tropicalis, a closely related diploid species8.
One way to visualize the vasculature in a live Xenopus embryo is to inject a fluorescent dye to label the blood vessels. Acetylated low density lipoprotein (AcLDL) labeled with a fluorescent molecule such as DiI is a very useful probe. Unlike unacetylated LDL, AcLDL does not bind to the LDL receptor9 but is endocytosed by macrophages and endothelial cells. The injection of DiI-AcLDL into the heart of a live animal results in the specific fluorescent labeling of endothelial cells, and the entire vasculature can be imaged by fluorescence microscopy in live or fixed embryos4.
Here, we present detailed protocols for the visualization and quantification of blood vessels using DiI-AcLDL in Xenopus tropicalis (Figure 1). We provide key practical points, with examples of successful and unsuccessful experiments. In addition, we provide a straightforward method for the quantitative analysis of vascular complexity, which might be useful in assessing the effects of genetic and environmental factors on the shaping of the vascular network.
Protocol
All experiments complied with protocols approved by the Yonsei University College of Medicine Institutional Animal Care and Use Committees.
1. Preparation of Xenopus tropicalis Embryos
NOTE: Xenopus tropicalis embryos were produced as previously described10, with slight modification. Xenopus tropicalis embryos were staged according to the tables of Nieuwkoop and Faber11 .
- Induction of ovulation
- To pre-prime, inject human chorionic gonadotropin (hCG) into the dorsal lymph sac of a pair of frogs (one male and one female) on the day prior to the day of mating. Inject 20 units per female frog and 10 units per male. House a male and a female in the same tank overnight (for more than 18 h).
- To prime, on the following day, inject 200 units of hCG to the pre-primed female and 100 units of hCG to the pre-primed male; the primed female will start to lay eggs in approximately 3 h and will continue to do so throughout the day. Keep the male and female pair together for natural mating or in separate tanks for in vitro fertilization (step 1.2).
- Fertilization NOTE: Eggs can be fertilized by natural mating or in vitro fertilization. In natural mating, eggs are fertilized as they are laid, and therefore, the timing of fertilization cannot be controlled. Alternatively, a primed female frog can be housed in a separate tank and unfertilized eggs can be collected for in vitro fertilization, which generates hundreds of embryos fertilized at the same time. The latter approach is useful when blastomere-targeted microinjection will be performed prior to vessel labeling. In this approach, however, a male is sacrificed.
- Natural mating
- Leave primed female and male pair in the same tank. The male will clasp the female, leading to immediate fertilization of the laid eggs. Collect the eggs with a glass or plastic pipette and transfer them to a Petri dish. To prevent the eggs from sticking to the pipette, coat the inner surface of the pipette with 0.5% bovine serum albumin (BSA).
- In vitro fertilization (IVF)
- Separate the primed female from the male after priming. The female will start to lay eggs spontaneously in approximately 3 h. Transfer a few laid eggs using a BSA-coated pipette to a Petri dish and check their quality under a microscope. If healthy eggs (clear pigment, elastic ball-shaped) were laid, prepare to dissect out the testes from the primed male.
- Make 0.4% Tricaine methanesulfonate (MS222) for euthanasia and inject 300 µL into the dorsal lymph sac of the primed male. In approximately 10 min, the male will lose his sense of balance and will stay afloat; confirm that the male is euthanized by pinching a hind leg with a pair of blunt forceps and observing no response.
- Dissect out the two testes, clean them by quickly tapping them on lint-free paper, and transfer them into a microtube containing 1 mL of 60% Leibovitz's L-15 Medium (60%) supplemented with 10% fetal bovine serum (FBS); a detailed procedure is described elsewhere10.
- Gently mince the testes with a pestle to extract the sperm. At this concentration (i.e., 2 testes in 1 mL of 10% FBS in 60% L-15), a few drops of the testis solution (0.1 mL) can fertilize approximately three hundred eggs. The remaining sperm can be stored at 4 °C in a tightly sealed tube and used for IVF the next day.
- Squeeze the eggs from the female into a small Petri dish. Hold the upper part of the female upside down on the palm and put the forefinger between its hind legs. Spread its hind legs with the forefinger and the other hand to expose the cloaca. Touch the cloaca to the Petri dish. Coat the Petri dish with 0.5% BSA to evenly distribute the eggs as a monolayer during IVF.
