

Abstract
Background
Blood clams (Tegillarca granosa) are one of the most commercial shellfish in China and South Asia with wide distribution in Indo-Pacific tropical to temperate estuaries. However, recent data indicate a decline in the germplasm of this species. Furthermore, the molecular mechanisms underpinning reproductive regulation remain unclear and information regarding genetic diversity is limited. Understanding the reproductive biology of shellfish is important in interpreting their embryology development, reproduction and population structure. Transcriptome sequencing (RNA-seq) rapidly obtains genetic sequence information from almost all transcripts of a particular tissue and currently represents the most prevalent and effective method for constructing genetic expression profiles.
Results
Non-reference RNA-seq, an Illumina HiSeq2500 Solexa system, and de novo assembly were used to construct a gonadal expression profile of the blood clam. A total of 63.75 Gb of clean data, with at least 89.46% of Quality30 (Q30), were generated which was then combined into 214,440 transcripts and 125,673 unigenes with a mean length of 1,122.63 and 781.30 base pairs (bp). In total, 27,325 genes were annotated by comparison with public databases. Of these, 2,140 and 2,070 differentially expressed genes (DEGs) were obtained (T05 T08 vs T01 T02 T04, T06 T07 vs T01 T02 T04; in which T01-T04 and T05-T08 represent biological replicates of individual female and male clams, respectively) and classified into two groups according to the evaluation of biological replicates. Then 35 DEGs and 5 sex-related unigenes, in other similar species, were investigated using qRT-PCR, the results of which were confirmed to data arising from RNA-seq. Among the DEGs, sex-related genes were identified, including forkhead box L2 (Foxl2), sex determining region Y-box (Sox), beta-catenin (β-catenin), chromobox homolog (CBX) and Sex-lethal (Sxl). In addition, 6,283 simple sequence repeats (SSRs) and 614,710 single nucleotide polymorphisms (SNPs) were identified from the RNA-seq results.
Conclusions
This study provided the first complete gonadal transcriptome data for the blood clam and allowed us to search many aspects of gene sequence information, not limited to gender. This data will improve our understanding of the transcriptomics and reproductive biology of the blood clam. Furthermore, molecular markers such as SSRs and SNPs will be useful in the analysis of genetic evolution, bulked segregant analysis (BSA) and genome-wide association studies (GWAS). Our transcriptome data will therefore provide important genetic information for the breeding and conservation of germplasm.
Introduction
The blood clam (Tegillarca granosa) belongs to the family Arcidae, and inhabits the intertidal zone of the Indian Ocean and the western Pacific Ocean. As a shellfish with significant economic value, the blood clam has been widely cultivated across countries in Southeast Asia over the years. This species has been farmed in China for many years and is distributed from the Shandong Province to Guangxi Province. Indeed, Zhejiang Province produced over 353 kilotons of blood clam in 2014, accounting for 38% of the total national production[1]. Blood clams are able to tolerate wider ranges of temperature and saline concentrations, and exhibit significant resistance to environmental stress. Consequently, this species is easily bred on a large scale.
Over the past two decades, a reduction in blood clam germplasm resources has become increasingly evident, predominantly due to artificial inbreeding and poor environmental conditions. Collectively, these factors have resulted in reduced genetic heterozygosity, poor resistance, precocious puberty and thin shells. In addition, studies have shown a reduction in sperm and egg quality, as well as a reduction in the fecundity of females, leading to a rapid decline in the number of offspring. Lastly, existing studies have only addressed the medicinal properties of blood clams, including their anti-cancer[2–4], antioxidant[5] and antibacterial[6–8] properties, while research targeting the protection of germplasm resources, such as its growth and reproduction, are scarce. Elucidating the reproductive biology of shellfish is important in understanding the embryonic and individual development, reproduction and population structure of this important commercial resource.
The process of sex determination regulates the differentiation of the original gonad into either testis or ovaries, and includes genetic sex determination (GSD) and environmental sex determination (ESD). Sex differentiation is the evolutionary process that causes genetic sex to create a series of gender characteristics. However, this process is controledmulti-factorial and involves sex chromosomes, chromosome ploidy, as well as other genetic factors. Furthermore, environmental factors, such as light exposure, temperature, nutritional conditions, and reproductive endocrinology can cause sex differentiation to deviate from original intent [9]. Moreover, genetic effects of sex determination have become increasingly obvious over evolutionary periods. Bivalves belonging to primitive species and show extraordinary diversification in terms of sex determination mechanisms, which vary among phylogenetically closely-related species, as well as within a single species[9–10]. Consequently, it is vital for blood clams to build up their own transcriptome, thus laying the foundation for further research in this important species.
All genetic and environmental factors are controlled by sex-related genes in order to regulate sexual development and sex differentiation. There have been many studies carried out examining the sex-related genes of vertebrates. In mammals, sex determining region Y (Sry)[11] and anti-Mullerian hormone (AMH)[11–13] are the two predominant promoters required for testis determination, and the sex determining region Y-box (Sox9)[14–15] is the only target identified for Sry. A number of genes have also been involved in the regulation of Sry and AMH expression, for example, steroidogenic factor 1 (Sf1)[16], GATA4[17–18], Wilms tumor 1 (WT1)[17,19] and Lim homeobox protein 9 (Lhx9)[20]. Indeed, research on a range of species from worms to mammals has shown that doublesex- and Mab-3-related transcription factors (DMRT) are the most well conserved male sex-determining genes[21]. The DMRT family shares a DM domain, which was first identified in Drosophila melanogaster (doublesex; dsx) and Caenorhabditis elegans (mab-3)[22–23] and later in Oryzias latipes (Y-linked DM domainGene: DMY)[24], Xenopuslaevis (W-linked DM domain gene: DMW, ovary-determinating)[25] and DMRT1 in fish, birds and mice[24,26,27]. Compared to testis-related genes, however, genes expressed by the ovary have been studied far less extensively. The most important pathway of ovary development is the Wnt/beta-catenin signaling pathway, including Wnt family member 4 (Wnt4)[28], beta-catenin (β-catenin)[29] and R-spondin1 (Rspo1)[30]. The roles of forkhead box L2 (Foxl2)[31] and the dosage-sensitive sex reversal-adrenal hypoplasia congenital (AHC) critical region on the X chromosomegene 1 (Dax1)[32] has also been extensively studied during ovarian development.
Since the discovery of Dsx and Mab in invertebrates, many other sex-related genes have been identified, such as fruitless (Fru)[33], sex-lethal (Sxl), transformer (Tra, Tra-2)[34] in D. melanogaster and XO lethal (Xol)[35], sex-determination and dosage compensation defect (Sdc)[36], hermaphroditization (Her)[37], Tra[38] and feminization (Fem)[39–40] in C. elegans. In D. melanogaster, Sxl expression in females is activated in a dose-dependent manner by the X chromosome (X:A = 1.0); the activated Sxl transcripts encode functional proteins which cannot be produced in males. Sxl proteins subsequently splice their own transcripts as well as those of Tra, and Tra functions in cooperation with Tra-2 to alternatively splice DsxF and FruF transcripts[34]. Finally, downstream genes encode transcription factors which promote female-specific development. In males, a single dose of X chromosome blocks this cascade of splice regulation, and as a result, DsxM is produced which causes the development of a male-specific pathway [34, 41]. In C. elegans, a double dose of X chromosome represses the activity of Xol-1, stimulating the expression of Sdc, which represents a classical pathway of X chromosome dosage compensation. In the female (hermaphroditic) sex-determination pathway, Sdc inhibits Her-1 to upregulate Tra-2 which is repressed by Her-1 protein. Fem forms a complex with Cullin-2-like ubiquitinligase (Cul-2), which targets Tra-1A for proteasome-mediated degradation. However, Fem is downregulated by Tra-2, and Tra-1A represses mab-3, leading to the transcription of hermaphroditic genes. In males, Fem can combine with Cul-2 and degrade the target Tra-1A, leading to the activation of male-specific genes[36, 38]. Nevertheless, studies involving non-model invertebrates are rare, especially in bivalves[42–45].
