

Abstract
Knowing how RNAs interact with themselves and with others is key to understanding RNA based gene regulation in the cell. While examples of RNA-RNA interactions such as microRNA-mRNA interactions have been shown to regulate gene expression, the full extent to which RNA interactions occur in the cell is still unknown. Previous methods to study RNA interactions have primarily focused on subsets of RNAs that are interacting with a particular protein or RNA species. Here, we detail a method named Sequencing of Psoralen crosslinked, Ligated, and Selected Hybrids (SPLASH) that allows genome-wide capture of RNA interactions in vivo in an unbiased manner. SPLASH utilizes in vivo crosslinking, proximity ligation, and high throughput sequencing to identify intramolecular and intermolecular RNA base-pairing partners globally. SPLASH can be applied to different organisms including bacteria, yeast and human cells, as well as diverse cellular conditions to facilitate the understanding of the dynamics of RNA organization under diverse cellular contexts. The entire experimental SPLASH protocol takes about 5 days to complete and the computational workflow takes about 7 days to complete.
Keywords: Genetics, Issue 123, RNA, genomics, interactome, sequencing, structure, human
Introduction
Studying how macromolecules fold and interact with each other is the key to understanding gene regulation in the cell. While much effort has been focused in the past decade on understanding how DNA and proteins contribute to gene regulation, relatively less is known about post-transcriptional regulation of gene expression. RNA carries information in both its linear sequence and in its secondary and tertiary structure1. Its ability to base pair with itself and with others is important for its function in vivo. Recent advances in high throughput RNA secondary structure probing has provided valuable insights into the locations of double and single stranded regions in the transcriptome2,3,4,5,6,7,8, however information on the pairing interaction partners is still largely missing. To determine which RNA sequence is interacting with another RNA region in the transcriptome, we need global pair-wise information.
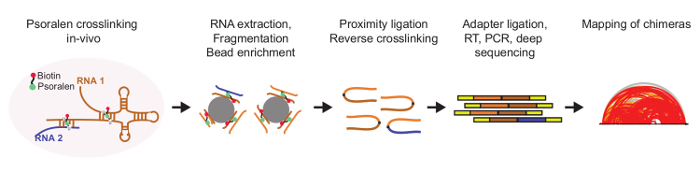
Mapping pair-wise RNA interactions in a global, unbiased manner has traditionally been a major challenge. While previous approaches, such as CLASH9, hiCLIP10 and RAP11, are used to identify RNA interactions in a large scale manner, these techniques typically map RNA base pairing for a subset of RNAs that either interact with a particular protein or RNA species. Recent developments in studying global RNA interactions include the method RPL12, which does not stabilize RNA interactions in vivo and hence may only capture a subset of in vivo interactions. To overcome these challenges, we and others developed genome-wide, unbiased strategies to map RNA interactomes in vivo, using modified versions of the crosslinker psoralen13,14,15. In this protocol, we describe the details for performing Sequencing of Psoralen crosslinked, Ligated, and Selected Hybrids (SPLASH), which utilizes biotinylated psoralen to crosslink base pairing RNAs in vivo, followed by proximity ligation and high throughput sequencing to identify RNA base-pairing partners genome-wide (Figure 1)15.
In this manuscript, we describe the steps to perform SPLASH using cultured adherent cells, in this case HeLa cells. The same protocol can be easily adapted to suspension mammalian cells and to yeast and bacteria cells. Briefly, HeLa cells are treated with biotinylated psoralen and irradiated at 365 nm to crosslink interacting RNA base pairs in vivo. The RNAs are then extracted from the cells, fragmented and enriched for crosslinking regions using streptavidin beads. Interacting RNA fragments are then ligated together using proximity ligation and made into a cDNA library for deep sequencing. Upon sequencing, the chimeric RNAs are mapped onto the transcriptome/genome to identify the RNA interacting regions that are paired to each other. We have successfully utilized SPLASH to identify thousands of RNA interactions in vivo in yeast and different human cells, including intramolecular and intermolecular RNA base pairing in diverse classes of RNAs, such as snoRNAs, lncRNAs and mRNAs, to glimpse into the structural organization and interaction patterns of RNAs in the cell.
Protocol
1. Treatment of HeLa Cells with Biotinylated Psoralen and RNA Extraction
Culture HeLa cells in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin streptomycin (PS) in a 10 cm plate.
Wash the HeLa cells twice with 5 mL of 1x PBS. Drain excess PBS completely from the dish by placing the dish vertically for 1 min.
Add 1 mL of PBS containing 200 µM of biotinylated psoralen and 0.01% w/v digitonin, to the cells uniformly and incubate at 37 °C for 5 min. While biotinylated psoralen can enter the cell, its permeability is much higher when the cells are exposed to low amounts of digitonin (0.01%) for 5 min
Remove the lid of the 10 cm plate containing the treated HeLa cells and place the dish flat on ice. Irradiate the dish containing the cells with 365 nm UV for 20 min on ice, at a distance of 3 cm from the UV bulbs, using the UV crosslinker.