- Add a few drops (0.1 mL) of sperm-containing L-15 medium to the eggs. For synchronous fertilization, gently stir the sperm solution across the dish. After 4 min at room temperature, fill the Petri dish with 0.01x Modified Barth's Saline (MBS), incubate for 10 min, and replace the solution with 0.1x MBS. The fertilized eggs will undergo the first cleavage in approximately 1 h at room temperature. 1x MBS is 88 mM NaCl, 1 mM KCl, 2.5 mM NaHCO3, 5 mM HEPES, 1 mM MgSO4, and 0.7 mM CaCl2, pH 7.5.
- (Optional) Injection of antisense morpholino oligonucleotides (MOs) and/or mRNAs
- To investigate the effect of a gene of interest on vessel formation, perform the microinjection of antisense MO and/or mRNAs.
- For such experiments, de-jelly the eggs with 2% L-cysteine in 1x MBS (pH 7.5-7.8). Replace 0.1x MBS in the dish containing the fertilized embryos with 2% L-cysteine in 1x MBS. Gently swirl the dish to mix the solution. NOTE: It usually takes about 5 min for the embryos to be de-jellied. During this step, monitor the eggs frequently to ensure that they are not over-exposed to the solution. If the eggs are over-exposed, they will lose elasticity and become flattened, which will cause aberrant development and lethality.
- Wash the de-jellied eggs five times with 0.1x MBS. Perform the microinjection in 4% Ficoll dissolved in 0.1x MBS. Typically, inject 4 ng of MO and 600 pg of mRNA per blastomere at the two-cell stage. Inject into one cell to manipulate gene expression on only one side of the embryo. Use the uninjected side of the same embryo as a control.
- As another control for the specificity of gene manipulation, inject the same amount of a control MO (CoMO) or control mRNA (e.g., beta-galactosidase mRNA). CoMO must have the same molecular weight as the gene-specific MO and must not target any gene in the Xenopus genome. For easy visualization of the injected side, use fluorescein-tagged MO or co-inject a tracer (e.g., EGFP mRNA).
- Leave the injected embryos in 4% Ficoll for 2 h and then transfer them to a new 60 mm Petri dish filled with 0.1x MBS.
- Raising
- Raise the embryos at 23 °C. Although the speed of development can be slowed down or accelerated by using temperatures below or above 23 °C, respectively (range: 16-26 °C), it is recommended to raise them at 23 °C.
- (Optional) Treatment with pharmacological reagents
- For pharmacological manipulations, administer drugs to the developing embryos by bath application.
2. Preparation of DiI-AcLDL Injection
Make a tapering glass pipette using a standard micropipette puller. Place a borosilicate glass tube in the micropipette puller and use the following settings: pressure, 200; heat, 30; pull, 30; velocity, 120; time, 200.
Store DiI-AcLDL stock solution at 4 °C. Take the upper clear part of the solution for injection, but not the precipitated particles on the bottom of tube. Any debris in the glass pipette will disturb the proper ejection of the solution.
Fill the prepared glass pipette with a suitable amount of DiI-AcLDL solution (~5 µL) using a microloader.
Clip off the tip of the glass pipette little by little using a fine pair of forceps (number 55). Set up the pressure-controlled injection system so that approximately 5 nL of solution is ejected at a time; 50-60 nL of DiI-AcLDL solution is enough to stain the vessels of one embryo.
Do not leave the DiI-AcLDL-loaded pipette in air for an extended period of time in order to avoid drying. Keep the tip of the pipette submerged in 0.04% MS222 in 0.1X MBS solution. Just before injecting it into the embryo (step 4.2.1), check to see whether the same amount of dye ejects.
3. Injection Setup
- Sylgard mold
- To prepare a small Sylgard mold, mix Sylgard base and curing agent with a weight ratio of 10 to 1. Fill approximately half of a small Petri dish (35 mm or 60 mm dish) with this mixed solution and then leave it to solidify for approximately 48 h at room temperature.
- Anesthesia
- Use 0.04% MS222 in 1x MBS (pH 7.5-8.0) for anesthesia. When long anesthesia is needed (e.g., during live imaging), use 0.04% MS222 in 0.1x MBS to reduce the osmotic imbalance. To adjust the pH, add a few drops of NaOH (~20 µL) and check the pH using pH paper.
- Wait until the transferred embryos stop moving. Touch the dorsal fins of the embryos and inspect whether they respond to confirm complete anesthesia.