RNA-seq is a technique arising from high-throughput sequencing and is commonly used for the analysis of differentially expressed genes (DEGs), functional gene mining, and transcriptional profile construction. This method has proven high throughput, low cost, high accuracy, and rapid processing time [46]. Research involving RNA-seq has focused mainly upon developmental regulation, environmental stress, and biotic stress. In recent years, a range of molluscs have been used in gonadal transcriptome studies[47–51]. However, unlike other families of bivalves, which have doubly uniparental inheritance (DUI) and sex reversal[52–55], the blood clam is a hermaphroditic shellfish, exhibiting 38 diploid chromosomes without sex chromosomes or sex reversal[56]. In the blood clam, sex is more likely to be dominated by the interaction of multiple genes.
In the present study, the transcriptomes of four mature males and females were sequenced in order to search for DEGs which could be used in subsequent research involving protein structure prediction and functional analysis. Such data could be used to identify the specific pathways of sex determination and establish a suitable foundation for population genetic breeding.
Materials and methods
Ethics statement
The blood clam is a new breed of shellfish referred to as ‘No. 1 Yueqing Bay’cultivated by Zhejiang Mariculture Research Institute. Samples were collected from Wenling (28°17’7.83”N, 121°14’25.56”E, Taizhou, China) on June 10th 2015 for scientific purposes. The shellfish were starved for two days in Qingjiang Station (Wenzhou, China) to eliminate effects of the hepatopancreas, and the gonadal tissues were dissected, immediately immersed in liquid nitrogen, packed with dry ice and sent to Biomarker Technologies Corporation for RNA-seq.
Sample collection and RNA extraction
The blood clams collected were mature and approximately two years old. Their shells had a mean length of 30.62±2.28 mm, mean height of 24.33±1.93 mm, mean width of 21.07±1.56 mm and a mean weight of 9.96±2.29 g. The testes of the blood clam were filled with white sperm, while the ovaries were filled with orange eggs, which can be easily identified with the naked eye. Gonadal tissues were dissected, and total RNA was isolated using an EASYspin Plus Tissue and Cell RNA Rapid Extraction Kit (Aidlab, Beijing, China), which also removed genomic DNA. RNA quality was then determined with a 2100 Bioanalyser (Agilent Technologies, CA, USA) and quantified using a NanoPhotometer spectrophotometer (Thermo Fisher Scientific, Wilmington, DE).
cDNA library preparation, Illumina sequencing and quality control
Four RNA samples from each group (males and females) were sent to Biomarker Technologies Corporation (Beijing, China) for cDNA library construction and sequencing. RNA sequencing libraries were generated using the NEBNext® Ultra RNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, MA, U.S.A.) with multiplexing primers, according to the manufacturer’s protocol. cDNA libraries were constructed with average inserts of 200 base pairs (bp). In brief, mRNA was purified from total RNA using NEBNext Oligo d(T)25 beads, and fragmentation was carried out for first strand cDNA and second strand cDNA synthesis. After fragment purification using AMPure XP Beads (Beckman Coulter, Inc.), the short cDNA fragments were subjected to end repair and adapter ligation. Then, ligation reaction was purified for PCR library enrichment by 12–15 PCR cycling. Sequencing was performed via paired-end 25 cycles rapid run on an Illumina HiSeq2500. In addition, Q20, Q30, GC-content and sequence duplication level were calculated to assess the quality of clean data.
De novo assembly, quality control and functional gene annotation
High-quality clean reads were obtained by removing the adaptor sequences, duplicated sequences, ambiguous reads and low-quality reads. De novo assembly was then accomplished using Trinity software. Clean reads were fragmented and recombined into long fragments by overlap named contigs. Related contigs were clustered using TGICL software[57] to yield unigenes that could not be extended on either end, and redundancies were removed to acquire non-redundant unigenes. We then assessed the quality of unigenes by testing the randomness of inserts, insert length and saturation measurement of the transcriptome data. Unigenes were then annotated using blastx against the Nr database (NCBI non-redundant protein sequences), KEGG (Kyoto Encyclopedia of Genes and Genomes), GO (Gene Ontology), and COG (Cluster of Orthologous Groups) to obtain protein functional annotation based upon sequence similarity. ESTScan software[58] was used to determine the sequence direction of unigenes which could not be aligned to any of the above databases. The Blast2GO program[59]was then used to retrieve GO annotations of unigenes with an E-value threshold of 1x105 for further functional categorization, including cellular components, molecular functions and biological processes. Finally, WEGO software[60] was used to plot the distribution of GO functional classification of the unigenes.
Gene expression quantification
Fragments per kilobase of transcript per million mapped reads (FPKM) was applied to evaluate gene expression levels in different samples, thus eliminating the influence of different gene lengths and sequencing levelson the calculation of gene expression.
Correlation assessment of biological replicates
Recent studies have demonstrated that there is biological variability in the expression of genes among different individuals[61]. In order to eliminate biological variability among different individuals, four males and four females (not pooled) were investigated using the same conditions. Pearson’s correlation coefficient (r)[62] was used as an evaluation index for biological replicates; a value of r2 close to 1 indicated a strong correlation between the two repeated samples.
Differentially expressed genes (DEGs)
The DESeq[63]package was used to identify DEGs and P values were corrected using the Benjamini-Hochberg method. An False Discovery Rate (FDR)<0.01 (corrected P values) and |log2FC (Fold Change)|≥1 were set as thresholds in order to identify significant DEGs between two samples. In our study, two groups (T05 T08 versus T01 T02 T04, T06 T07 versus T01 T02 T04; where T01-T04 were females and T05-T08 were males; T03 was eliminated from analysis and males were grouped into T05 + T08 and T06 + T07 according to r2 value) were assigned to acquire DEGs according to the correlation assessment of biological replicates. A volcano plot was created to show the significance of differential genes and a Venn diagram was created to identify similarities and differences between groups. Finally, a hierarchical cluster analysis was performed to display differential expression patterns of genes in different experimental conditions.