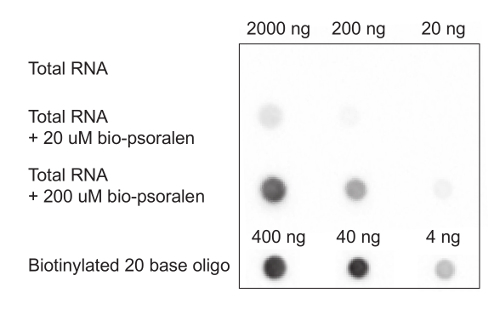
Isolate total RNA from HeLa cells by guanidinium thiocyanate-phenol-chloroform extraction following the manufacturer's protocol. Add 1:1 volume of isopropanol to precipitate the RNA at room temperature for 30 min. Pause Point: RNA can be stored at -20 °C in isopropanol indefinitely. A dot blot can be performed at this step to check that biotinylated psoralen has successfully entered the cells and has crosslinked RNAs in vivo (Figure 2).
Centrifuge the precipitated RNA at 20,000 x g for 30 min at 4 °C to recover the RNA. Remove the supernatant and add 1 mL of 70% cold ethanol to the RNA pellet to wash the RNA.
Centrifuge the samples at 20,000 x g for 15 min at 4 °C. Remove the supernatant and air dry the pellet for 5 min at room temperature.
Add nuclease free water to the RNA pellet and quantify the concentration of RNA using UV spectrometer. Pause Point: RNA can be stored at -80 °C after flash freezing in liquid nitrogen.
2. RNA Fragmentation
Preheat 15 µL of 2x RNA fragmentation buffer and 10 µL of the RNA sample (1 µg/µL) individually in 0.2 mL PCR tubes, at 95 °C for 1 min. Mix 10 µL of the heated 2x RNA fragmentation buffer with 10 µL RNA sample by pipetting up and down. Incubate the sample at 95 °C for 5 min. Stop the reaction by snap cooling the reaction on ice.
Transfer and pool the 2 fragmentation reactions to a 1.5 mL microfuge tube. Add 40 µL of nuclease free water to the reaction, 10 µL of 3 M sodium acetate, 1 µL of glycogen and 300 µL of 100% ethanol to the fragmentation reaction. Incubate the RNA at -20 °C for at least 1 h to precipitate the RNA.
Centrifuge the precipitated RNA at 20,000 x g for 30 min, remove the supernatant and wash the RNA using 1 mL of 70% ethanol.
Centrifuge the RNA at 20,000 x g for 15 min, remove the supernatant and dissolve the RNA in 20 µL of nuclease free water.
3. RNA Size Selection and Elution
Add 20 µL of RNA loading dye to the recovered RNA in the microfuge tube. Heat the RNA with the RNA loading dye at 95 °C for 3 min and place it on ice for 2 min.
Add 3 µL of RNA loading dye to 0.25 µL of 10 nt DNA ladder in a microfuge tube. Heat the ladder at 95 °C for 3 min and place it on ice for 2 min.
Load the denatured ladder and samples onto an 8.6 cm x 6.8 cm 6% TBE urea gel and size fractionate by electrophoresis for 40 min at 180 V. Stain the gel in 10 mL of TBE buffer containing 1:10,000 of nucleic acid gel stain for 5 min in the dark.
Puncture a clean 0.6 mL microfuge tube at the bottom using a 24 G needle and place it in a 2 mL microfuge tube.
Visualize the bands on the post-stained gel using a transilluminator and cut a gel slice corresponding to 90-110 nt. Transfer the gel slice into the punctured 0.6 mL microfuge tube in the 2 mL microfuge tube. This size selection is performed to allow the chimeric fragments to have a minimum length to map to the transcriptome and to distinguish between proximity ligated products versus unligated products in downstream size selection steps.
Centrifuge the gel slices at 12,000 x g at room temperature for 2 min to shred the gel slice and collect it in the 2 mL microfuge tube. Discard the empty 0.6 mL microfuge tube. Add 700 µL of elution buffer to the shredded gel slice in the 2 mL microfuge tube and incubate at 4 °C overnight with constant rotation, to allow diffusion of the samples into the buffer.
Transfer the gel slices and the elution buffer to a centrifuge tube filter and centrifuge at 20,000 x g for 2 min. The gel slices will be trapped at the top compartment of the filter. Discard the gel slices and the top compartment.
Add 1 µL of glycogen and 700 µL of 100% isopropanol to the filtrate in the microfuge tube and precipitate the RNA at -20 °C for at least 1 h or overnight. Pause Point: RNA can be precipitated in isopropanol indefinitely at -20 °C.
Centrifuge the precipitated RNA at 20,000 x g for 30 min at 4 °C to recover the RNA. Remove the supernatant and add 1 mL of 70% cold ethanol to wash the RNA pellet. Centrifuge the washed pellet at 20,000 x g for 15 min at 4 °C.
Remove the wash buffer and add 20 µL of nuclease free water to resuspend the RNA. Quantitate the concentration of RNA using an UV spectrometer. Pause Point: RNA can be stored at -80 °C after flash freezing in liquid nitrogen.
