

Abstract
The mitochondrial electron transport chain (ETC) transduces the energy derived from the breakdown of various fuels into the bioenergetic currency of the cell, ATP. The ETC is composed of 5 massive protein complexes, which also assemble into supercomplexes called respirasomes (C-I, C-III, and C-IV) and synthasomes (C-V) that increase the efficiency of electron transport and ATP production. Various methods have been used for over 50 years to measure ETC function, but these protocols do not provide information on the assembly of individual complexes and supercomplexes. This protocol describes the technique of native gel polyacrylamide gel electrophoresis (PAGE), a method that was modified more than 20 years ago to study ETC complex structure. Native electrophoresis permits the separation of ETC complexes into their active forms, and these complexes can then be studied using immunoblotting, in-gel assays (IGA), and purification by electroelution. By combining the results of native gel PAGE with those of other mitochondrial assays, it is possible to obtain a completer picture of ETC activity, its dynamic assembly and disassembly, and how this regulates mitochondrial structure and function. This work will also discuss limitations of these techniques. In summary, the technique of native PAGE, followed by immunoblotting, IGA, and electroelution, presented below, is a powerful way to investigate the functionality and composition of mitochondrial ETC supercomplexes.
Keywords: Biochemistry, Issue 124, Mitochondria, electron transport chain, respirasomes, synthasomes, clear native electrophoresis, in-gel assays, electroelution
Introduction
Mitochondrial energy in the form of ATP is not only essential for cell survival, but also for the regulation of cell death. The generation of ATP by oxidative phosphorylation requires a functional electron transport chain (ETC; Cx-I to IV) and mitochondrial ATP synthase (Cx-V). Recent studies have shown that these large protein complexes are organized into supercomplexes, called respirasomes and synthasomes1,2. It is challenging to analyze the assembly, dynamics, and activity regulation of these massive complexes and supercomplexes. While oxygen consumption measurements taken with an oxygen electrode and enzyme assays conducted using a spectrophotometer can give valuable information about ETC complex activity, these assays cannot provide information regarding the presence, size, and subunit composition of the protein complex or supercomplexes involved. However, the development of blue and clear native (BN and CN, respectively) PAGE3 has created a powerful tool for revealing important information about complex composition and assembly/disassembly and about the dynamic regulation of the supramolecular organization of these vital respiratory complexes under physiological and pathological conditions4.
The assembly of these complexes into higher-order supercomplexes appears to regulate mitochondrial structure and function5. For example, respirasome assembly increases the efficiency of electron transfer and the generation of the proton motive force across the mitochondrial inner membrane5. In addition, the assembly of synthasomes not only increases the efficiency of ATP production and the transfer of energy equivalents into the cytoplasm2, but it also molds the mitochondrial inner membrane into the tubular cristae6,7. Studies of supercomplex assembly during cardiac development in mouse embryos show that the generation of Cx-I-containing supercomplexes in the heart begins at about embryonic day 13.58. Others have shown that the amount of Cx-I-containing supercomplexes decreases in the heart due to aging or ischemia/reperfusion injuries9,10 or may play a role in the progression of neurodegenerative diseases11.
This protocol describes methods for native gel PAGE that can be used to investigate the assembly and activity of the ETC complexes and supercomplexes. The approximate molecular weight of mitochondrial supercomplexes can be assessed by separating the protein complexes in CN or BN polyacrylamide gels. CN PAGE also allows for the visualization of the enzymatic activity of all mitochondrial complexes directly in the gel (in-gel assays; IGA)12. This work demonstrates the activity of respirasomes by highlighting the ability of Cx-I to oxidize NADH through IGA and the presence of synthasomes due to the ATP-hydrolyzing activity of Cx-V by IGA. The multiple complexes and supercomplexes containing Cx-I and Cx-V can also be demonstrated by transferring the proteins onto nitrocellulose membranes and performing immunoblotting. The advantage of this approach is that BN or CN PAGE generally separates protein complexes based on their physiological size and composition; the transfer to a membrane preserves this pattern of bands. Analyzing protein complexes in a BN or CN PAGE can also be done using 2D-PAGE (see Fiala et al.13 for a demonstration) or by sucrose density centrifugation14,15. To further analyze a specific band, it can be excised from the BN PAGE, and the proteins from this protein complex can be purified by electroeluting them under native conditions. Native electroelution can be performed within a few hours, which could make a significant difference to the passive diffusion (as used in Reference 16) of proteins from a gel into the surrounding buffer.
In summary, these methods describe several approaches that allow for the further characterization of high-molecular-weight supercomplexes from mitochondrial membranes.
Protocol
All experiments were performed using hearts from C57BL/6N mice (wild type). Mice were anesthetized with CO2 prior to cervical dislocation, and all procedures were performed in strict accordance with the Division of Laboratory Animal Medicine at the University of Rochester and in compliance with state law, federal statute, and NIH policy. The protocol was approved by the Institutional Animal Care and Use Committee of the University of Rochester (University Committee on Animal Resources).
1. CN and BN PAGE
NOTE: All equipment used for BN and CN PAGE must be free of detergent. To ensure this, wash all equipment with 0.1 M hydrochloric acid, followed by extensively rinsing with deionized H2O.
- Preparation
- Prepare anode buffer consisting of 25 mM imidazole, pH 7.0, at 4 °C; store at 4 °C.
- Prepare cathode buffer for CN or BN PAGE.
- For CN PAGE, use 7.5 mM imidazole and 50 mM tricine; add 0.5 g of sodium deoxycholate and 0.2 g of lauryl maltoside per liter of buffer. Adjust the pH to 7.0 at 4 °C and store at 4 °C.
- For BN PAGE, use 7.5 mM imidazole and 50 mM tricine and adjust the pH to 7.0 at 4 °C. Store at 4 °C.
- For light-blue BN cathode buffer, use 7.5 mM imidazole and 50 mM tricine. Adjust the pH to 7.0 at 4 °C and add 20 mg of Coomassie per liter of buffer. Store at 4 °C.
- Prepare extraction buffer (EB) with 50 mM NaCl, 50 mM imidazole, 2 mM aminocaproic acid, and 1 mM EDTA. Adjust the pH to 7.0 at 4 °C and store at 4 °C.
- Prepare 3x gel buffer (used for BN and CN gels) with 75 mM imidazole and 1.5 M aminocaproic acid. Adjust the pH to 7.0 at 4 °C and store at 4 °C.