Quantitative real-time PCR validation
FDR<0.001 and |log2FC|≥1 were set as thresholds for identifying positive and significant DEGs from transcriptome sequencing data. In total, 40 unigenes, consisting of 16 up-regulated ovarian unigenes and 19 down-regulated testicular unigenes and 5 sex-related unigenes in other similar species, were investigated. 18SrRNA (F:5'-CTTTCAAATGTCTGCCCTATCAACT-3', R:5'-TCCCGTATTGTTATTTTTCGTCACT-3')[64] and β-actin (F:5'-GCCGCTTCTTCATCCTCAT-3', R:5'- GTCGGCAATACCTGGGAAC-3')[65] were used as reference genes. RNA templates were extracted from three mature males and three mature females and were then reverse transcribed to cDNA using the PrimeScriptTMRT reagent kit with gDNA Eraser (TakaRa, Japan) and stored at -20°C to await qRT-PCR.
qRT-PCR was carried with a StepOnePlusTMreal-time PCR system using SYBR®Green I (TakaRa, Japan) according to the manufacturer’s instructions. cDNAs were diluted five-fold for the final amplified templates of target and reference genes. Two-step qRT-PCR cycles were as follows: 95°C for 30s, followed by 40 cycles of 95°C for 5s and 60°C for 30s. The specificity of amplification was measured by melt curve analysis, which needed to be a single peak. Relative expression profiles of target genes were analyzed using the 2-ΔΔCт Meanmethod, where CT values of reference genes were calculated with a geometrical mean. Significant differences (P<0.05) were determined by the Student’s t-test using SPSS17.0.
Simple sequence repeats (SSR) and single nucleotide polymorphism (SNP)
MicroSAtellite (MISA) software[66] was used to detect SSR markers, including mono-, di-, tri-, quad-, penta-, and hexa-nucleotide repeats. Tool Kit (GATK)[67] was used to select SNP loci from our transcriptome using the following parameters: a continuous single base mismatch was not ≤3 within the range of 35bp, while the quality value of SNP was >2.0 after sequence standardization.
Results
Evaluation of biological replicates
Biological replicates were designed to acquire more reliable DEGs and r2 was calculated to evaluate correlation of the samples. Based upon the r2 values illustrated in Table 1, two groups of males versus females (T05 T08 versus T01 T02 T04, T06 T07 versus T01 T02 T04) were constructed, eliminating significantly differential individuals.
Table 1. Correlation analysis between two selected individual blood clams.
| T01 | T02 | T03 | T04 | T05 | T06 | T07 | |
|---|---|---|---|---|---|---|---|
| T02 | 0.8843 | ||||||
| T03 | 0.7863 | 0.8641 | |||||
| T04 | 0.8557 | 0.9085 | 0.7593 | ||||
| T05 | 0.6415 | 0.7548 | 0.7108 | 0.6454 | |||
| T06 | 0.5396 | 0.4673 | 0.4125 | 0.4078 | 0.7390 | ||
| T07 | 0.5991 | 0.5164 | 0.4338 | 0.4442 | 0.7004 | 0.8368 | |
| T08 | 0.4502 | 0.5410 | 0.5555 | 0.4406 | 0.8801 | 0.7267 | 0.6601 |
Values shown represent r2 value. T01-T04 are females and T05-T08 are males. The condition (r2>0.82) was used to eliminate differential individuals. T01, T02, T04 were similar, thus eliminating T03 which was significantly different. T05 is similar to T08 while T06 is similar to T07. However, T05 was not similar to T06. Therefore, we constructed two groups for analysis (T05 T08 versus T01 T02 T04, T06 T07 versus T01 T02 T04) to search for more differentially-expressed genes (DEGs).
De novo assembly and functional gene annotation
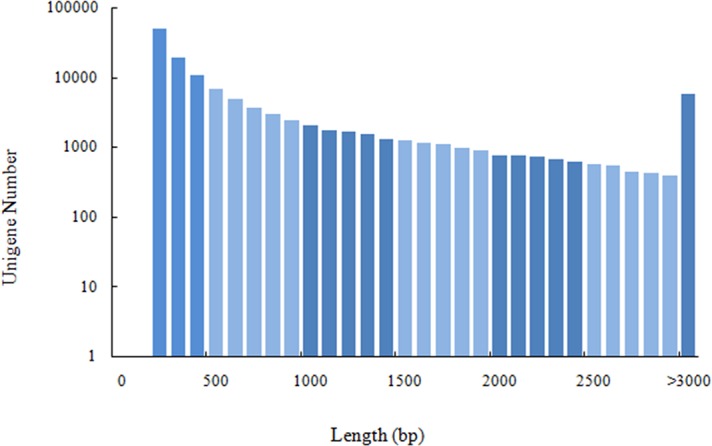
Eight mature blood clam gonads were sequenced, generating 63.75 Gb of clean data with at least 89.46% of Q30 from each sample (Table 2), the data had been submitted to NCBI (accession number: SRR5512703, SRR5515063). All clean data were combined into 214,440 transcripts and 125,673 unigenes with a mean length of 1,122.63 and 781.30 bp. The length distribution of unigenes is shown in Fig 1. Bioinformatic analysis of unigenes produced 27,325 annotated genes, featuring in Nr (97.7%), Swissprot (57.6%), PFAM (65.0%), KOG (53.8%), KEGG (25.1%), GO (24.3%) and COG (25.9%) databases in which there was 48% functional annotations from Crassostrea gigas.
Table 2. Quality control analysis for RNA-seq data.
| Sample | ID | GC (%) | Q20 (%) | Q30 (%) |
|---|---|---|---|---|
| ♀1 | T01 | 37.48 | 94.06 | 89.46 |
| ♀2 | T02 | 37.05 | 94.20 | 89.78 |
| ♀3 | T03 | 36.86 | 94.22 | 89.77 |
| ♀4 | T04 | 37.65 | 94.22 | 89.77 |
| ♂1 | T05 | 36.82 | 94.50 | 90.22 |
| ♂2 | T06 | 37.04 | 94.51 | 90.25 |
| ♂3 | T07 | 37.29 | 94.28 | 89.84 |
| ♂4 | T08 | 36.25 | 94.45 | 90.16 |
Q (Quality) -score represents the accuracy of base recognition, Q-score = -10*log P where P represents the probability of an error in base recognition (Q30>85%), and GC (%) represents the proportion of GC bases of the entire database for each individual blood clam (T01-T08).
Fig 1. Length distribution of unigenes showing assembly quality of the blood clam transcriptome.
DEG analysis
Several groups (T05 T08 versus T01 T02 T04, T06 T07 versus T01 T02 T04) were constructed to analyze DEGs using an FDR<0.01 and a |log2FC|≥1.The former group (T05 T08 versus T01 T02 T04) was identified to have 2,140 DEGs, containing 516 up-regulated and 1,624 down-regulated genes, while the latter (T06 T07 versus T01 T02 T04) had 2,070 DEGs, of which 795 were up-regulated and 1,275 were down-regulated. The relationship between the two groups is shown in Fig 2. In this study, we used the former group for further research, and created a volcano plot, shown in Fig 3. The volcano plot showed that male-biased DEGs were much more prevalent than female-biased DEGs, and that the expression level of male-biased DEGs varied significantly but were very stable in females. Hierarchical cluster analysis showed that the clustering branch displayed the similarity of genes or samples, which conformed to the evaluation of biological replicates (Fig 4).
Fig 2. Venn diagrams for DEG relationships between two groups of blood clam showing the common differentially expressed genes (DEGs) and specific DEGs in each gene set.