4. Enrichment of RNA Crosslinking Regions
Vortex streptavidin coated magnetic beads vigorously to resuspend the beads homogenously in the tube. Transfer 100 µL of beads to a 1.5 mL microfuge tube and place it on a magnetic stand for 1 min to allow the beads to stick to the magnetic side of the tube. Remove the storage buffer from the beads carefully and take the tube out of the magnet stand.
Add 1 mL Lysis Buffer to the beads in the microfuge tube and pipette up and down. Place the microfuge containing beads on the magnetic stand for 1 min to separate the beads from the wash buffer. Repeat this for a total of 3 washes.
Remove the wash buffer from the beads by placing the microfuge containing beads on the magnetic stand for 1 min. Add RNase inhibitor (1:200) to the lysis buffer and add 100 µL of lysis buffer to the washed beads in the microfuge tube.
Add 2 mL of freshly prepared Hybridization Buffer, 1 mL of supplemented lysis buffer, 1.5 µg of size fractionated RNA and 100 µL of resuspended beads to a 15 mL conical centrifuge tube. Vortex the tube gently.
Incubate the 15 mL conical centrifuge tube at 37 °C for 30 min with end to end rotation. Pre-warm the wash buffer at 37 °C.
Place the 15 mL conical centrifuge tubes on a magnetic stand for 15 mL tubes for 2 min to separate the beads from the buffer. Pipette the buffer carefully out of the tube and discard it.
Add 1 mL of pre-warmed wash buffer to the beads, pipette up and down and transfer the beads to a 1.5 mL microfuge tube. Incubate at the beads at 37 °C for 5 min, with end to end rotation.
Briefly centrifuge down the contents of the microfuge tube. Place the 1.5 mL washed beads in microfuge tubes on a magnetic stand for 2 mL tubes for 1 min, to separate the beads from the wash buffer. Remove the buffer from the beads by gently pipetting the buffer out of the microfuge tube.
Add 1 mL of pre-warmed wash buffer to the beads and pipette up and down to repeat the wash. Perform a total of 5 washes. At the end of the 5th wash, remove the wash buffer by placing the microfuge containing beads on the magnet stand for 1 min.
5. Proximity Ligation
Add 1 mL of cold T4 polynucleotide kinase (PNK) buffer to the washed beads and incubate the beads for 5 min at 4 °C, with end to end rotation. Place the microfuge tube containing the beads on the magnetic strip for 1 min and gently remove the T4 PNK buffer. Repeat this step for a total of 2 washes and remove the T4 PNK buffer after the last wash.
Add buffers to the beads, according to Table 1. To enable the 3' ends of the RNA fragment to be ligated to 3' adapters below, incubate the beads in the buffer at 37 °C for 4 h with constant agitation to convert the 3' cyclic phosphate group at the end of the RNA to 3' OH.
Add buffers to the previous reaction, according to Table 2. To enable the 5' end of the RNA fragments to be ligation competent, incubate the reaction at 37 °C for 1 h with constant agitation in the presence of ATP, to convert 5' OH on the RNA to 5' phosphate.
Add buffer to the previous reaction to perform proximity ligation, according to Table 3. Incubate the reaction at 16 °C for 16 h, with constant agitation,
Place the microfuge containing the ligated RNA onto a magnet stand for 1 min. Remove the supernatant carefully and add 1 mL of room temperature wash buffer to the beads. Resuspend by pipetting up and down.
Place the microfuge tube onto the magnet stand for 1 min and remove the wash buffer carefully. Perform a total of 2 washes, and remove the wash buffer from the beads at the end of the second wash.
Add 100 µL of RNA PK Buffer to the beads and resuspend by pipetting up and down. Heat the samples at 95 °C for 10 min on a heat block.
Chill the sample on ice for 1 min and add 500 µL of guanidinium thiocyanate-phenol-chloroform to the sample. Mix by vortexing vigorously for 10 s. Incubate the mixture at room temperature for 10 min. Pause Point: RNA can be stored in guanidinium thiocyanate-phenol-chloroform at -80 °C for a couple of months.
Add 100 µL of chloroform to the guanidinium thiocyanate-phenol-chloroform extracted samples. Vortex vigorously for 10 s. Centrifuge the samples at 20,000 x g for 15 min at 4 °C.
Transfer 400 µL of the aqueous layer to a new 1.5 mL microfuge tube. Add 800 µL (2 volumes) of 100% ethanol and mix by pipetting up and down.
Transfer the RNA solution to RNA cleanup columns and recover the RNA following manufacturer's instructions, ensuring that even small RNAs are retained. Elute the RNA in 100 µL of nuclease-free water. Pause Point: RNA can be stored at -80 °C after flash freezing in liquid nitrogen.
6. Reverse Crosslinking of Biotinylated Psoralen
Transfer the 100 µL of the eluted samples into a well of a 24 well plate. Remove the cover and irradiate the sample under UV 254 nm, for 5 min on ice.
Transfer the reverse crosslinked sample to a clean microfuge tube. Add 10 µL of sodium acetate, 1 µL of glycogen and 300 µL of 100% ethanol and precipitate the RNA at -20 °C for at least 1 h or overnight. Pause Point: RNA can be precipitated in ethanol indefinitely at -20 °C.
Centrifuge the precipitated RNA at 20,000 x g for 30 min at 4 °C to recover the RNA. Remove the supernatant and add 1 mL of 70% cold ethanol to wash the RNA pellet.