- Prepare a loading buffer (LB) of 0.01 g of Ponceau S, 5 g of glycerol, and 5 mL of H2O. Store at room temperature.
- Prepare Coomassie blue by adding 50 mg of Coomassie to 1 mL of 500 mM 6-aminohexanoic acid. Adjust the pH to 7.0 at 4 °C and store at 4 °C.
- Prepare 20 mM and 200 mM lauryl maltoside in H2O. Make aliquots of 200 µL and store them frozen. Thaw the detergent before use.
| 3 % to 8 % (mini) | 4 % to 10 % (maxi) | |||
| 0.5 gels (light) | 0.5 gels (heavy) | 0.5 gels (light) | 0.5 gels (heavy) | |
| AAB (mL) | 0.42 | 1.3 | 2.5 | 7.7 |
| CN/BN buffer (mL) | 1.6 | 1.6 | 8.5 | 8.5 |
| H2O (mL) | 2.7 | 1.4 | 14 | 6.3 |
| Glycerol (g) | 0 | 0.47 | 0 | 2.5 |
| Volume (mL) | 4.72 | 4.77 | 25 | 25 |
| APS (µL) | 27 | 27 | 65 | 65 |
| TEMED (µL) | 4 | 4 | 10 | 10 |
Table 1: Quantities of Ingredients Needed to Pour 1 Mini- or Maxi-PAGE. The volumes used in this table are calculated for 1 mini- or 1 maxi-gel, 1.5 mm thick. The volume of AAB is based on a 40% stock solution. Light and heavy refer to the concentration of AAB. APS and TEMED are added after each column of the gradient mixer is filled with AAB solution.
- Pouring and running gels NOTE: Use 3-8% or 4-10% acrylamide/bisacrylamide (AAB) gradient for CN or BN gels, respectively. Table 1 summarizes the quantities of buffer, AAB, H2O, glycerol, ammoniumpersulfate (APS) and tetramethylethylenediamine (TEMED) used for a mini-gel (85 mm wide x 73 mm high x 1.5 mm thick) or maxi-gel (160 mm wide x 200 mm high x 1.5 mm thick). Assemblies of glass plates with CN or BN gels can be refrigerated in a bag with a few mL of 1x gel buffer or wrapped in paper towel wetted with 1x gel buffer for storage. The gels are stable for use for up to a week.
- To pour the gel, place the gradient mixer on an elevated stirring plate to ensure that the gel will flow by gravity into the prepared gel chamber.
- Fill the outflow chamber of the gradient mixer with 4.77/25 mL (mini/maxi) of the heavy solution (with a higher concentration of AAB).
- Gently open the stop-cock connection between the heavy and light chamber and allow a drop of solution to go through to the other side. NOTE: This pushes air bubbles from the connecting tube and stop-cock, which will prevent flow between the two chambers. This cannot be done if both sides have already been filled, because the equal pressure will prevent the bubble from moving through.
- Fill the other chamber of the gradient mixer with 4.72/25 mL (mini/maxi) of the light solution.
- Place a stir bar in the outflow chamber with the heavy solution and begin stirring. Use a stir bar speed that does not cause bubbling.
- Quickly add APS and TEMED to each chamber to initiate polymerization.
- Open the connection between the two chambers of the gradient mixer and allow mixing for a few seconds before opening the outflow chamber to pour the gel. NOTE: Gravity will drain both chambers equally, and mixing of the light into the heavy solution will slowly decrease the acrylamide density from the bottom to the top of the gel. Use the entire content that is in the gradient mixer to pour the gel.
- At the end, carefully mount the comb, with the wells inside the gel to avoid bubbles and layer mixing.
- Immediately wash the gradient mixer with ethanol to rinse out any gel. Rinse with water and pour the second gel. Let the gels polymerize (usually less than 20 min is needed for mini-gels).
- To run the gels, mount them into the electrode assembly clamp and fill the center/upper chamber with CN or light-blue BN cathode buffer. Wait a few minutes to check for leaks before adding anode buffer to the outer/lower chamber.
- Gently pull out the well combs and wash the wells with cathode buffer using a syringe or pipette.
- Run the gels in a cold room (4 °C) or completely packed in ice.
- For CN mini-PAGE, use 100 V for the first hour and 200 V until finished, usually an additional 1-1.5 h. Alternatively, run the CN mini-PAGE at 30-40 V overnight. NOTE: The focus here is on high-molecular-weight protein complexes, so protein complexes with a molecular weight of less than 140 kDa will run out of the gel. Shorter running times for electrophoresis can be used to retain low-molecular-weight complexes.
- For BN maxi-PAGE, use 100 V and run the gels overnight (about 18 h). NOTE: The current will be very low (< 15 mA), so a power supply that can handle these conditions is needed. At this point, the gels can be used for IGA or immunoblotting. In some cases, bands or lanes can be cut out of the gels using a razor blade on a glass plate for electroelution.
- Sample preparation NOTE: Membrane-bound mitochondrial supercomplexes must be extracted from the inner mitochondrial membrane. To preserve mitochondrial supercomplexes, use either freshly isolated mitochondria or samples that have been frozen and thawed only once. The calculations/volumes below are given for mini-gels (the well of a 10-well comb in a gel 1.5 mm thick holds up to 35-40 µL) and maxi-gels (the well of a 15-well comb holds up to 200 µL). In addition, save an aliquot of each sample (usually 10 µL), to be run on a denaturing SDS gel for the detection of the voltage-dependent anion channel (VDAC), as a loading control.
- Place an appropriate amount (e.g., 10-50 µg of protein for mini-gels and 50 - 200 µg for maxi-gels) of isolated mitochondria or tissue homogenate in microtubes and centrifuge at 17,000 x g for 10-15 min at 4 °C. NOTE: This step removes some of the soluble mitochondrial matrix and/or cytosolic proteins.
- Aspirate and discard the supernatant and add extraction buffer to the amount desired to load onto the gel. Gently resuspend the sediment on ice. If desired, add a general protease inhibitor mix at this point. NOTE: Based on the equipment used here, 30 µL was used for a mini-gel and 100 µL for a maxi-gel, limiting the protein/buffer ratio to not more than 2 µg of protein to 1 µL of buffer.
- Add detergent (e.g., 2 µg of lauryl maltoside/1 µg of protein; see the Representative Results and Discussion for more information). NOTE: Generally, lauryl maltoside is used, but digitonin may also be used.