- Positioning embryos for injection
- Indent the surface of the hardened Sylgard mold with a thin and sharp blade. Make a V-shaped concave indentation in which to place the embryos (see Figure 2D). Place an anesthetized embryo, ventral-side up, in a slightly oblique fashion (approximately 30°) (Figure 2D).
- Fill the mold with MS222 solution and place the embryos in the mold.
4. DiI-AcLDL Injection
- Incision of skin overlying the heart
- Prepare two pins (Minutiens, 0.10 mm) to cut the skin overlying the heart. Tightly fix each pin to a pin-holder; the heart should be readily identifiable as it pulsates (Figure 2A-2C, pink).
- Puncture the skin of an embryo with one pin. Insert the pin through the hole in the space between the heart and the skin (Figure 2C, solid arrow). Do not puncture the heart. Position the inserted part of this pin in the region where the incision will be made.
- Make a linear incision by gently rubbing the other pin held outside the skin against the inserted pin. Gently widen this incision with two pins,exposing the heart (Figure 2B' and 2D', arrow). The pipette loaded with DiI-AcLDL can now directly approach the heart (Figure 2D').
- Injection
- Insert the tip of the loaded glass pipette into the heart and inject 50-60 nL of DiI-AcLDL solution in approximately 10 ejection pulses. Do not inject an excessive amount of solution, as it causes the heart to rupture. Check whether the heart continues to beat after the injection. After every injection, check whether the same amount of DiI-AcLDL is ejected; otherwise, the tip of the pipette may be blocked (see step 2.5).
- Recovery and visual screening
- Transfer the injected embryos to a new Petri dish filled with 0.1x MBS for recovery. After 5-10 min, the tadpoles should regain consciousness and start to move. At this time, the labeled vessels can be imaged. It is recommended to visually screen successfully labeled embryos under a fluorescence microscope (Figure 3A and 3B). If an insufficient amount of DiI-AcLDL is injected into an embryo, it can be injected again (Figure 3C).
- Discard the tadpoles that have other organs labeled (Figure 3D). Continue until the required number of successfully labeled embryos is obtained.
- (Optional) Fixation of the embryos
- For more thorough imaging of blood vessels, use confocal microscopy; it might be easier to fix the labeled embryos for confocal microscopy. First anesthetize the labeled embryos in 0.04% MS222 in 1x MBS (pH 7.5-8.0) and then fix them in 4% paraformaldehyde in 1X phosphate-buffered saline (PBS) for 1 h at room temperature. Fixed embryos can be stored for several days at 4 °C; protect the injected samples from light to avoid photobleaching of the DiI.
5. Imaging of DiI-AcLDL
- Stereoscopic imaging
- Take photographs under a fluorescence stereoscopic microscope (Figure 3) to assess the overall morphology of the vasculature.
- Melt 1% agarose in 1x PBS for fixed embryos or in 1x MBS for live imaging. Pour melted agarose into a Petri dish and wait until it solidifies. Make a shallow indentation on the surface of agarose gel that will fit the embryo to be imaged when lying on its lateral side. Fill the dish with buffer (MS222 solution for anesthetized embryos or 1x PBS for fixed embryos).
- Fluorescence microscopy (Rhodamine filter set)
- As DiI is a green-excited and red-emitting dye, image using a standard filter set for rhodamine or spectrally related fluorophores (maximum excitation at 554 nm and emission at 571 nm). Place the injected embryos in the indentations on the agarose mold and take photographs (Figure 3).
- Confocal microscopy NOTE: For a closer analysis of vascular architecture, use confocal microscopy.
- If using an inverted microscope, position the embryos on a glass-bottom dish. Add a few drops of 1x PBS and immobilize them by placing a cover slip on top. Remove excess PBS. If live embryos are to be imaged, use anesthetized embryos and use 0.015% MS222 in 0.1x MBS instead of PBS.
- For low-magnification imaging, use a 4X to 10X objective to find the area of interest; the rostral part of the posterior cardinal vein (PCV) is a useful region to investigate developmental angiogenesis (Figure 4, dashed boxes).
- Use an acquisition setting for a red emission fluorophore (such as Rhodamine).
- Make z-stacked images by imaging the lateral half of an embryo. First, focus on the PCV on the lateral side near the objective and then set the lower limit of the focus at the position where the vessels nearest to the objective are in focus. Set the upper limit at the position where the medial part of the PCV being imaged can be focused. Image this entire lateral half of the embryo. Use software that stores metadata on the acquisition settings, such as x, y, and z scales, as they are required to calculate the vessel lengths.