Centrifuge the washed pellet at 20,000 x g for 15 min at 4 °C. Remove the wash buffer and add 4.25 µL of nuclease free water to resuspend the RNA. Transfer the RNA to a 0.2 mL PCR tube. Pause Point: RNA can be stored at -80 °C after flash freezing in liquid nitrogen.
7. Reverse Transcription and cDNA Circularization
Add 0.75 µL of RNA linker (0.5 µg/µL) to the resuspended sample in PCR tube and heat at 80 °C for 90 s. Place the sample on ice for 2 min.
Add the buffers to the denatured sample for 3' adapter ligation, according to Table 4. Incubate the reaction at 25 °C for 2.5 h.
Transfer the reaction to a 1.5 mL microfuge tube. Add 90 µL of nuclease free water to the reaction, followed by 10 µL of sodium acetate, 1 µL of glycogen and 300 µL of 100% ethanol. Precipitate the RNA at -20 °C for at least 1 h or overnight. Pause Point: RNA can be precipitated in ethanol indefinitely at -20 °C.
Centrifuge the precipitated RNA at 20,000 x g for 30 min at 4 °C to recover the RNA. Remove the supernatant and add 1 mL of 70% cold ethanol to wash the RNA pellet.
Centrifuge the washed pellet at 20,000 g for 15 min at 4 °C. Remove the wash buffer resuspend the pellet in 5 µL of nuclease free water.
Add 5 µL of RNA loading dye to the recovered RNA in the microfuge tube. Heat the RNA with the RNA loading dye at 95 °C for 3 min and place it on ice for 2 min.
Add 3 µL of RNA loading dye to 0.25 µL of 10 nt DNA ladder in a microfuge tube. Heat the ladder at 95 °C for 3 min and place it on ice for 2 min.
Perform size fractionation using 8.6 cm x 6.8 cm 6% TBE urea gel as in steps 3.3 and 3.4.
Visualize the gel stained with nucleic acid stain using the transilluminator and cut a gel slice corresponding to 110-140 nt on the ladder. Transfer the gel slice into the punctured 0.6 mL microfuge tube in the 2 mL microfuge tube and follow the protocol from steps 3.6- 3.9.
Add 5 µL of nuclease free water to resuspend the RNA pellet. Transfer the RNA to a 0.2 mL PCR tube. Pause Point: RNA can be stored at -80 °C after flash freezing in liquid nitrogen.
Add 1 µL of RT primer at 1.25 µM to the RNA in PCR tube and heat at 80 °C for 2 min. Place the sample on ice for 2 min.
Add the buffers for reverse transcription according to Table 5. Incubate the reaction at 50 °C for 30 min.
Add 1.1 µL of 1 N NaOH to the reaction and heat at 98 °C for 20 min. Place the reaction on ice.
Transfer the reaction to a 1.5 mL microfuge tube. Add 90 µL of nuclease free water to the reaction, followed by 10 µL of sodium acetate, 1 µL of glycogen and 300 µL of 100% ethanol. Precipitate the DNA at -20 °C for at least 1 h or overnight. Pause Point: DNA can be precipitated in ethanol indefinitely at -20 °C.
Centrifuge the precipitated DNA at 20,000 g for 30 min at 4 °C to recover the DNA. Remove the supernatant and add 1 mL of 70% cold ethanol to wash the DNA pellet. Centrifuge the washed pellet at 20,000 x g for 15 min at 4 °C. Remove the wash buffer resuspend the pellet in 5 µL of nuclease free water.
Perform size fractionation using 8.6 cm x 6.8 cm 6% TBE urea gel as in steps 3.3 and 3.4.
Visualize the post-stained gel using the transilluminator and cut a gel slice corresponding to 200-240 nt on the ladder. The reverse transcribed cDNA is now 96 bases longer than the RNA fragment due to the addition of the 96 base RT primer. Transfer the gel slice into the punctured 0.6 mL microfuge tube in the 2 mL microfuge tube.
Centrifuge the gel slices at 12,000 x g at room temperature for 2 min to shred the gel slice and collect it in the 2 mL microfuge tube. Discard the empty 0.6 mL microfuge tube.
Add 700 µL of elution buffer to the shredded gel slice in the 2 mL microfuge tube and incubate at 37 °C for 2 h with constant rotation. Follow the protocol as described in steps 3.7- 3.8.
Add 6 µL of nuclease free water to resuspend the DNA pellet. Transfer the DNA to a 0.2 mL PCR tube. Pause Point: DNA can be stored at -80 °C after flash freezing in liquid nitrogen.
Add the buffers to the reaction for cDNA circularization, according to Table 6. Incubate the reaction at 60 °C for 1 h and heat at 80 °C for 10 min to inactivate the reaction.
Transfer the reaction to a 1.5 mL microfuge tube. Purify the circularized cDNA using a DNA cleanup column following the manufacturer's instructions. Add 20 µL of nuclease free water to the column to elute the purified cDNA. Pause Point: DNA can be stored at -20 °C for a couple of months.
8. PCR Amplification (Small Scale PCR)
Add the buffers to the reaction according to Table 7 for PCR amplification to test the minimal number of cycles need for library amplification.