- Incubate on ice for 20 min. Gently mix at the beginning and occasionally during incubation by triturating and/or agitating the tube.
- Centrifuge at 17,000 x g for 10 min at 4 °C to remove any membrane and tissue fragments.
- Transfer the supernatant to a new tube. For CN samples, add 1 µL of LB for every 10 µL of sample volume; the total volume of the sample should be approximately 40 µL for a mini-gel and 130 µL for a maxi-gel. For BN samples, add Coomassie to the samples so that the ratio of dye to detergent is 1:4 (w/w).
- Load 30 and 120 µL of the samples into the wells of the mini- or maxi-gel, respectively. Use the remaining 10 µL from each sample for the denaturing SDS gel to detect VDAC as a loading control.
- To prepare the molecular weight markers, dissolve 1 vial of a high-molecular-weight calibration mix (see the Table of Materials for more information) in 60 µL of BN/CN gel buffer and add 120 µL of H20 and 20 µL of LB. Load 15 µL per lane.
- Run the gels as outlined in step 1.2.9.2.
2. In-gel Assays for Cx-I and Cx-V
NOTE: The assays are performed at room temperature. Take photos, scans, or images of the developing bands for documentation. (Important) Proteins cannot not be transferred onto nitrocellulose membranes after completing an IGA.
- Cx-I assay
- Preparation.
- Prepare an assay buffer of 5 mM Tris in H2O, with a pH of 7.4; store at room temperature.
- Dissolve 10 mg of NADH in 1 mL of assay buffer. Store in aliquots of 100 µL at -20 °C until use; avoid freezing and thawing.
- Weigh 25 mg of Nitroblue tetrazolium into a microtube.
- Prepare the fixative by diluting 5 mL of acetic acid in 95 mL of H2O; store at room temperature.
- Performing the assay.
- Combine 10 mL of assay buffer with 25 mg of Nitroblue tetrazolium (final concentration: 2.5 mg/mL) and 100 µL of 10 mg/mL NADH (0.1 mg/mL). Add this to the entire gel, a lane, or an area of interest excised from a CN gel. NOTE: This can be done in a clear plastic or glass container. Please note that this assay cannot be performed after BN PAGE.
- Follow the development of blue bands after gentle agitation (rocker) for > 3 min.
- Fix the gel in 10 - 20 mL of acetic acid solution or wash in 5 mM Tris, pH 7.4 to stop the reaction. Take photographs for documentation (see the Table of Materials).
- Cx-V assay NOTE: The Cx-V assay should be done using duplicate CN or BN gels, where one is incubated with 5 µg/mL oligomycin (a Cx-V inhibitor) to demonstrate Cx-V-independent activity.
- Preparation.
- Prepare an assay buffer with 35 mM Tris and 270 mM glycine; adjust the pH to 8.3 at room temperature. Store the buffer frozen in 50-mL aliquots, but check the pH after freezing and thawing.
- Prepare 1 M MgSO4 in H2O; store at 4 °C until use.
- Weigh 27.28 mg of Pb(NO3)2 into a microtube.
- Weigh 60 mg of ATP into a microtube.
- Dissolve 1 mg of oligomycin in 1 mL of ethanol. Store at -20 °C until use.
- Prepare a fixative by mixing 50 mL of methanol with 50 mL of H2O; store at room temperature.
- Performing the assay.
- Incubate a gel, a lane, or an area of interest from a BN or CN gel for 2 h with gentle agitation (rocker) in 10-20 mL of assay buffer ± 5 µg/mL oligomycin (50-100 µL of 1 mg/mL in ethanol) at room temperature.
- After incubation, replace the buffer with 14 mL of fresh assay buffer and add, in order, 190 µL of 1 M MgSO4 (14 mM), 27.28 mg of Pb(NO3)2 (5 mM), 60 mg of ATP (8 mM), and 75 µL of oligomycin (where necessary).
- Incubate with gentle agitation (rocker) and watch for a white precipitate, as bands on oligomycin-treated gels will give non-Cx-V-dependent bands. NOTE: The appearance of the precipitate may take several hours.
- Fix the gel in methanol-based fixative (e.g., 50% methanol), because acidic solutions will dissolve the lead precipitate. Photograph the results. NOTE: The lead precipitate does not interfere with the Coomassie staining of the gels.
3. Protein Transfer to Nitrocellulose or Polyvinylidene Difluoride (PVDF) Membranes
- Prepare a transfer buffer of 25 mM Tris and 200 mM glycine. Adjust the pH to 8.3 and add 0.0005 g/L SDS and 200 mL/L methanol. Store at room temperature.
- Cut a nitrocellulose or PVDF membrane (pore size: 0.45 µm) with a razor blade or scissors to a size slightly larger than that of the gel. NOTE: Nitrocellulose membranes allow for Ponceau S staining to assess protein loading, while PVDF membranes yield more sharply defined bands.
- Protocol.
- Soak the nitrocellulose membrane, the filter paper, and the sponges for at least 10 min in transfer buffer. Place the PVDF membrane in 100% methanol for 15 s or according to the recommendation of the manufacturer before placing it in transfer buffer. Place the open cassette of the transfer kit into a flat bowl with transfer buffer.
- Place the sponge and 1-2 layers of filter paper on the back side of the cassette. Remove the bubbles.
- Place the gel on the filter paper. Indicate the orientation of the gel by clipping a corner of the gel and/or the membrane.
- Place the membrane on top of the gel. Remove all bubbles.
- Place filter paper and a sponge on top of the membrane. Remove all bubbles in this sandwich.
- Close the cassette and place it into the cassette holder of the transfer kit.
- Transfer the proteins at 25 V for about 12-18 h.
4. Immunoblotting
- Preparation.
- Prepare a Ponceau Stain (500 mL) by adding 25 mL of acetic acid and 0.5 g of Ponceau S to 475 mL of H2O; store at room temperature (can be reused).
- Prepare Tris-buffered saline (TBS) with 200 mM NaCl, 25 mM Tris-Base, and 2.7 mM KCl. Adjust the pH to 8.0 and store at room temperature.
- Prepare TBS-tween (TBST) by adding 0.5 mL/L Tween 20 to TBS; store at room temperature.
- Prepare milk solids/TBST by dissolving 5 g of milk solids in 100 mL of TBST. Store at 4 °C and use within 3 days.
- Prepare BSA/TBST by dissolving 3 g of bovine serum albumin (BSA, fraction V) in 100 mL of TBST. Store at 4 °C and use within 3 days.