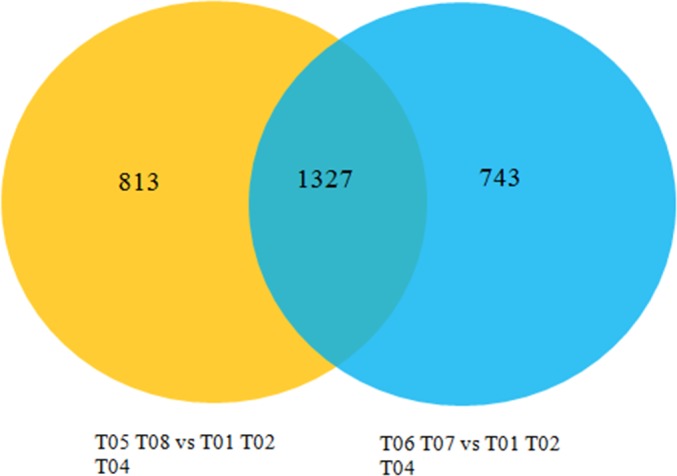
Fig 3. Volcano plot for group analysis of blood clams (T05 T08 versus T01 T02 T04).
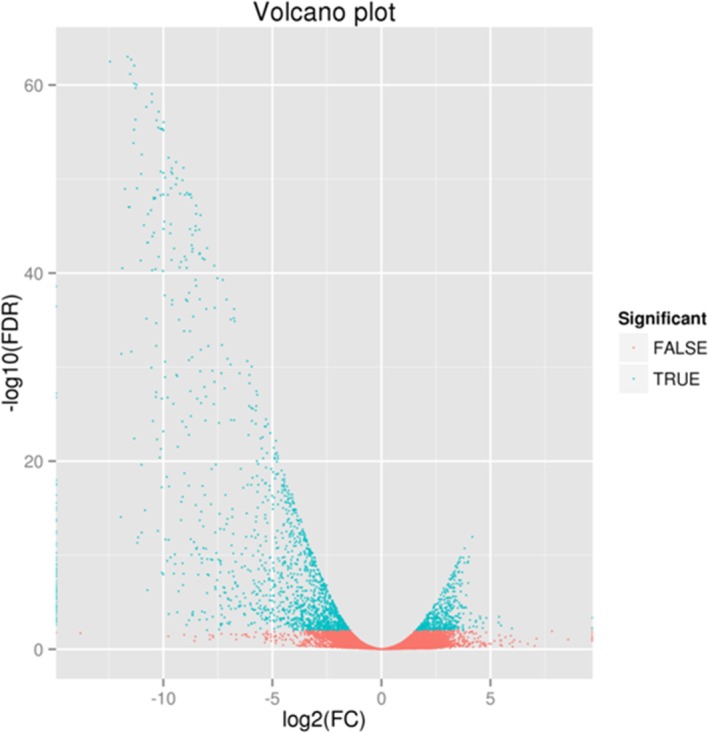
We used specific criteria to identify the significance of differential expression: an FDR (False Discovery Rate)<0.01 and |log2FC (Fold Change)|≥1. The left side of 0 on the x-axis represents male-biased differentially expressed genes (DEGs) while the right side of 0 represents female-biased DEGs. The green region shows significant differences (an FDR<0.01), while the red region shows non-significant differences (an FDR≥0.01).
Fig 4. Hierarchical cluster analysis of blood clams (T01 T02 T03 T04 versus T05 T06 T07 T08).
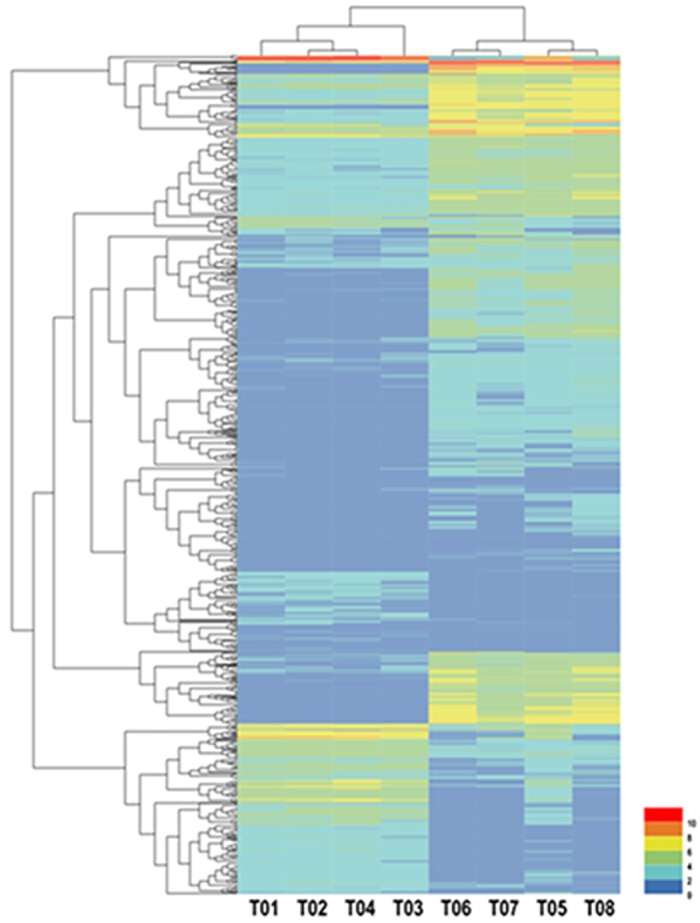
Each column represents a sample, each row represents a gene, and each different color represents log2 (Fragments per kilobase of transcript per million mapped reads (FPKM)) to indicate different expression levels. The clustering branch indicates similarity between genes or samples.
DEG functional annotation (T05 T08 versus T01 T02 T04)
DEGs were aligned to several public databases to obtain functional annotations (E-value≤1×10−5). Consequently, 1346, 910, 971, 726, 307, 332 and 365 DEGs were annotated to Nr, Swissprot, PFAM, KOG, KEGG, GO, and COG databases, respectively.
Analysis of COG annotation
COG, constructed by comparisons from a large number of prokaryotic protein sequences, is an early database for the identification and classification of orthologs. A total of 365 COG annotated DEGs were classified into 25 categories, among which general function prediction only (30.14%) was in the majority, followed by replication, recombination and repair (18.36%), signal transduction mechanisms (14.25%) and transcription (11.51%). COG annotation is shown in Fig 5.
Fig 5. Cluster of Orthologous Groups (COG) functional classification of differentially expressed genes (DEGs) in blood clam (T05 T08 versus T01 T02 T04).
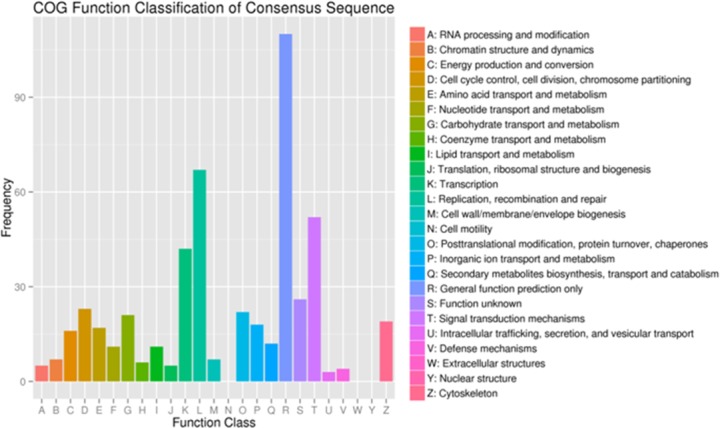
The x-axis shows 25 categories while the y-axis shows the number of DEGs corresponding to each category.