Set up the PCR cycling conditions according to Table 8. Pause the reaction and transfer 5 µL of the PCR reaction at 10, 15 and 20 cycles, into separate 1.5 mL microfuge tubes.
- Add 1 µL of 6x DNA gel loading dye to each of the 5 µL PCR reaction at the different cycles. Perform gel electrophoresis on a 3% agarose gel, at 120 V, for 1 h to visualize the PCR products.
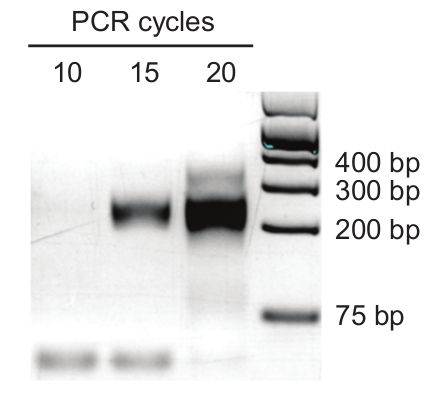
- Use the lowest number of PCR cycles (Y) that show amplification at around 250 bases (In Figure 3, there is a strong band at 15 cycles of amplification and a faint band at 10 cycles. 12 cycles of large scale PCR amplification is likely to generate enough material for deep sequencing as the amount of cDNA used is 10 times that of small scale PCR amplification).
9. PCR Amplification (Large Scale PCR) and Purification
Add the buffers to the reaction according to Table 9 for PCR amplification for large scale PCR.
Set up the PCR cycling conditions according to Table 10.
Add 5 µL of 6x DNA gel loading dye to the 25 µL of PCR reaction and load into a well of a 3% agarose gel. Load 1 kb plus DNA ladder into a separate well. Perform gel electrophoresis at 100 V, for 1.5 h.
Visualize the completed gel using a UV Transilluminator.
Size extract a gel slice containing PCR products from 200- 300 bases and transfer the gel to a clean 2 mL microfuge tube.
Add 1 mL of gel solubility buffer, from a DNA gel extraction kit, to the gel slice and incubate at room temperature for 30 min, or until the gel dissolves, with constant agitation.
Add 200 µL of isopropanol to the dissolved gel and mix well by pipetting. Transfer 700 µL of the sample to a DNA gel extraction cleanup column and centrifuge at 13,000 x g for 30 s. Keep transferring the dissolved sample to the column until all of the sample has been loaded onto the column.
Add 500 µL gel solubility buffer, followed by 700 µL of column wash buffer, to the column, according to the manufacturer's protocol. Add 12 µL of nuclease-free water to the center of the column to elude the DNA. Pause Point: DNA can be stored at -20 °C.
Quantitate the concentration of the DNA using fluorometric quantification. If multiple libraries are constructed and pooled together for sequencing, add equal amount of each sample to the pool, for high throughput sequencing.
Representative Results
Figure 1 depicts the schematic of the SPLASH workflow. Upon the addition of biotinylated psoralen in the presence of 0.01% digitonin, and UV crosslinking, total RNA is extracted from the cells and a dot blot is performed to ensure that crosslinking of biotinylated to the RNA has happened efficiently (Figure 2). We use biotinylated 20 base oligos as positive controls to titrate the amount of biotinylated psoralen to be added to the cells, such that approximately 1 in every 150 bases are crosslinked.
As more PCR duplication events tend to occur with increased PCR amplification cycles, we perform a small scale PCR amplification using different PCR cycles to determine the lowest number of amplification cycles that will provide enough material for deep sequencing. In an efficient library preparation process, we are able to amplify a cDNA sequencing library from a 1.5 µg of size selected RNA input in less than 15 cycles of PCR amplification (Figure 3). The library is then sequenced using 2x 150 base pair reads on a high throughput sequencing machine and the sequencing reads are then processed according to the computational pipeline in Figure 4. The end result is a list of filtered chimeric interactions that includes both intramolecular and intermolecular RNA-RNA interactions in the transcriptome (Table 11).
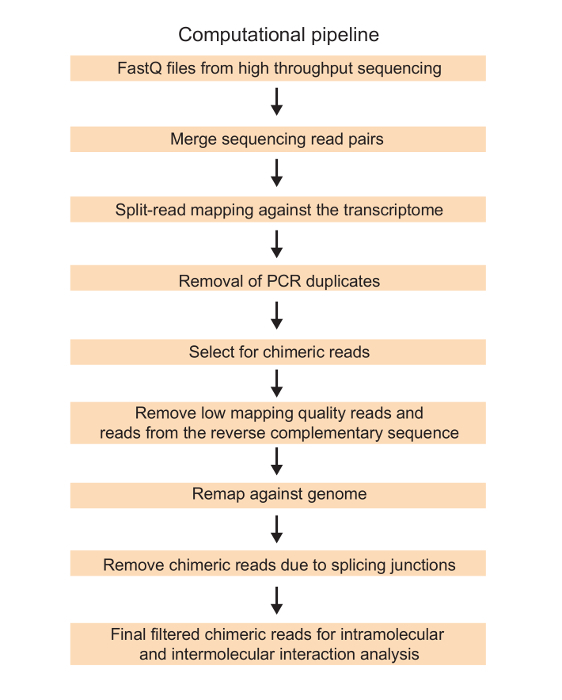
Figure 1: Schematic of the experimental workflow of SPLASH15. Pair-wise RNA interactions inside the cell are crosslinked using biotinylated psoralen under UV light at 365 nm. Crosslinked RNA is then extracted and fragmented to around 100 bases. Interacting regions that contain the biotinylated psoralen crosslinks are enriched by binding to streptavidin beads and ligated together. Upon reverse crosslinking under UV light at 265 nm, the chimeric RNAs are then cloned into a cDNA library for deep sequencing. Please click here to view a larger version of this figure.