- If needed, use the tile scan mode to image the entire vasculature of an embryo. Empirically determine the number of horizontal and vertical tiles.
- For high-magnification imaging, follow the procedures above to image the rostral part of the PCV and 4 rostral-most intersomitic veins (ISVs) (Figure 4, dashed box). Adjust the number of stacks (z-stack) and tiles (tile scan) appropriately.
6. Quantification of DiI-labeled Vessels
NOTE: DiI-labeled vessels can be traced manually or using software. We use "Simple neurite tracer," a free ImageJ (NIH) plugin, which allows semi-automatic tracing of tube-like structures such as blood vessels and neurites12. Using this free software, the following parameters can be calculated. A detailed procedure for the use of this software is described elsewhere (https://imagej.net/Simple_Neurite_Tracer)12. Below, we use examples of the vasculature of embryos in which Tie2 signaling is inhibited or enhanced (Figure 4A-4C)5. Antisense Tie2MO-injected embryos exhibit reduces angiogenesis and shortens ISVs (Figure 4B), whereas the co-injection of constitutively active Tie2 mutant mRNA (caTie2 mRNA) over-rescues this phenotype, resulting in exuberant ISV branches (Figure 4C).
- ISV lengths
- Using Simple neurite tracer, calculate the length of each traced vessel. Calculate the lengths of the 4 rostral-most ISVs.
- ISV branches NOTE: In wildtype embryos, only a few short branches sprout from the rostral-most ISVs at stage 42.
- In Simple neurite tracer, trace these small vessels after making the ISV from which they originate the primary branch. Calculate the following parameters of these branches originating from the ISVs.
- Total branch number
- Once all of the ISVs and the associated branches are traced, calculate the total number of branches (Figure 5B-5D).
- Branch order
- As Simple neurite tracer records the order of each branch (Figure 5A), calculate the number of branches for each order. The majority of vessels in wildtype embryos should be primary branches (i.e., ISVs).
- Vein complexity index (VCI)
- As VCI is a useful parameter for vessel complexity, which gives more weight to higher-order branches, calculate the VCI for each embryo using the formula in Figure 5A. For example, Tie2MO-injected embryos have a lower VCI compared to control (Figure 5B and 5C), and caTie2 mRNA-injected embryos have a higher VCI (Figure 5D).
Representative Results
Timeline of experiments (Figures 1 and 2)
Shortly after fertilization, targeted microinjection can be performed to modulate gene expression. For example, an antisense MO that specifically binds to the initiation codon of the endogenous Tie2 mRNA can be injected, inhibiting the translation of Tie2 target mRNA by steric hindrance. A MO can be conjugated to fluorescein for the easy visual screening of successfully injected embryos. Alternatively, a tracer mRNA (e.g., mRNA encoding EGFP) can be co-injected with MO. Additionally, drugs can be treated by bath application after hatching (early tailbud stage, around NF stage 26). Dissolve the drug in 0.1x MBS and add it to the embryos. For earlier treatment, embryos can be freed from the membranes with a pair of forceps. DiI-AcLDL can be injected into the heart around stage 33/34 to investigate the dynamics of vessel formation, but typically we inject at stage 37/38 or later (Figure 2, colored regions).
Typical examples of successful and unsuccessful injections (step 4.3) (Figure 3)
Stereoscopic imaging is appropriate for visual screening. If a sufficient amount of DiI-AcLDL is injected into the heart, the PCV should be immediately visible under a fluorescence stereoscope (Figure 3A and 3B). Insufficiently or incorrectly injected embryos are easy to identify (Figure 3C and 3D). Embryos with an insufficient amount of DiI-AcLDL (Figure 3C) can be injected again with the same dye until the vessels are clearly visible. Image successfully injected embryos under a confocal microscope, live or after fixation.
Development of the posterior cardinal vein (PCV) and the intersomitic veins (ISVs)
The PCV extends in the rostal-to-caudal direction, and this extension ends around stage 37/38. ISVs emerge dorsally from the PCV in an anterior-to-posterior wave. This wave of ISV formation begins at stage 36 and ends around stage 40/41; ISVs continue to grow and become lightly branched until stage 43 (Figure 3A and 3B).