Analysis of GO annotation
GO is an international standardized database to describe features and functions of gene products. A total of 332 DEGs were assigned to 16 cellular components, 17 molecular functions and 19 biological processes. In the cellular component, most of the functions of DEGs were focused within the cell part (28.01%), cell (27.41%), and organelle (23.19%). In terms of molecular function, catalytic activity (55.42%) and binding (49.70%) contributed the largest proportion. With regard to biological processes, metabolic processes (57.23%) and cellular processes (48.80%) represented were the most prevalent. GO annotation is shown in Fig 6.
Fig 6. Gene Ontology (GO) functional classification of differentially expressed genes (DEGs) in blood clam (T05 T08 versus T01 T02 T04).
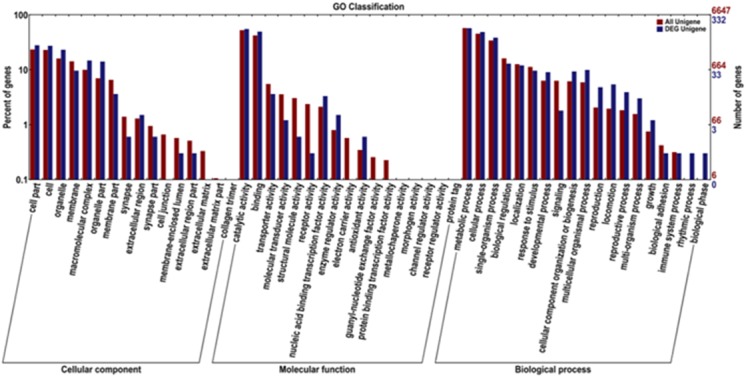
The x-axis shows three terms and 52 sub-terms while the y-axis shows the proportion of DEGs and unigenes corresponding to each subcategory. The red column represents annotation of all genes, while the blue column represents annotation of DEGs. Under the background of the total genes and DEGs, a term having a large number of DEGs may be related to sexual differences.
Analysis of KEGG annotation
KEGG is a database used to analyze the metabolic pathways and function of gene products, which integrates genomics, along with chemical, molecular and biochemical systems. A total of 307 DEGs were noted from KEGG. Of these, 139 DEGs were assigned to six categories, including organismal systems, human disease, metabolism, environmental information processing, genetic information processing and cellular processes. These were then mapped onto 104 pathways including purine metabolism (10.07%), citrate cycle (7.91%) and glycolysis/gluconeogenesis (7.19%), which represented the top three pathways (Fig 7). These data indicated that metabolic pathways play an important role in sex differentiation.
Fig 7. Partial Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway of differentially expressed genes (DEGs) (T05 T08 versus T01 T02 T04).
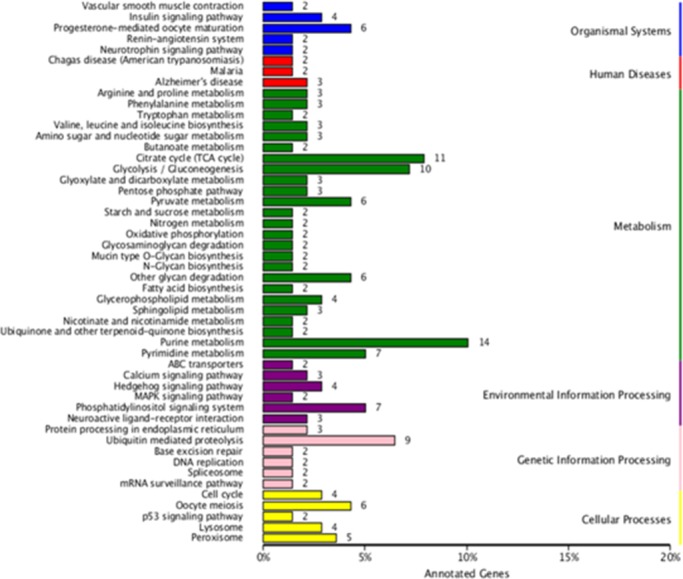
The x-axis shows 50 out of 104 pathways which contain more than one DEG while the y-axis shows the proportion of DEGs corresponding to each pathway.
Quantitative real-time PCR validation
As depicted in Fig 2, both of the two groups owned 1,327 DEGs when an FDR<0.001 and a |log2FC|≥1 were set as screening conditions. In this study, 35 DEGs and five sex-related unigenes in other similar species were investigated. Of the 40 unigenes, we were able to amplify all except for three down-regulated testicular unigenes. Of the 16 male-biased unigenes, M10 was a testis-specific gene while the other 15 genes were down-regulated with a fold change ranging from 3.825 to 40,322.961. Of the 16 female-biased unigenes, F4, F5, F10 were ovary-specific genes while the other 13 were up-regulated genes with a fold change varying from 3.310 to 177.465. Additionally, five sex-related unigenes were tested; in these, the fold change varied between 1.412 and 4.045, except for A5 (29.468) (Table 3). In brief, the qRT-PCR results substantially conformed to those of transcriptome sequencing, except for a few genes which were expressed at levels lower than expected, which may be related to the stability of β-actin and the amplification efficiency of our primers.
Table 3. qRT-PCR validation of transcriptome sequencing.