- Protocol.
- When the transfer is finished, place the membrane into Ponceau S solution to visualize all transferred proteins. Label the position of the markers on the membrane with a pencil and document the Ponceau-S-stained membrane by photograph or scan.
- Wash the membrane 3 times for 10 min each with TBS under gentle agitation.
- Block the membrane with milk solids/TBST for 1-2 h at room temperature or overnight in a cold room with gentle agitation.
- Wash the membrane for 10 min with TBST under gentle agitation.
- Incubate with primary antibody overnight in the cold room under gentle agitation. Dilute the antibody (e.g., 1:1,000 for anti-ATP5A and -NDUFB6) in BSA/TBST. NOTE: Most antibodies give a better signal on membranes from native gels when incubated overnight.
- Wash the membrane for 10 min with TBST under gentle agitation.
- Incubate with secondary antibody for at least 60 min at room temperature uner gentle agitation. Dilute the antibody (1:5,000 to 1: 50,000) in milk solids/TBST.
- Wash the membrane 3 times for 10 min at room temperature with TBST uner gentle agitation.
- During the last wash, prepare the enhanced chemoluminescence (ECL) substrate according to the instructions of the manufacturer.
- Incubate the membrane with ECL substrate using instructions provided by the manufacturer.
- Detect the signal on the film using instructions provided by the manufacturer of the ECL substrate.
5. Electroelution
- Preparation.
- Prepare an elution buffer of 25 mM tricine, 3.75 mM imidazole (pH 7.0 at 4 °C), and 5 mM 6-aminohexanoic acid. NOTE: Wear gloves at all times while handling the electroeluter and membrane caps to prevent contamination with external proteins.
- On the day before using the electroeluter for native electroelution, perform the following:
- Soak the membrane caps (cut-off: 3.5 kDa) in elution buffer for 1 h at 60 °C. Transfer the caps to fresh elution buffer and soak them for 12 h or longer in the refrigerator. NOTE: A cut-off molecular weight of 3.5 kDa prevents the loss of small proteins that may easily dissociate from the protein complex of interest.
- Wash the electroeluter module, glass tubes, tank, and lid thoroughly with ethanol. Rinse with water and let the equipment dry.
- On the day of native electroelution, do the following.
- Place a frit in the bottom (frosted) of each glass tube to be used. If necessary, place the glass tube in elution buffer and push the frit from the inside to the bottom of the tube.
- Push the glass tube with the frit into the module of the electroeluter. Wet the grommet with elution buffer and slide the glass tube into place. Ensure that the tops of the glass tubes are even with the grommet.
- Close the empty grommets with stoppers.
- Place a wet membrane cap at the bottom of the silicone adapter and fill the adapter with elution buffer. Slowly pipette the buffer in the adapter up and down to remove any air bubbles around the dialysis membrane.
- Slide the buffer-filled adapter to the bottom of the glass tube. Remove all bubbles that appear on the frit inside the glass tube.
- Fill each glass tube with elution buffer.
- Place the excised bands of the BN PAGE into the glass tubes (see step 1.2.9.3). Cut large pieces into smaller pieces. Ensure that the fill height within the glass tube is around 1 cm.
- Place the entire module into the tank.
- Add about 600 mL of cold elution buffer to the tank. Ensure that the silicon adaptor caps are in the buffer to prevent bubbles at the dialysis membrane.
- Place a stir bar at the bottom of the tank. NOTE: Stirring will prevent bubbles from sticking to the bottom of the dialysis membrane.
- Elute the proteins for 4 h at 350 V in a cold room.
- After the elution is completed, remove the electroeluter module from the buffer tank and place it into a sink or bowl.
- If a stopper was used, remove it to drain the upper buffer chamber. Otherwise, use a large pipette to remove the buffer.
- Remove the buffer from each glass tube and discard. Make sure that the silicone adapter stays in place and that the liquid below the frit is not disturbed or shaken.
- Carefully remove the silicone adapter, together with the membrane cap from the bottom of the glass tube. Pipette the content (about 400 µL) of the silicone cap into a microtube. With another 200 µL of elution buffer, rinse the silicon cap and add to the microtube. Repeat for all glass tubes.
- As the membrane caps may be reused, preserve the membrane caps by placing them into elution buffer containing 0.5 mg/mL sodium azide. Refrigerate them. NOTE: The eluate has low protein concentration and can be concentrated using a centrifugal filter devices.
Representative Results
To visualize mitochondrial supercomplexes, freshly isolated mitochondria from mice were used17,18. Mitochondrial supercomplexes are sensitive to repeated cycles of freezing and thawing, leading to their disintegration, although this may be tolerable for some researchers. If freezing is necessary for storage, to ensure best results, samples should not undergo more than one cycle of freezing and thawing.
To visualize the mitochondrial ETC complexes with BN PAGE, 100 µg of protein from isolated heart mitochondria were loaded onto a 4-10% gel (Figure 1A). The Coomassie stain in the loading and cathode buffer is sufficient to label the protein complexes during the run. Supercomplexes appear after increasing the contrast digitally (not shown). For CN PAGE, two samples of 20 µg of protein from isolated mitochondria were loaded onto a 3 - 8% CN gel and separated (Figure 1B). The CN PAGE was stained with Coomassie and destained to visualize the protein complexes. After increasing the contrast digitally, several protein complexes with a molecular weight greater than the monomer of Cx-I appeared (Figure 1B, right). The concentrations of AAB used allowed for the largest supercomplexes to just enter the gel from the well. However, a gel ending with a gradient of less than 3% AAB is not stable enough to manipulate for transfer or for excising a band or lane. In addition, the low concentration of AAB in the upper parts of the 3-8% CN gels maintains some mobility of the protein complexes, which is important if considering native electroelution19.