Tie2 signaling controls developmental ISV branching (Figure 4)
The PCV and ISVs grow in a stereotypical pattern, and their development is under tight control (Figure 4A). The formation of the ISVs from the PCV can be described as angiogenesis, and therefore it is a useful model to investigate molecular mechanisms underlying angiogenesis. We introduce the result of our recent study showing that developmental signals inhibit Tie2 signaling in ISVs to limit their branching5. The knockdown of Tie2 signaling by antisense MO decreases the length and complexity of ISVs (Figure 4B), and expressing the constitutive active form of Tie2 (caTie2) causes excessive ISV branching (Figure 4C).
Quantification of ISV complexity (Figure 5)
In addition to the lengths and branch numbers of the ISVs, the "vein complexity index" (VCI) can be calculated to assess their complexity (Figure 5A). We adopted the "complexity index" developed by Cohen-Cory and colleagues, which was designed to quantify the complexity of neuronal axonal or dendritic arbors13. In this index, an embryo with more high-order branches receives a higher score than an embryo with the same number of total branches but with more low-order branches. Therefore, a higher VCI indicates that this embryo has a more complex venous network. "Simple neurite tracer" is useful software to keep track of the order and length of individual branches. It is also possible to generate "rendered paths," which is a useful way to visualize the overall architecture of the vasculature (Figures 5B-5D, right panels).
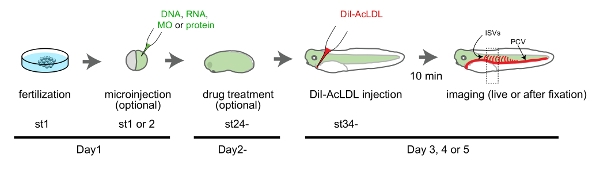 Figure 1: Timeline of the experiments. On the day of fertilization, morpholinos (MOs), DNA, RNA, or protein can be injected into fertilized eggs at NF stage 1 (st1) or 2 (st2). If injected only into the one blastomere at st2, the injected reagent will affect only one lateral side. The uninjected side can be used as a control. Co-inject a tracer (e.g., EGFP mRNA) to identify the injected side. On the second day, early tailbud-stage (~st24) embryos can be treated with pharmacological reagents by bath application. The heart is clearly visible on the third day. DiI-AcLDL can be injected into the heart and blood vessels can be imaged shortly after the injection. Use st33-37 embryos to image the dynamics of vessel growth or st42 embryos to image fully developed vessels. Please click here to view a larger version of this figure.
Figure 1: Timeline of the experiments. On the day of fertilization, morpholinos (MOs), DNA, RNA, or protein can be injected into fertilized eggs at NF stage 1 (st1) or 2 (st2). If injected only into the one blastomere at st2, the injected reagent will affect only one lateral side. The uninjected side can be used as a control. Co-inject a tracer (e.g., EGFP mRNA) to identify the injected side. On the second day, early tailbud-stage (~st24) embryos can be treated with pharmacological reagents by bath application. The heart is clearly visible on the third day. DiI-AcLDL can be injected into the heart and blood vessels can be imaged shortly after the injection. Use st33-37 embryos to image the dynamics of vessel growth or st42 embryos to image fully developed vessels. Please click here to view a larger version of this figure.
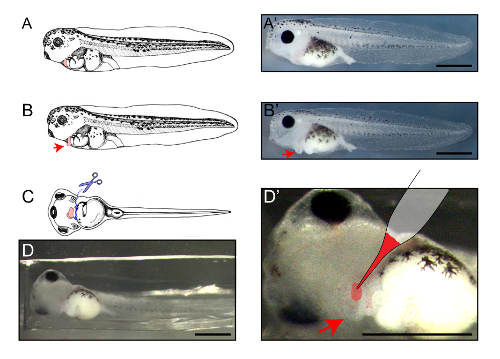 Figure 2: Location of the DiI-AcLDL injection at st42. Lateral (A-B') and ventral (C-D) views of the stage 42 embryo. The heart is colored in pink in A-C, in images taken from Xenbase (www.xenbase.org). Representative stereoscopic images are shown in A', B', and D. For injection, puncture the skin overlying the heart (C, solid arrow), make a linear incision, and open the skin laterally to expose the heart (red arrows). After making the incision, the heart will be clearly distinguishable for injection (D'). Scale bars = 200 µm. Please click here to view a larger version of this figure.