Figure 2: Testing biotinylated crosslinking efficiency by dot blotting15. Dot blot of biotinylated psoralen crosslinked RNA in vivo in the presence of 1% digitonin. Different concentrations of crosslinked RNA (20-2,000 ng) are spotted on the membrane. Different concentrations of biotinylated 20 base oligo are spotted as positive controls. Please click here to view a larger version of this figure.
Figure 3: Small scale library amplification for SPLASH. 1 µL of cDNA from the RT reaction is used for small scale PCR. Reactions are taken out at 10, 15 and 20 cycles and run on a 3% agarose gel to determine the minimum number of amplification cycles required for library generation. In this specific example, 10 cycles of amplification using 10 µL of cDNA in a large scale PCR reaction will generate enough amplicons for deep sequencing.
Figure 4: Schematic of the computational workflow of SPLASH. Sequencing reads from high throughput sequencing are mapped to the transcriptome and filtered against poor quality reads, PCR duplicates, duplicate mapping and splicing junction reads using the analysis pipeline. The final output is a list of chimeras that represent both intra- and intermolecular interactions in the transcriptome. Please click here to view a larger version of this figure.
| Reagents | Volume (µL) | Final |
| Nuclease free water | 64 | |
| 10x T4 PNK buffer | 8 | 1x |
| Rnase Inhibitor (20 U/µL) | 4 | 1 U/µL |
| T4 PNK (10 U/µL) | 4 | 0.5 U/µL |
| Total | 80 |
Table 1: Reagents for 3' end repair.
| Reagents | Volume (µL) | Final |
| PNK reaction | 80 | |
| Nuclease free water | 3 | |
| 10x T4 PNK buffer | 2 | 1x |
| 10 mM ATP | 10 | 1 mM |
| T4 PNK (10 U/µL) | 5 | 0.5 U/µL |
| Total | 100 |
Table 2: Reagents for 5' end repair.
| Reagents | Volume (μL) | Final |
| PNK reaction | 100 | |
| 10x T4 RNA ligase buffer | 6 | 1x |
| 10 mM ATP | 6 | 1 mM |
| Rnase inhibitor (20 U/µL) | 4 | 0.5 U/µL |
| T4 Rnl1 (10 U/µL) | 40 | 2.5 U/µL |
| Nuclease free water | 4 | |
| Total | 160 |
Table 3: Reagents for proximity ligation.
| Component | Amount per reaction (µL) | Final |
| RNA and linker | 5 | |
| T4 Rnl2 buffer (10x) | 1 | 1x |
| PEG 8000 (50%, wt/vol) | 3 | 15% (wt/vol) |
| Rnase inhibitor (20 U/µL) | 0.5 | 1 U/µL |
| T4 Rnl2 (tr) (200U/µL) | 0.5 | 10 U/µL |
| Total | 10 |
Table 4: Reagents for adaptor ligation.
| Component | Amount per reaction (µL) | Final |
| Ligation and primer | 6 | |
| First-strand buffer (5x) | 2 | 1x |
| dNTPs (10 mM) | 0.5 | 0.5 mM |
| DTT (0.1 M) | 0.5 | 5 mM |
| Rnase inhibitor (20 U/µL) | 0.5 | 1 U/µL |
| Reverse transcriptase (200 U/µL) | 0.5 | 10 U/µL |
| Total | 10 |
Table 5: Reagents for reverse transcription.
| Component | Amount per reaction (µL) | Final |
| First-strand cDNA | 6 | |
| Single strand DNA ligase buffer (10x) | 1 | 1x |
| Betaine (5 M) | 2 | 1 M |
| MnCl2 (50 mM) | 0.5 | 2.5 mM |
| Single strand DNA ligase (100U/µL) | 0.5 | 5 U/µL |
| Total | 10 |
Table 6: Reagents for circularization of cDNA.
| Component | Amount per reaction (µL) | Final |
| Ligated DNA Product | 1 | |
| Nuclease free Water | 10.5 | |
| High-Fidelity DNA polymerase 2x master mix | 12.5 | 1x |
| Universal PCR Primer (10 µM) | 0.5 | 0.02 µM |
| Index (X) Primer (10 µM) | 0.5 | 0.02 µM |
| Total | 25 |
Table 7: Reagents used for small scale PCR.