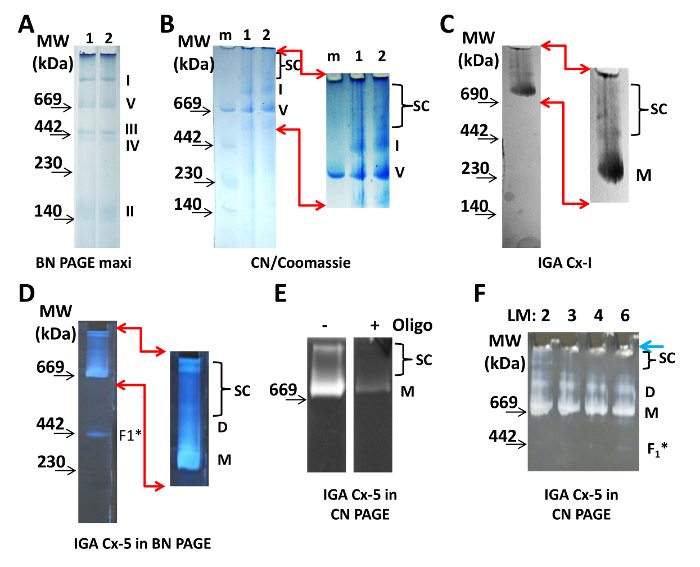
Monomers and supercomplexes of Cx-I and monomers, dimers, and supercomplexes of Cx-V are enzymatically active and can be visualized by IGA (Figure 1C-F). The assays show that, in isolated heart mitochondria, Cx-I and Cx-V are present in protein complexes greater than their respective monomers. In the IGA assay for Cx-I, NADH is oxidized and electrons are transferred to reduce nitroblue tetrazolium. This results in a localized blue color at the molecular weight of the Cx-I monomers and the Cx-I-containing respirasomes/supercomplexes (Figure 1C). The activity of Cx-V is assessed from the ability of the F1 subunit to hydrolyze ATP and can be done using CN or BN gels (Figure 1D-F). The ADP generated from this reaction interacts with lead and results in a white precipitate at the level of Cx-V monomers, dimers, synthasomes, and subcomplexes (most likely the unassembled F1 portion of Cx-V). Note that oligomycin eliminates the labeling of these bands, confirming that they contain Cx-V (Figure 1E).
For all experiments described here, the zwitterionic detergent, lauryl maltoside, was used at a concentration of 2 µg/1 µg of protein, which is the highest possible concentration that preserves the supercomplexes while providing consistent and reproducible results (Figure 1F). However, the effectiveness of lauryl maltoside depends on the lot number, storage conditions, and age. Thus, the exact concentration used in one laboratory is not necessarily the same as reported in manuscripts. The proper concentration of detergent will solubilize the membranes but keep the complexes and supercomplexes intact and must be determined by using a variety of concentrations of lauryl maltoside (Figure 1F). For CN PAGE, a total sample volume of 40 µL per well was prepared here, resulting in a protein/detergent ratio of 1 µg/2 µg or a detergent/buffer ratio of 1 µg/1 µL (equal to 1.9 mM). From the 40 µL, 30 µL was applied per well of the mini-gel; the remaining was used as an aliquot for the detection of VDAC, a loading control (Figure 2D).
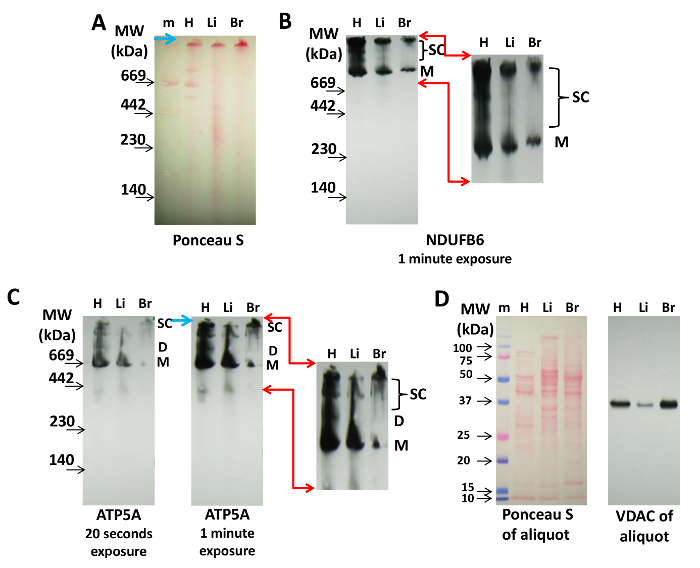
For immunoblotting, CN is preferred to BN PAGE because the proteins are not loaded with Coomassie, which may interfere with detection by antibodies. Figure 2 shows the detection of supercomplexes containing the Cx-I and Cx-V proteins NDUFB6 and ATP5A from isolated heart, liver, and brain mitochondria. Ponceau S labeling after transfer and before immunoblotting can be used to mark the molecular weight markers and to control for protein loading (Figure 2A). This will visualize the monomers of the protein complexes of the ETC on nitrocellulose membranes, but Ponceau S labeling is not always sufficient for the visualization of mitochondrial supercomplexes (Figure 2A), which can be achieved by immunoblotting.
Cx-I does not assemble into dimers and tetramers, per se, but forms increasingly higher molecular-weight supercomplexes with Cx-III and Cx-IV14,20,21. Here, using an antibody against NDUFB6 shows that the sample from the heart contained more Cx-I monomer and high-molecular-weight supercomplexes (top bands) that the mitochondria from the liver or the brain (Figure 2B). The amount of mid-range respirasome supercomplexes was also much higher in the heart than in the other tissues (Figure 2B).
Using anti-ATP5A antibodies, monomers of Cx-V are detectable in mitochondria from all tissues, while a distinct pattern of bands representative of dimers (D) and larger supercomplexes (SC), which are clearly visible in heart mitochondria, are not as prominent in liver and brain mitochondria (Figure 2C). Overexposure (1 min versus 20 s) of the immunoblot shows several Cx-V-containing supercomplexes, which could represent tetramers and synthasomes (Figure 2C). These patterns of Cx-V-containing protein complexes in heart, liver, and brain mitochondria show differences that may be tissue-specific and have not yet been explored.
Protein loading of these blots can be followed by Ponceau S staining and VDAC detection of the abovementioned aliquot by SDS PAGE, as demonstrated in Figure 2D.
Not all antibodies are suitable to detect a protein within the quaternary or tertiary structure of a protein complex after native PAGE. To overcome this problem, entire and partial lanes from the native gel can be mounted on a denaturing gel for a second dimension (2D gels, see Reference 13 for a demonstration). 2D electrophoresis is a valuable tool for visualizing proteins in a supramolecular complex. However, as shown in Figure 2, supercomplexes are present in variable amounts, and the signal of individual proteins from supercomplexes may be hard to visualize in the second, denaturing dimension. To overcome this problem, the electroelution of protein complexes from native gels was used here. This isolates supercomplex bands from multiple lanes to yield more material for further study.