Figure 2: Location of the DiI-AcLDL injection at st42. Lateral (A-B') and ventral (C-D) views of the stage 42 embryo. The heart is colored in pink in A-C, in images taken from Xenbase (www.xenbase.org). Representative stereoscopic images are shown in A', B', and D. For injection, puncture the skin overlying the heart (C, solid arrow), make a linear incision, and open the skin laterally to expose the heart (red arrows). After making the incision, the heart will be clearly distinguishable for injection (D'). Scale bars = 200 µm. Please click here to view a larger version of this figure.
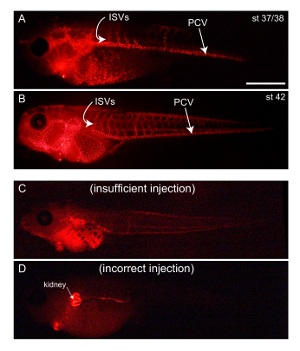 Figure 3: Expected results (stereoscopic images). Representative fluorescence images from stage 37/38 (A) and stage 42 (B) embryos. The left sides of the embryos are shown. The posterior cardinal vein (PCV) extends caudally, from which intersomitic veins (ISVs) branch dorsally in a rostral-to-caudal wave. Only rostral ISVs are visible at stage 37/38 (A), and most ISVs have formed by stage 42 (B). If an insufficient amount of DiI-AcLDL is injected, the PCV will not be clearly visible, even after 30 min (C). In this case, more DiI-AcLDL can be injected. If DiI-AcLDL is accidentally injected into other organs (D), discard the embryos. Scale bar = 500 µm. Please click here to view a larger version of this figure.
Figure 3: Expected results (stereoscopic images). Representative fluorescence images from stage 37/38 (A) and stage 42 (B) embryos. The left sides of the embryos are shown. The posterior cardinal vein (PCV) extends caudally, from which intersomitic veins (ISVs) branch dorsally in a rostral-to-caudal wave. Only rostral ISVs are visible at stage 37/38 (A), and most ISVs have formed by stage 42 (B). If an insufficient amount of DiI-AcLDL is injected, the PCV will not be clearly visible, even after 30 min (C). In this case, more DiI-AcLDL can be injected. If DiI-AcLDL is accidentally injected into other organs (D), discard the embryos. Scale bar = 500 µm. Please click here to view a larger version of this figure.
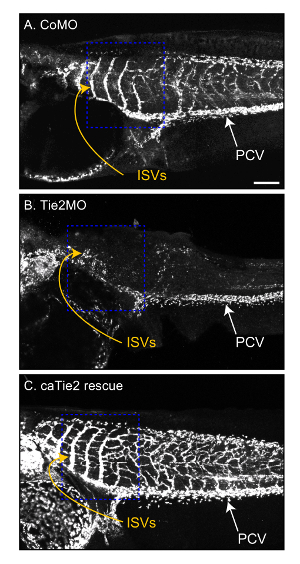 Figure 4: Effects of Tie2 signaling on ISV development. Tie2 gene expression was knocked down by the blastomere injection of translation-blocking antisense morpholino oligonucleotides (Tie2MO). The control morpholino (CoMO) has the same molecular weight but does not target any gene in Xenopus tropicalis. mRNA-encoding constitutively active Tie2 mutant (caTie2) was co-injected to rescue the Tie2 knockdown phenotype. In contrast to the control (A), theTie2MO injection led to a dramatic decrease in the length and number of ISVs (B). This phenotype was over-rescued by caTie2, which showed ISVs with exuberant collateral branches (C). The complexity of ISVs in regions indicated by blue dashed boxes is quantified in Figure 5. Scale bar = 200 µm. Please click here to view a larger version of this figure.
Figure 4: Effects of Tie2 signaling on ISV development. Tie2 gene expression was knocked down by the blastomere injection of translation-blocking antisense morpholino oligonucleotides (Tie2MO). The control morpholino (CoMO) has the same molecular weight but does not target any gene in Xenopus tropicalis. mRNA-encoding constitutively active Tie2 mutant (caTie2) was co-injected to rescue the Tie2 knockdown phenotype. In contrast to the control (A), theTie2MO injection led to a dramatic decrease in the length and number of ISVs (B). This phenotype was over-rescued by caTie2, which showed ISVs with exuberant collateral branches (C). The complexity of ISVs in regions indicated by blue dashed boxes is quantified in Figure 5. Scale bar = 200 µm. Please click here to view a larger version of this figure.