| Functional classification | ID | Function | RNA-seq | qRT-PCR |
|---|---|---|---|---|
| Transcription | M1 | Sox-14 | 166.664 | 170.386 |
| M2 | Armadillo repeat-containing protein 4 | 11.295 | 53.269 | |
| M3 | Armadillo repeat-containing protein 3 | 6.678 | 14.705 | |
| M4 | Sox-8 | 533.812 | 2987.417 | |
| M5 | forkhead box J1 protein, partial | 79.224 | 3.825 | |
| F1 | Foxl2 | 129.687 | 146.537 | |
| F2 | Forkhead box protein N2 | 115.624 | 133.897 | |
| F3 | Forkhead box E protein, partial | 47.888 | 117.538 | |
| F4 | Spermatogenesis- and oogenesis -specific protein 2 |
23.519 | — | |
| A1 (M) | Sox2 | 5.920 | 1.412 | |
| A2 (F) | β-catenin | 2.203 | 4.045 | |
| A3 (F) | Dax1 | 1.344 | 1.766 | |
| A4 (M) | Sox 9 | 1.593 | 2.174 | |
| A5 (M) | DMRTA2 | 2.468 | 29.468 | |
| Signal transduction mechanisms |
M6 | Testis-specific serine/threonine-protein kinase 1 | 2264.369 | 1482.427 |
| M7 | Troponin C, skeletal muscle | 411.277 | 5803.698 | |
| M8 | Testis-specific serine/threonine-protein kinase 1 | 1939.053 | 1056.228 | |
| M9 | Sperm motility kinase X | 334.901 | 3955.033 | |
| M10 | Testis-specific serine/threonine-protein kinase 5 | 280.606 | — | |
| M11 | Testis-specific serine/threonine-protein kinase 4 | 1832.153 | 4594.291 | |
| Carbohydrate, Lipid, Amino acid transport and metabolism | M12 | Glycogen phosphorylase, muscle form | 1523.181 | 40322.961 |
| M13 | Tax1-binding protein 1-like protein B | 163.139 | 73.838 | |
| F5 | Vitellogenin-6 | 2090.712 | — | |
| F6 | Chymotrypsin-like elastase family member 2A | 17.829 | 2.962 | |
| F7 | Chymotrypsin-like serine proteinase | 17.423 | 7.342 | |
| F8 | Chymotrypsin-like elastase family member 2A | 14.892 | 4.283 | |
| F9 | Chymotrypsin-like serine proteinase | 16.022 | 5.073 | |
| Egg coated protein | F10 | vitelline envelope zona pellucida domain 4 | 129.305 | — |
| F11 | vitelline envelope zona pellucida domain 10 | 12.964 | 7.139 | |
| F12 | vitelline envelope zona pellucida domain 10 | 597.534 | 177.465 | |
| F13 | vitelline envelope zona pellucida domain 10 | 12.382 | 3.310 | |
| Immune-related protein | M14 | Sperm-associated antigen 6 | 20.916 | 41.411 |
| F14 | placenta-specific gene 8 protein-like | 20.263 | 31.953 | |
| F15 | Placental protein 11 | 7.582 | 10.440 | |
| Cell cycle control | M15 | F-box only protein 39 | 350.894 | 6.619 |
| F16 | G2/mitotic-specific cyclin-B | 193.215 | 159.333 | |
| Chromatin structure and dynamics | M16 | Sperm-specific protein PHI-2B/PHI-3 | 716.255 | 2678.482 |
‘–’ represents male or female-specific genes, M represents males, F represents females and fold change indicates the differential change in expression between the two genders.
Sex determination and differentiation genes
We carried out a search of the known sex determination/differentiation genes in animals, focusing mostly on Mus musculus (mouse), Danio rerio (fish), Drosophila melanogaster (fly), Caenorhabditis elegans (worm) and Crassostrea hongkongensis (oyster) (Table 4). In total, 23 out of 43 genes were detected in blood clam transcriptome, and were similar to those of C. hongkongensis[48]. Of these 23 genes, CBX8 and Foxl2 were female-biased, while the family of Sox, armadillo-catenin and Sxl were male-biased. Other common sex determination genes, such as ATRX, DMRT, Dax1, Wnt4, Tra-2, and Fem-1, showed no significant difference between males and females and appeared to express at the highest level during early stages of development and recover to normal levels as animals matured.
Table 4. Candidate genes for sex determination and differentiation in model organism.
| Gene source | C.hongkongensis | T.granosa(homologues) | |
|---|---|---|---|
| WT1 | Mouse | ||
| Sf1 | Mouse | ||
| LHX9 | Mouse | Y | LHX9 |
| EMX2 | Mouse | EMX1 | |
| GATA4 | Mouse | Y | GATA4 |
| SRY | Mouse | ||
| Sox | Mouse | Y |
Sox2*(M), Sox8**(M), Sox9 Sox14**(M) |
| RSP01 | Mouse | ||
| FOG2 | Mouse | ||
| AMH | Mouse | ||
| DMRT | Mouse | DMRTA2 | |
| MAP3K | Mouse | Y | MAP3K1, MAP3K4 |
| ATRX | Mouse | Y | ATRX |
| FGF9 | Mouse | FGF18 | |
| Gadd45g | Mouse | Y | Gadd45g |
| Hhat | Mouse | Y | |
| Kdm3a | Mouse | ||
| Dax1 | Mouse | Y | Dax1 |
| Six | Mouse | Y | Six1, Six3, Six4 |
| GSDF | Mouse | ||
| PDGF | Mouse | ||
| AR | Mouse | ||
| SRD5A1 | Mouse | Y | |
| WNT4 | Mouse | Y | Wnt4 |
| FOXL2 | Mouse | Y | Foxl2**(F) |
| β-catenin | Mouse | Y |
Armadillo/beta-catenin Armadillo/beta-catenin**(M) |
| FST | Mouse | Y | FST-like |
| cyp11b | Fish | ||
| cyp19A1 | Fish | ||
| ERa | Fish | Y | ERa |
| Fhl2 | Fish | Fhl2 | |
| Ixl | Fish | ||
| Xol-1 | Worm | ||
| Sdc | Worm | ||
| Her | Worm | ||
| Tra | Worm | Y | Tra-2 |
| Fem | Worm | Y | Fem-1 |
| CBX | Fly | CBX5, CBX8**(F) | |
| Fru | Fly | ||
| Sis | Fly | ||
| Runt | Fly | Y | Runt |
| Sxl | Fly | Sxl**(M) | |
| Doa | Fly | Y | Doa |
Y represents the presence of these genes or their homologues in C. hongkongensis while the fourth column represents the homologues of these genes in T. granosa. M indicates significant expression in males while F indicates significant expression in females.
* indicates a significant difference (P<0.05) while
** indicates a highly significant difference (P<0.01).
SSRs and SNPs
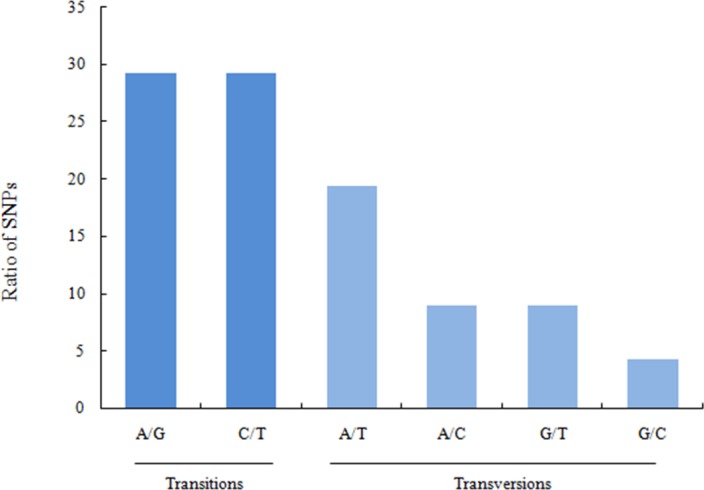
Structural analysis of unigenes detected 6,283 SSR markers, of which di -nucleotide was the most predominant (62.41%), followed by tri- (21.36%), tetra- (2.05%), penta- (0.10%), and hexa-nucleotides (0.03%) (Table 5). In addition, 614,710 SNPs were also detected, including 58.53% transitions (A/G 29.30% and T/C 29.23%) and 41.47% transversions (A/T 19.33%, A/C 8.93%, G/T 8.92%, G/C 4.30%) (Fig 8).