| Step | Temperature | Time | Cycles |
| Initial Denaturation | 98 °C | 30 s | 1 |
| Denaturation | 98 °C | 10 s | |
| Annealing | 65 °C | 30 s | 25 |
| Extension | 72 °C | 30 s | |
| Final Extension | 72 °C | 5 min | 1 |
| Hold | 4 °C | ∞ |
Table 8: Conditions used for small scale PCR.
| Component | Amount per reaction (µL) | Final |
| Ligated DNA Product | 5 | |
| Nuclease free Water | 6.5 | |
| High-Fidelity DNA polymerase 2x master mix | 12.5 | 1x |
| Universal PCR Primer (10 µM) | 0.5 | 0.02 µM |
| Index (X) Primer (10 µM) | 0.5 | 0.02 µM |
| Total | 25 |
Table 9: Reagents used for large scale PCR.
| Step | Temperature | Time | Cycles |
| Initial Denaturation | 98 °C | 30 s | 1 |
| Denaturation | 98 °C | 10 s | |
| Annealing | 65 °C | 30 s | Y |
| Extension | 72 °C | 30 s | |
| Final Extension | 72 °C | 5 min | 1 |
| Hold | 4 °C | ∞ |
Table 10: Conditions used for large scale PCR.
 Table 11: Analysis output of the computational pipeline. "SAM flag split read 1" = 0 indicates that the read is mapped to the positive strand of the transcriptome. "RNA 1 identity" refers to the identity of the RNA of the left of the chimera. "RNA 1 start position" refers to the start position to which the read is mapped along RNA 1. "RNA 1 end position" refers to the end position to which the read is mapped along RNA 1. "Mapping score split read 1" refers to the mapping score of the read that was mapped to RNA 1. "Cigar split read 1" indicates the number of bases that was clipped from the read "S" and the number of bases that was mapped to the read "M". "Split read 1" indicates the sequence that was mapped to RNA 1. The same nomenclature was used for the right side of the chimera, which is named as RNA 2. Please click here to view a larger version of this table.
Table 11: Analysis output of the computational pipeline. "SAM flag split read 1" = 0 indicates that the read is mapped to the positive strand of the transcriptome. "RNA 1 identity" refers to the identity of the RNA of the left of the chimera. "RNA 1 start position" refers to the start position to which the read is mapped along RNA 1. "RNA 1 end position" refers to the end position to which the read is mapped along RNA 1. "Mapping score split read 1" refers to the mapping score of the read that was mapped to RNA 1. "Cigar split read 1" indicates the number of bases that was clipped from the read "S" and the number of bases that was mapped to the read "M". "Split read 1" indicates the sequence that was mapped to RNA 1. The same nomenclature was used for the right side of the chimera, which is named as RNA 2. Please click here to view a larger version of this table.
Discussion
Here, we describe in detail the experimental and computational workflow for SPLASH, a method that allows us to identify pair-wise RNA interactions in a genome-wide manner. We have successfully utilized SPLASH in bacterial, yeast and human cultures and anticipate that the strategy can be widely applied to diverse organisms under different cellular states. One of the critical steps in the protocol is to start with at least 20 µg of crosslinked RNA to have adequate material for downstream processes. The RNA is then fragmented to 100 bases and PAGE-size-selected. These steps are important for us to preferentially enrich for ligated chimeras, rather than monomers during the library generation process. We typically recover around 1.5 µg of RNA after fragmentation and the first size selection, and the RNA is then converted into a cDNA library according to the SPLASH workflow. We find that this amount of starting RNA allows us to generate enough material for high throughput sequencing using less than 15 cycles of PCR amplification. As the number of PCR duplication events increases dramatically with increased PCR cycles, keeping the number of amplification cycles low is critical to be able to extract useful, and unique chimeras in the downstream analysis.
In contrast to the other genome-wide psoralen based strategies, SPLASH utilizes a biotinylated version of psoralen to crosslink base-paired RNA fragments to each other. As we titrated the amount of crosslinking to approximately one crosslink per hundred and fifty bases, we can enrich for crosslinked RNA regions by using streptavidin beads after RNA fragmentation. Furthermore, performing enzymatic reactions while the RNAs are bound on beads also allowed us to perform buffer exchanges and washes conveniently. However, one of the limitations of the strategy is that biotinylated psoralen is less efficient at penetrating into cells than psoralen or 4'-aminomethyl trioxsalen (AMT). As such, it is critical to ensure that biotinylated psoralen has entered the cells and crosslinked the RNAs efficiently. We routinely perform dot blots on crosslinked, extracted RNAs, together with biotinylated oligos as positive controls, to ensure that our RNAs are properly crosslinked. In the event that crosslinking is weak, strategies to permeate the cellular membrane, such as adding low concentrations of digitonin (to 0.01%) for 5 min, can be used to allow biotinylated psoralen to enter the cells efficiently. As psoralen absorbs at the same wavelength as nucleic acids (UV 260 nm), we typically use fluorometric quantification systems, rather than UV absorption for quantification of crosslinked RNAs.