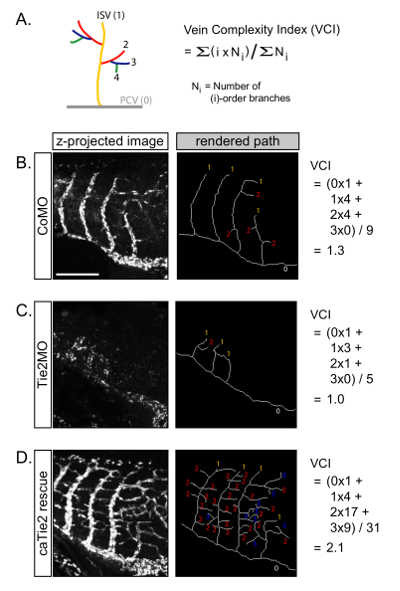 Figure 5: Quantification of vein complexity. (A) The complexity of ISV can be represented by the vein complexity index (VCI). (B-D) VCIs were calculated from the embryos shown in Figure 4. Scale bar = 200 µm. Please click here to view a larger version of this figure.
Figure 5: Quantification of vein complexity. (A) The complexity of ISV can be represented by the vein complexity index (VCI). (B-D) VCIs were calculated from the embryos shown in Figure 4. Scale bar = 200 µm. Please click here to view a larger version of this figure.
Discussion
The protocol presented here was first developed by Ali H. Brivanlou and colleagues to investigate developmental events during vascular formation in Xenopus laevis4, but, as shown in this manuscript, it can be applied to other small animals. Dye injection into the heart is simple to perform, and the entire vascular network can be imaged under a fluorescence dissection microscope, as well as a confocal microscope. If the dye is injected into the heart during vessel development, the dynamics of vessel growth and branching can be imaged in real time4. Using the well-defined venous network, particularly the PCV and rostral-most ISVs, the effects of genetic or environmental perturbation on angiogenesis can be quantitatively assessed.
The most critical step in this protocol is the injection of DiI-AcLDL (step 4). We provide examples of successful and unsuccessful injections (Figure 3). Successfully injected embryos should be screened under a fluorescence microscope, as described in step 4.3. If the PCV is not visible 30 min after injection, not enough DiI-AcLDL was injected (Figure 3C). Unsuccessfully injected embryos may be anesthetized and injected again. In some cases, DiI-AcLDL may be injected into an incorrect location, in which case other organs will be labeled (Figure 3D). Discard incorrectly injected embryos.
The blood vessels of a successfully injected embryo will be visible shortly after injection, and the fluorescence lasts for several hours. Therefore, if the injection is performed before the formation of ISVs, their development can be imaged in a live embryo, providing a powerful tool to investigate development of the cardiovascular system in real time and in vivo4. As Xenopus has been successfully used as a model to screen vascular disrupting agents in mammals14, the experimental approach described here might be combined with pharmacological perturbation experiments and used as a screening platform to find drugs that enhance or inhibit angiogenesis.
Blood vessels can be visualized by genetic tools, as well as by the dye-based labeling methods described here. For example, transgenic Xenopus15 or zebrafish16 that express fluorescence proteins under the control of cell type-specific promoters enable the live imaging of blood and lymphatic vessels, and it is even possible to discriminate arteries from veins16. Although genetic approaches are superior and generate more consistent labeling efficiency, dye-based methods are easily accessible to most laboratories without access to such genetically engineered animals. In Xenopus, the transcardial perfusion of DiI-AcLDL results in the labeling of most endothelial cells4, and arteries and veins are not distinguishable in fluorescence images. Intriguingly, tomato lectin-fluorescein selectively labels arteries in mice, and when co-injected with DiI-AcLDL, arteries and veins can be discriminated17. Although the application was not tested in other tissues or animals, this provides a promising direction to investigate the dynamic development of arteries and veins in Xenopus.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This study was inspired by the work of Levine et al., which described this experimental method and provided a comprehensive description of vascular development in Xenopus laevis. We thank the members of our laboratory for their input. This study was supported by the Yonsei University Future-leading Research Initiative of 2015 (2015-22- 0095) and the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the ministry of Science, ICT & Future Planning (NRF-2013M3A9D5072551)