Table 5. Types of simple sequence repeats (SSR) identified in the gonadal transcriptome of the blood clam.
| Repeat motif | Number | Percentage (%) |
|---|---|---|
| Di-nucleotide | ||
| AC/CA/GT/TG | 347/348/424/378 | |
| AG/GA/CT/TC | 200/270/119/162 | |
| AT/TA/GC/CG | 872/800/0/1 | |
| Total | 3921 | 62.41% |
| Tri-nucleotide | ||
| AAC/AAG/AAT(N≥5) | 43/16/139 | |
| ACA/ACC/ACG/ACT | 40/22/3/2 | |
| AGA/AGC/AGG/AGT | 13/1/3/5 | |
| ATA/ATC/ATG/ATT | 95/34/48/88 | |
| CAA/CAC/CAG/CAT | 46/10/8/33 | |
| CCA/CCT/CGC | 16/4/1 | |
| CTA/CTC/CTG/CTT | 4/6/6/6 | |
| GAA/GAC/GAG/GAT | 20/3/6/48 | |
| GCA/GCT/GGA/GGT | 10/6/3/11 | |
| GTA/GTC/GTG/GTT | 3/2/12/18 | |
| TAA/TAC/TAG/TAT | 63/9/3/81 | |
| TCA/TCC/TCG/TCT | 52/4/3/14 | |
| TGA/TGC/TGG/TGT | 66/7/11/36 | |
| TTA/TTC/TTG | 108/15/36 | |
| Total | 1342 | 21.36% |
| Tetra-nucleotide | ||
| AAAC/AAAT | 3/9 | |
| AACA/AATA/AATC/AATT | 3/10/5/1 | |
| ACAG/ACAT/ACGC/ACTG | 1/2/1/1 | |
| AGAA/AGAT/AGTG | 1/1/1 | |
| ATAA/ATAC/ATAG/ATGT | 5/3/2/3 | |
| ATTA/ATTG/ATTT | 1/2/8 | |
| CAAA/CAAC/CTAT/CTGT | 1/2/1/1 | |
| GAAA/GAAT/GACA/GATA | 1/2/1/1 | |
| GTAT/GTCA/GTCC/GTCT | 1/1/1/2 | |
| GTGC/GTTG/GTTT | 1/1/1 | |
| TAAA/TAAT/TACA/TACT | 4/1/1/1 | |
| TATC/TATG/TATT | 3/2/6 | |
| TCAA/TCAT/TCTT | 1/2/1 | |
| TGAC/TGTA/TGTC | 1/1/2 | |
| TTAA/TTAT/TTGA/TTGT | 1/7/1/1 | |
| TTTA/TTTC/TTTG | 7/2/3 | |
| Total | 129 | 2.05% |
| Penta-nucleotide | ||
| AAAAT/AATCC/TGAGT | 1/1/1 | |
| CAAAG/CAGGC/CCAGC | 1/1/1 | |
| Total | 6 | 0.10% |
| Hexa-nucleotide | ||
| TTTTTC/TTATAA | 1/1 | |
| Total | 2 | 0.03% |
| others | 883 | 14.05% |
‘Number’ indicates the number of different types of SSR detected in unigenes while‘Percentage’ indicates the relative proportion of SSRs with different repeat motifs among the total number of SSRs.
Fig 8. Singlenucleotidepolymorphism (SNP) types showing polymorphism of the sexual transcriptome sequence of the blood clam.
Discussion
Blood clams are one of the most economically important marine bivalves. In recent years, the sex ratio of this shellfish has become distorted and female fecundity has decreased, resulting in gamete deformity and a decline in yield. Consequently, elucidating the reproductive biology of shellfish is important in understanding embryonic and individual development, reproduction and population structure. In addition, the sex differentiation and sex regulation mechanisms of bivalves vary significantly, and existing studies on sex-related genes remain insufficient. Therefore, it was necessary to construct a sexual transcriptome for this important species.
Elucidation of the blood clam transcriptome
To build up a gonadal expression profile from the blood clam, 6G of each sample was sequenced using an Illumina HiSeq2500 high-throughput sequencing platform; this identified a total of 6,772,406 contigs, 214,440 transcripts and 125,673 unigenes. Furthermore, 6,283 SSRs and 614,710 SNPs were also determined. Compared with other bivalves, this was the largest gonadal transcriptome data so far; For the blood clam, there was only one existing resource, a microRNA transcriptome associated with resistance response to Cd2+; the present study identified the first transcriptome relating to gonadal expression profile in blood clams, which will provide a useful resource for the future study of mechanisms underlying the function of sex-related genes in bivalves.
DEGs analysis of males and females
DEGs were allocated to 104 pathways by KEGG; the most dominant pathways related to purine metabolism, followed by the citrate cycle and glycolysis/gluconeogenesis. Purine can be converted into ATP which is used for the storage and supply of energy while purines may also be transformed into cAMP and used as a second messenger to regulate metabolism and physiological activities. Glycolysis/gluconeogenesis are processes used to metabolize glycogen into pyruvate and thus produce energy. The DEGs identified in our study which related to these three processes were predominantly male-biased. Therefore, we may draw the conclusion that energy metabolism in males may be higher than females.
The validated DEGs are functionally classified into 7 categories, including Transcription, Signal transduction mechanisms, Carbohydrate, lipid, amino acid transport and metabolism, Egg coated protein, Immune-related protein, Cell cycle control and Chromatin structure and dynamics. Transcription factors serve as sex determination genes which determine the direction of gender differentiation. Genes annotated in signal transduction pathway mostly are kinase with the ability to transfer the different gender development signals intercellularly. Genes annotated in signal transduction pathway mostly are kinase with the ability to transfer the different gender development signals intercellularly. The transport and metabolism genes of Carbohydrate, lipids and amino acids, three important metabolites, are essential for hormone synthesis. Moreover, the egg coated protein and immune-related protein, the cell cycle genes, the chromatin structure and dynamics genes play an irreplaceable roles in protection of gametes and organs, the controlling of cell apoptosis process, and the compression of chromatin, respectively.
Candidate sex-related genes
Sox is a family of transcription factors with a high mobility group (HMG) domain which can bind and bend DNA. The Sox family has more than 20 homologues ranging across different species. In vertebrates, Sry is the main promoter of sexual differentiation and functions only in mammals. Sox9 is the only known target of Sry and over-expression of Sox9 can substitute for the function of Sry during testis determination. Sox8 can reinforce Sox9 function during testis differentiation and can even replace Sox9 when Sox9 is either not expressed or is expressed too late. Moreover, Sox8 and Sox9 are critical for the maintenance of male fertility[14–15]. Other members of the Sox family, such as Sox5, Sox6, and Sox13 also play roles in spermatogenesis. In invertebrates, we were able to identify Sox100B (ID: 45039) in Drosophila melanogaster and Sox8 (SoxE, ID: 105340517) in Crassostrea gigas from NCBI. In our present study, we identified Sox2, Sox8, Sox9, and Sox14, and showed, with the exception of Sox9, that these genes have a significant difference in gene expression when compared between males and females. Although the detailed function of Sox9 is unknown, we speculate that this gene may function during earlier stages, and that Sox8 is more likely to replace the function of Sry in the determination of testes in the blood clam.