When using electroelution, only the band of interest, which has been identified by IGA and/or visualized on a BN PAGE, is excised; the proteins from this piece of gel are further purified by elution from the gel. Figure 3A shows a lane of a BN PAGE from which bands representing the monomer were excised for electroelution. To assess the Cx-V activity of the monomer after electroelution, the eluate was applied to a second CN PAGE. The eluate of the monomer still contains enzymatically active Cx-V, but subcomplexes also appear (Figure 3B). Silver staining of the eluate after native CN PAGE (Figure 3C) or denaturing SDS PAGE (Figure 3D) indicates the presence of proteins in the eluate, and immunoblotting against ATP5A indicates the presence of Cx-V in both samples (Figure 3D).
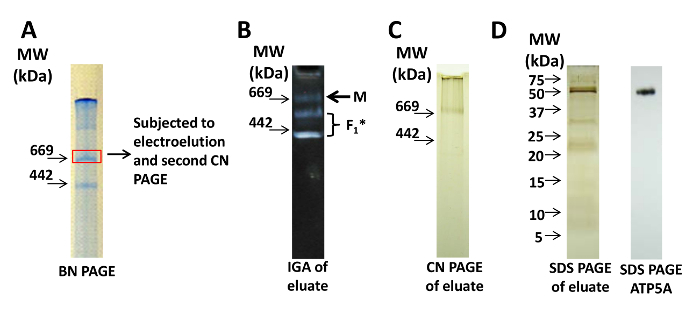
Figure 1: Visualization of Mitochondrial Supercomplexes. (A) Two lanes of a 4-10% BN maxi-gel, with samples of isolated heart mitochondria. Aliquots of the same sample were run in both lanes, at 100 µg of protein per well. The monomers of protein complexes I, II, III, IV, and V of the ETC are clearly visible and are labeled to the right of the gel. (B) Two samples of heart mitochondria (1 and 2, 20 µg of protein/lane) were separated on a CN PAGE and visualized by Coomassie staining. SC indicates the position of the supercomplexes after magnification and digital enhancement of the upper part of the gel (the area is indicated by red arrows). (C) 20 µg of mitochondrial protein were separated on a 3-8% CN PAGE and were processed for Cx-I IGA. Magnified and digitally enhanced images demonstrate bands of Cx-I reaction product. (D) 20 µg of mitochondrial protein were separated on a 3-8% BN PAGE and processed for Cx-V IGA. Magnified and digitally enhanced images demonstrate bands of Cx-V reaction product. (E) 50 µg of mitochondrial protein were separated on a 5-15% CN PAGE and processed for Cx-V IGA without (-) and with (+) 5 µg/mL oligomycin (Oligo). (F) Mitochondrial protein (20 µg/lane) was solubilized with 2 - 6 µg of lauryl maltoside/1 µg of protein, as indicated at the top of the gel, and separated on a 3-8% CN gel followed by Cx-V IGA. All images were photographed using either a light table (A-C) or a black surface (D-F). Camera specifications are in the Table of Materials. The location of the molecular weight markers (MW) are indicated on the left side of all panels. Abbreviations: D = dimers; F1* = subcomplex of Cx-V; LM = lauryl maltoside; m = molecular weight marker; M = monomers; SC = supercomplexes. Please click here to view a larger version of this figure.
Figure 2: Detection of Mitochondrial Supercomplexes by Immunoblotting. 20 µg of protein from the heart (H), liver (Li), and brain (B) mitochondria were separated on a 3-8% CN PAGE and transferred onto nitrocellulose membrane.(A) Ponceau S staining of the membrane indicates the presence of protein complexes and molecular weight markers (m). The blue arrow points to the top of the gel. (B) The Cx-I protein, NDUFB6, was immunolabeled on the blot shown in (A) (1 min exposure time). (C) Cx-V was visualized with anti-ATP5A antibody (20 s and 1-min exposure, as indicated). The red arrows in (B) and (C) indicate the area magnified and digitally enhanced for the visualization of Cx-I- and Cx-V-containing supercomplexes, and the blue arrow points to the top of the gel. (D) Ponceau S and immunolabeling of the VDAC aliquot of each extract used in (A), (B), and (C), which were separated by SDS PAGE and transferred to nitrocellulose. Abbreviations: M = monomers of Cx-I or Cx-V, D = dimers of Cx-V, SC = supercomplexes containing Cx-I or Cx-V. Please click here to view a larger version of this figure.
Figure 3: Electroelution of Cx-V. (A) BN PAGE of a mitochondrial sample. The boxed band represents the monomer of Cx-V that was excised and electroeluted. (B) The eluate was subjected to CN PAGE, and subsequent IGA for Cx-V demonstrates monomers (M) and subcomplexes containing F1 of Cx-V. (C) Silver staining of the CN PAGE of the eluate demonstrates monomers (M). (D) Silver staining (left panel) and immunoblot for ATP5A (right panel) of a denaturing sodium dodecyl sulfate (SDS) PAGE of the eluate indicates the presence of Cx-V. For SDS PAGE, please refer to protocols published elsewhere. Please click here to view a larger version of this figure.
Discussion
A functional ETC is necessary for mitochondrial ATP generation. The complexes of the ETC are able to form two types of supercomplexes: the respirasomes (Cx-I, -III, and -IV)1 and the synthasomes (Cx-V)2. The assembly of each complex is required for an intact ETC, while the organization of the ETC into supercomplexes is thought to increase overall ETC efficiency5,22. How these supercomplexes assemble and disassemble is not well understood, but the protocols presented here may allow for a better understanding of these processes.
The major challenge of studying ETC assembly is the size of these protein complexes. For example, mitochondrial respirasomes may consist of Cx-I (about 880 kDa) and one or more molecules of Cx-III (460 kDa) and Cx-IV (200 kDa, active as a dimer), resulting in a supercomplex with a molecular weight of about 2,000 kDa. In addition, Cx-V has a molecular weight of about 600 kDa but has been shown to assemble into dimers, tetramers, oligomers, and ribbons of dimers7,23, resulting in supercomplexes with a molecular weight of at least 2,000 kDa. Considering the enormous size of these supercomplexes, several partly related approaches have traditionally been used to identify and to characterize these supercomplexes, by this lab and by others8,14,24.