References
- Herbert SP, Stainier DY. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat Rev Mol Cell Biol. 2011;12(9):551–564. doi: 10.1038/nrm3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustin HG, Koh GY, Thurston G, Alitalo K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat Rev Mol Cell Biol. 2009;10(3):165–177. doi: 10.1038/nrm2639. [DOI] [PubMed] [Google Scholar]
- Lawson ND, Weinstein BM. Arteries and veins: making a difference with zebrafish. Nat Rev Genet. 2002;3(9):674–682. doi: 10.1038/nrg888. [DOI] [PubMed] [Google Scholar]
- Levine AJ, Munoz-Sanjuan I, Bell E, North AJ, Brivanlou AH. Fluorescent labeling of endothelial cells allows in vivo, continuous characterization of the vascular development of Xenopus laevis. Dev Biol. 2003;254(1):50–67. doi: 10.1016/s0012-1606(02)00029-5. [DOI] [PubMed] [Google Scholar]
- Yang C, et al. Calmodulin Mediates Ca2+-Dependent Inhibition of Tie2 Signaling and Acts as a Developmental Brake During Embryonic Angiogenesis. Arterioscler Thromb Vasc Biol. 2016;36(7):1406–1416. doi: 10.1161/ATVBAHA.116.307619. [DOI] [PubMed] [Google Scholar]
- Moody SA. Fates of the blastomeres of the 32-cell-stage Xenopus embryo. Dev Biol. 1987;122(2):300–319. doi: 10.1016/0012-1606(87)90296-x. [DOI] [PubMed] [Google Scholar]
- Elliott KL, Houston DW, Fritzsch B. Transplantation of Xenopus laevis tissues to determine the ability of motor neurons to acquire a novel target. PLoS One. 2013;8(2):55541. doi: 10.1371/journal.pone.0055541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grainger RM. Xenopus tropicalis as a model organism for genetics and genomics: past, present, and future. Methods Mol Biol. 2012;917:3–15. doi: 10.1007/978-1-61779-992-1_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisgraber KH, Innerarity TL, Mahley RW. Role of lysine residues of plasma lipoproteins in high affinity binding to cell surface receptors on human fibroblasts. J Biol Chem. 1978;253(24):9053–9062. [PubMed] [Google Scholar]
- Showell C, Conlon FL. Egg collection and in vitro fertilization of the western clawed frog Xenopus tropicalis. Cold Spring Harb Protoc. 2009;2009(9):5293. doi: 10.1101/pdb.prot5293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwkoop PD, Faber J. Normal Table of Xenopus laevis (Daudin) Amsterdam: North-Holland Publishing Company. Guilders; 1956. [Google Scholar]
- Longair MH, Baker DA, Armstrong JD. Simple Neurite Tracer: open source software for reconstruction, visualization and analysis of neuronal processes. Bioinformatics. 2011;27(17):2453–2454. doi: 10.1093/bioinformatics/btr390. [DOI] [PubMed] [Google Scholar]
- Marshak S, Nikolakopoulou AM, Dirks R, Martens GJ, Cohen-Cory S. Cell-autonomous TrkB signaling in presynaptic retinal ganglion cells mediates axon arbor growth and synapse maturation during the establishment of retinotectal synaptic connectivity. J Neurosci. 2007;27(10):2444–2456. doi: 10.1523/JNEUROSCI.4434-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha HJ, et al. Evolutionarily repurposed networks reveal the well-known antifungal drug thiabendazole to be a novel vascular disrupting agent. PLoS Biol. 2012;10(8):1001379. doi: 10.1371/journal.pbio.1001379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ny A, et al. A transgenic Xenopus laevis reporter model to study lymphangiogenesis. Biol Open. 2013;2(9):882–890. doi: 10.1242/bio.20134739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussmann J, et al. Arteries provide essential guidance cues for lymphatic endothelial cells in the zebrafish trunk. Development. 2010;137(16):2653–2657. doi: 10.1242/dev.048207. [DOI] [PubMed] [Google Scholar]
- Li XM, Hu Z, Jorgenson ML, Slayton WB. High levels of acetylated low-density lipoprotein uptake and low tyrosine kinase with immunoglobulin and epidermal growth factor homology domains-2 (Tie2) promoter activity distinguish sinusoids from other vessel types in murine bone marrow. Circulation. 2009;120(19):1910–1918. doi: 10.1161/CIRCULATIONAHA.109.871574. [DOI] [PMC free article] [PubMed] [Google Scholar]