One limitation of psoralen based crosslinking strategies is that psoralen preferentially crosslinks at uridines (U). As such, base pairing regions that are U poor might be missed during crosslinking. Hence while detecting a crosslinking event between two strands provide evidence that an interaction is occurring, a lack of crosslinking does not equate to a lack of interaction. In our SPLASH protocol, we capture very little microRNA-mRNA interactions, and low amounts of lncRNA interactions. As microRNA bound mRNAs are likely to be downregulated in gene expression and lncRNAs are typically lowly expressed, their poor representation in our data is primarily due to insufficient sequencing depth. We typically sequence at least 200 million paired end reads per human transcriptome library for each replicate. At this depth, most of our sequencing reads fall on fairly abundant RNAs. We anticipate that the usage of enrichment strategies for specific populations of RNAs will greatly enhance the signal for these relatively low abundant RNAs. One other challenge that we observed in analyzing chimeric data is to distinguish between true interacting chimeric events versus chimeras that are generated from splicing. To prevent contamination of splicing reads in our chimeric list, we filter away all reads that are close to known annotated splicing sites in the transcriptome. We anticipate that with more complete splicing annotations, the final list of chimeras will become even more accurate.
Different from previous strategies that focused on RNA interactions that are specific to a single RNA species or a single RNA binding protein, the ability of SPLASH to map RNA-RNA interactions in a genome-wide manner for all RNAs enables us to study large scale RNA interaction networks and to identify novel intramolecular and intermolecular RNA interactions for the first time. Using SPLASH, we obtained thousands of intramolecular and intermolecular interactions in the human and yeast transcriptomes, allowing us to glimpse into the organization and dynamics of the RNA interactome in vivo. Advances in integrating this long range RNA interaction data into RNA structure modeling algorithms is likely to refine our current models of RNA organization in vivo. Incorporating SPLASH into intermolecular RNA prediction algorithms, such as snoRNA prediction programs, can also improve the accuracy of these predictions. We anticipate that future uses of SPLASH on other complex organisms and dynamic systems will continue to shed light on the intricacies of RNA based gene regulation in biology.
Disclosures
The authors do not have competing financial interests.
Acknowledgments
We thank members of the Wan lab and the Nagarajan lab for informative discussions. N.Nagarajan is supported by funding from A*STAR. Y.Wan is supported by funding from A*STAR and Society in Science-Branco Weiss Fellowship.
References
- Wan Y, Kertesz M, Spitale RC, Segal E, Chang HY. Understanding the transcriptome through RNA structure. Nat.Rev.Genet. 2011;12(9):641–655. doi: 10.1038/nrg3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kertesz M, et al. Genome-wide measurement of RNA secondary structure in yeast. Nature. 2010;467(7311):103–107. doi: 10.1038/nature09322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y, et al. Genome-wide Measurement of RNA Folding Energies. Mol.Cell. 2012;48(2):169–181. doi: 10.1016/j.molcel.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y, Qu K, Ouyang Z, Chang HY. Genome-wide mapping of RNA structure using nuclease digestion and high-throughput sequencing. Nat.Protoc. 2013;8(5):849–869. doi: 10.1038/nprot.2013.045. [DOI] [PubMed] [Google Scholar]
- Wan Y, et al. Landscape and variation of RNA secondary structure across the human transcriptome. Nature. 2014;505(7485):706–709. doi: 10.1038/nature12946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitale RC, et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature. 2015;519(7544):486–490. doi: 10.1038/nature14263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, et al. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature. 2014;505(7485):696–700. doi: 10.1038/nature12756. [DOI] [PubMed] [Google Scholar]
- Rouskin S, Zubradt M, Washietl S, Kellis M, Weissman JS. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature. 2014;505(7485):701–705. doi: 10.1038/nature12894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudla G, Granneman S, Hahn D, Beggs JD, Tollervey D. Cross-linking, ligation, and sequencing of hybrids reveals RNA-RNA interactions in yeast. Proc.Natl.Acad.Sci.U.S.A. 2011;108(24):10010–10015. doi: 10.1073/pnas.1017386108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto Y, et al. hiCLIP reveals the in vivo atlas of mRNA secondary structures recognized by Staufen 1. Nature. 2015;519(7544):491–494. doi: 10.1038/nature14280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engreitz JM, et al. RNA-RNA interactions enable specific targeting of noncoding RNAs to nascent Pre-mRNAs and chromatin sites. Cell. 2014;159(1):188–199. doi: 10.1016/j.cell.2014.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani V, Qiu R, Shendure J. High-throughput determination of RNA structure by proximity ligation. Nat.Biotechnol. 2015. [DOI] [PMC free article] [PubMed]
- Lu Z, et al. RNA Duplex Map in Living Cells Reveals Higher-Order Transcriptome Structure. Cell. 2016;165(5):1267–1279. doi: 10.1016/j.cell.2016.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma E, Sterne-Weiler T, O'Hanlon D, Blencowe BJ. Global Mapping of Human RNA-RNA Interactions. Mol.Cell. 2016;62(4):618–626. doi: 10.1016/j.molcel.2016.04.030. [DOI] [PubMed] [Google Scholar]
- Aw JG, et al. In Vivo Mapping of Eukaryotic RNA Interactomes Reveals Principles of Higher-Order Organization and Regulation. Mol.Cell. 2016;62(4):603–617. doi: 10.1016/j.molcel.2016.04.028. [DOI] [PubMed] [Google Scholar]