This work has demonstrated the technique of native gel PAGE, where active protein complexes are gently extracted from the mitochondrial membrane using mild detergents. The complexes migrate in the gels primarily due to their size and intrinsic charge (CN PAGE) or due to the size and negative charge of protein-bound Coomassie blue (BN PAGE). These gels can be stained using Coomassie blue (e.g., in CN gels) or silver stain (e.g., in BN and CN PAGE) to reveal proteins bands.
Lauryl maltoside was used for the experiments demonstrated here because this detergent worked most reliably. Alternatively, digitonin can be used to extract protein complexes12. Data from mitochondria isolated from adult hearts, livers, and brains have been shown here, but these techniques have been performed on embryonic and adult heart homogenates8. Others have performed this technique using isolated mitochondria from cultured cells24. However, when using tissue homogenates or cultured cells with a high DNA content, it may be useful to add a nuclease to prevent streaking during electrophoresis25.
The activity of the different complexes can be assayed directly in the gel, as demonstrated here for Cx-I and Cx-V, and techniques are also available to examine the activities of Cx-II, -III, and -IV12. However, analyzing these IGAs must be done carefully. First, the enzymatic reaction could be due to non-ETC enzymes or incompletely assembled complexes. For example, it is routine to perform the Cx-V IGA with a parallel gel that has been treated with oligomycin to inhibit intact Cx-V (Figure 3). In addition, other NADH oxidases may account for Cx-1 in-gel activity. One might perform a parallel IGA in the presence of a Cx-I inhibitor, such as rotenone. However, in Figure 1, isolated mitochondria were used, so cytoplasmic NADH oxidases were not present; thus, the data most likely represents Cx-I-containing monomers and supercomplexes. In addition, the quantification of these results is possible by measuring the signal intensity of the bands on photographs or scans. Drawbacks to this approach include the differences between and within gels, the mobility of the reaction product, the inability to adequately quantify the product throughout the depth of the gel, and the potential non-linearity of the reaction27. To overcome the latter, some have suggested obtaining rates of reactions using serial photographs, but this has not been tried here.
The protein in native gels can also be transferred to membranes, and the protein composition of the bands can be examined by immunoblotting (demonstrated here) or 2D PAGE (demonstrated in reference13). Monomers of the ETC complexes have traditionally been identified by their abundance in native gels of isolated mitochondria, their location in the gel, and their position relative to the molecular marker protein (Figure 1). Furthermore, the labeling of subsequent immunoblots with antibodies specific for subunits of different complexes helps to identify the complex and the composition of supercomplexes. Therefore, anti-NDUFB6 and -ATP5A identify monomers of and supercomplexes containing Cx-1 and Cx-V, respectively, as demonstrated here. Antibodies to Cx-II to IV can be used for the same purpose. In some cases, antibodies that work well in denaturing gels may not work well in native gels due to the fact that some antibodies are specific to denatured protein or that an epitope can be masked by other proteins in a native complex.
The exact determination of the molecular weight of these supercomplexes is difficult, since most available molecular weight markers lie below the size of Cx-V monomers. The migration of protein complexes in a native gel depends on the size, intrinsic charge, and detergent used28. In the examples here, monomers of the complexes were identified based on gels, markers, and immunoblotting. Dimers of Cx-V were identified based on relative migration compared to monomers of Cx-I and -V and on immunoblotting. Everything else above the monomers and dimers in gels, IGAs, and immunoblots are considered to be supercomplexes.
Native immunoblots may also be quantified for supercomplexes by analyzing band density using standard techniques. This method was not shown here, but a recent publication demonstrates this technique8. Normalization of protein loading may be done using the Ponceau red staining of the lane or by saving a sample of the mitochondrial extract to measure the density of the VDAC band on a denaturing immunoblot (Figure 2A and D). Furthermore, examining the ratio of supercomplexes to monomers in the same immunoblot is another way to quantify band density, but one must be careful to take pictures at different times during blot development so that the bands are not over-exposed.
Finally, bands from native gels can be electroeluted to generate purified, active protein complexes that can assemble into higher-order complexes and can be used for further studies of complex function. For example, Cx-V monomers were previously electroeluted and reconstituted into liposomes to demonstrate the electrical functionality29. An alternative approach to native electroelution is elution by passive diffusion into the surrounding buffer16, but this is slow compared to native electroelution. Finally, a major problem with electroelution is maintaining the enzymatic function of the electroeluted protein complex, as the complex may dissociate during purification. Therefore, any assay of function after isolation by this technique would have to be rigorously tested.
By combining these protocols with enzymatic assays and oxygen consumption measurements, which probe the function of individual ETC complexes and the activity of the entire ETC30, and other methods, such as the crystallographic31 and electron microscopic32 evaluation of the structure of complexes and supercomplexes, a completer picture of the inner workings of the ETC can and has emerged. We are closer to understanding how the complexes are assembled, how electrons flow down the chain, and how protons are pumped across the membrane to generate the gradient and then flow back to the matrix through Cx-V to generate ATP. Undoubtedly, these techniques will be further refined to provide additional details about the structure, function, and dynamic regulation of the ETC.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by grants from the American Heart Association Founder's Affiliate [12GRNT12060233] and the Strong Children's Research Center at the University of Rochester.
References
- Lenaz G, Genova ML. Supramolecular organisation of the mitochondrial respiratory chain: a new challenge for the mechanism and control of oxidative phosphorylation. Adv Exp Med Biol. 2012;748:107–144. doi: 10.1007/978-1-4614-3573-0_5. [DOI] [PubMed] [Google Scholar]
- Saks V, et al. Intracellular Energetic Units regulate metabolism in cardiac cells. J Mol Cell Cardiol. 2012;52(2):419–436. doi: 10.1016/j.yjmcc.2011.07.015. [DOI] [PubMed] [Google Scholar]
- Schagger H, Cramer WA, von Jagow G. Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal Biochem. 1994;217(2):220–230. doi: 10.1006/abio.1994.1112. [DOI] [PubMed] [Google Scholar]
- Wittig I, Schagger H. Native electrophoretic techniques to identify protein-protein interactions. Proteomics. 2009;9(23):5214–5223. doi: 10.1002/pmic.200900151. [DOI] [PubMed] [Google Scholar]
- Genova ML, Lenaz G. Functional role of mitochondrial respiratory supercomplexes. Biochim Biophys Acta. 2014;1837(4):427–443. doi: 10.1016/j.bbabio.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Hahn A, et al. Structure of a Complete ATP Synthase Dimer Reveals the Molecular Basis of Inner Mitochondrial Membrane Morphology. Molecular cell. 2016;63(3):445–456. doi: 10.1016/j.molcel.2016.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss M, Hofhaus G, Schroder RR, Kuhlbrandt W. Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J. 2008;27(7):1154–1160. doi: 10.1038/emboj.2008.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutner G, Eliseev RA, Porter GA. Initiation of electron transport chain activity in the embryonic heart coincides with the activation of mitochondrial complex 1 and the formation of supercomplexes. PloS one. 2014;9(11):e113330. doi: 10.1371/journal.pone.0113330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genova ML, Lenaz G. The Interplay Between Respiratory Supercomplexes and ROS in Aging. Antioxid Redox Signal. 2015;23(3):208–238. doi: 10.1089/ars.2014.6214. [DOI] [PubMed] [Google Scholar]
- Rosca MG, et al. Cardiac mitochondria in heart failure: decrease in respirasomes and oxidative phosphorylation. Cardiovasc Res. 2008;80(1):30–39. doi: 10.1093/cvr/cvn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuter K, et al. Adaptation within mitochondrial oxidative phosphorylation supercomplexes and membrane viscosity during degeneration of dopaminergic neurons in an animal model of early Parkinson's disease. Biochim Biophys Acta. 2016;1862(4):741–753. doi: 10.1016/j.bbadis.2016.01.022. [DOI] [PubMed] [Google Scholar]
- Wittig I, Karas M, Schagger H. High resolution clear native electrophoresis for in-gel functional assays and fluorescence studies of membrane protein complexes. Mol Cell Proteomics. 2007;6(7):1215–1225. doi: 10.1074/mcp.M700076-MCP200. [DOI] [PubMed] [Google Scholar]
- Fiala GJ, Schamel WW, Blumenthal B. Blue native polyacrylamide gel electrophoresis (BN-PAGE) for analysis of multiprotein complexes from cellular lysates. J Vis Exp. 2011. [DOI] [PMC free article] [PubMed]
- Acin-Perez R, Fernandez-Silva P, Peleato ML, Perez-Martos A, Enriquez JA. Respiratory active mitochondrial supercomplexes. Molecular cell. 2008;32(4):529–539. doi: 10.1016/j.molcel.2008.10.021. [DOI] [PubMed] [Google Scholar]
- Dudkina NV, Eubel H, Keegstra W, Boekema EJ, Braun HP. Structure of a mitochondrial supercomplex formed by respiratory-chain complexes I and III. Proc Nat Acad Sci USA. 2005;102(9):3225–3229. doi: 10.1073/pnas.0408870102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgio V, et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Nat Acad Sci USA. 2013;110(15):5887–5892. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutner G, Sharma VK, Giovannucci DR, Yule DI, Sheu SS. Identification of a ryanodine receptor in rat heart mitochondria. J Biol Chem. 2001;276(24):21482–21488. doi: 10.1074/jbc.M101486200. [DOI] [PubMed] [Google Scholar]
- Rehncrona S, Mela L, Siesjo BK. Recovery of brain mitochondrial function in the rat after complete and incomplete cerebral ischemia. Stroke. 1979;10(4):437–446. doi: 10.1161/01.str.10.4.437. [DOI] [PubMed] [Google Scholar]
- Schagger H. Blue-native gels to isolate protein complexes from mitochondria. Methods Cell Biol. 2001;65:231–244. doi: 10.1016/s0091-679x(01)65014-3. [DOI] [PubMed] [Google Scholar]
- Althoff T, Mills DJ, Popot JL, Kuhlbrandt W. Arrangement of electron transport chain components in bovine mitochondrial supercomplex I1III2IV1. EMBO J. 2011;30(22):4652–4664. doi: 10.1038/emboj.2011.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer E, et al. Architecture of active mammalian respiratory chain supercomplexes. J Biol Chem. 2006;281(22):15370–15375. doi: 10.1074/jbc.M513525200. [DOI] [PubMed] [Google Scholar]
- Wittig I, Schagger H. Supramolecular organization of ATP synthase and respiratory chain in mitochondrial membranes. Biochim Biophys Acta. 2009;1787(6):672–680. doi: 10.1016/j.bbabio.2008.12.016. [DOI] [PubMed] [Google Scholar]
- Davies KM, et al. Macromolecular organization of ATP synthase and complex I in whole mitochondria. Proc Nat Acad Sci USA. 2011;108(34):14121–14126. doi: 10.1073/pnas.1103621108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapuente-Brun E, et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science. 2013;340(6140):1567–1570. doi: 10.1126/science.1230381. [DOI] [PubMed] [Google Scholar]
- Antonioli P, Bachi A, Fasoli E, Righetti PG. Efficient removal of DNA from proteomic samples prior to two-dimensional map analysis. J Chromatogr A. 2009;1216(17):3606–3612. doi: 10.1016/j.chroma.2008.11.053. [DOI] [PubMed] [Google Scholar]
- Wittig I, Carrozzo R, Santorelli FM, Schagger H. Functional assays in high-resolution clear native gels to quantify mitochondrial complexes in human biopsies and cell lines. Electrophoresis. 2007;28(21):3811–3820. doi: 10.1002/elps.200700367. [DOI] [PubMed] [Google Scholar]
- Glancy B, Balaban RS. Protein composition and function of red and white skeletal muscle mitochondria. Am J Physiol Cell Physiol. 2011;300(6):C1280–C1290. doi: 10.1152/ajpcell.00496.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittig I, Beckhaus T, Wumaier Z, Karas M, Schagger H. Mass estimation of native proteins by blue native electrophoresis: principles and practical hints. Mol Cell Proteomics. 2010;9(10):2149–2161. doi: 10.1074/mcp.M900526-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alavian KN, et al. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Nat Acad Sci USA. 2014;111(29):10580–10585. doi: 10.1073/pnas.1401591111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. IV. The respiratory chain. J Biol Chem. 1955;217(1):429–438. [PubMed] [Google Scholar]
- Zickermann V, et al. Structural biology. Mechanistic insight from the crystal structure of mitochondrial complex I. Science. 2015;347(6217):44–49. doi: 10.1126/science.1259859. [DOI] [PubMed] [Google Scholar]
- Zhu J, Vinothkumar KR, Hirst J. Structure of mammalian respiratory complex I. Nature. 2016;536(7616):354–358. doi: 10.1038/nature19095. [DOI] [PMC free article] [PubMed] [Google Scholar]