Foxl2 is a member of the fork head (FKH) family with a winged helix domain, which was originally identified in Drosophila. Foxl2 functions as an important transcription factor which is indispensable for ovarian development and the growth and maturation of ovarian follicles[31]. In vertebrates, Foxl2 localizes to the granulosa cells and the early ovarian stroma, and knockout of this gene triggers disorder in ovarian follicular formation and partial ovary-to-testis sex reversal. In addition, Foxl2 can upregulate the expression of the P450 aromatase gene which converts androstenedione and testosterone into estrone and estradiol[68], and acts for extended periods of time throughout ovarian development. Homologues of Foxl2 have also been reported in invertebrates[43,69,70]. We also detected Foxl2 in the transcriptome of blood clam. Foxn2 and Foxe, belong to the FKH family and show significant differences among individuals from the two genders, suggesting that these three genes may be involved in ovarian determination, although the precise mechanisms involved remain uncertain.
β-catenin plays a key role in the Rspo1/Wnt signaling pathway which has been associated with ovarian determination. β-catenin has three components: an N-terminal used for GSK-3β phosphorylation, a central region consisting of 12 armadillo (ARM) repeats, and a C-terminal which has a transactivation domain. In mice, the activation of β-catenin by Wnt4 and Rspo1 effectively blocks the testis pathway, leading to male-to-female sex-reversal. Moreover, β-catenin is antagonistic to Sox9, resulting in differentiation towards the female pathway[29]. In mollusks, the Armadillo repeat region is a β-catenin ortholog and has been reported in C.gigas, C.hongkongensis and C. farreri[48, 71–72]. β-catenin is expressed at much higher levels in mature female gonads than those in male gonads. However, many Armadillo repeats are found in the blood clam transcriptome, some of which show no significant difference between males and females, while others are expressed at higher levels in males than in females. Of the genes containing Armadillo repeats, there are many genes recongised as sperm-associated antigens, suggesting that Armadillo repeats may play an important role in spermatogenesis. Therefore, we speculate that Armadillo repeats are more essential in male blood clams compared to females.
CBX (chromobox homolog) genes are members of the PcG family, which are major epigenetic regulators. CBX8 has been described as an epigenetic transcriptional repressor involved in the inhibition of cell senescence, proliferation and metastasis of cancer cells[73–74] while CBX7 is involved in the modulation of cell apoptosis and gene transcription in several cell types[75]. Beyond this, CBX2/M33 functions as a critical factor in controlling the meiotic process of male germ cells[76]. CBX2 is a female-biased gene in the mouse, and the targeted ablation of this gene leads to male-to-female sex reversal[77]. However, CBX2 also functions during testis differentiation by regulating genetic expression of Sry[78]. In the present study, we demonstrated CBX5 and CBX8 to be significantly expressed in the female blood clam. We therefore hypothesize that these two genes are regulators of the developmental process of germ cells, although the specific mechanisms involved remain unknown.
The Sxl gene in Drosophila melanogaster encodes an RNA-binding protein, which controls the regulation of sex determination pathways. In brief, gender in D. melanogaster is determined by the X:A signal; double doses of X in females initiates Sxl expression, and the Sxl protein regulates the splicing of TraFmRNAs into a female-specific form. In addition, the Sxl protein also establishes an auto-regulatory feedback loop to dominate the splicing of SxlFmRNAs, thus maintaining sex stability throughout development. In contrast, a single dose of X in males cannot activate Sxl protein expression, and the subsequent regulatory strategy will be different[41]. In the blood clam transcriptome, a Sxl-like gene was significantly expressed in males, which is contrary to that of D. melanogaster. We speculate that the Sxl regulatory strategy is perhaps not applicable to T. granosa which has no sex chromosomes, and the precise meaning of this key difference is still being investigated.
Our transcriptome is the most complete database for blood clam thus far. We not only identified many homologues of sex-related genes which have been reported in other species, but also found many other unigenes in our current transcriptome which related to our specific commercial needs, such as genes associated with immunity and growth. Our new database provides a relatively complete gene sequence for further analysis and represents a firm foundation for a range of further research studies.
Conclusions
In conclusion, our study provided the first gonadal transcriptome data for blood clam, a commercially important shellfish along the southeast coastline of China. We used non-reference transcriptional sequencing due to the insufficient amount of genetic information available for this species. Based upon COG, GO and KEGG classifications, we were able to elucidate the function of DEGs in specific pathways. This data will be beneficial in improving our understanding of the transcriptomics of blood clams, while SSRs and SNPs will be useful for genetic evolution analysis, bulked segregant analysis (BSA) and genome-wide association studies (GWAS), thus providing a theoretical basis for genetic breeding and the conservation of germplasm.
Acknowledgments
We thank Guangxi Liu (Zhejiang University, China) and Baozhong Liu (Institute of Oceanology, Chinese Academy of Sciences) for providing suggestions, and Biomarker Technologies for RNA-seq and technical consultation.
Data Availability
All transcriptome files of Tegillarca granosa are available from the SRA database (accession number SRR5512703, SRR5515063).
Funding Statement
This work was supported by the earmarked fund for Modern Agro-industry Technology Research System (CARS-47) received by XL Chai; Wenzhou public welfare science and technology project (N20160006) received by SS Teng; Zhejiang public welfare science and technology projuct (2015C33090) received by GQ Xiao; The Seed Science and Technology Innovation Project of Wenzhou City (N20120017) received by GQ Xiao. The funders had a role in study design and decision to publish.
References
- 1.Ministry of Agriculture, Bureau of Fisheries (2015) China fishery statistical yearbook Beijing: China Agriculture Press. [Google Scholar]
- 2.Lv SS, Gao JJ, Liu T, Zhu JH, Xu J, Song L, et al. (2015) Purification and partial characterization of a new antitumor protein from Tegillarca granosa. Mar Drugs 13: 1466–1480. doi: 10.3390/md13031466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu WH, Kong XY, Jiang CQ, Liu XY, Xu L (2015) The anti-tumor effect of a polypeptide extracted from Tegillarca granosa Linnaeus on renal metastatic tumor OS-RC-2 cells. Arch Med Sci 11: 849–855. doi: 10.5114/aoms.2015.53305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen X, Han Y, Zhan S, Wang C, Chen S (2015) Tegillarca granosa extract Haishengsu induces apoptosis in human hepatocellular carcinoma cell line BEL-7402 via Fas-signaling pathways. Cell Biochem Biophys 71: 837–844. doi: 10.1007/s12013-014-0271-3 [DOI] [PubMed] [Google Scholar]
- 5.Li CH, He JJ, Su XR, Li TW (2011) A manganese superoxide dismutase in blood clam Tegillarca granosa: molecular cloning, tissue distribution and expression analysis. Comp Biochem Physiol B Biochem Mol Biol 159: 64–70. doi: 10.1016/j.cbpb.2011.02.003 [DOI] [PubMed] [Google Scholar]
- 6.Bao YB, Li L, Ye MX, Dong YH, Jin WX, Lin ZH, et al. (2013) Expression of glutamine synthetase in Tegillarca granosa (Bivalvia, Arcidae) hemocytes stimulated by Vibrio parahaemolyticus and lipopolysaccharides. Genet Mol Res 12: 1143–1154. doi: 10.4238/2013.April.10.9 [DOI] [PubMed] [Google Scholar]
- 7.Bao YB, Wang Q, Lin ZH (2011) Hemoglobin of the bloody clam Tegillarca granosa (Tg-HbI) is involved in the immune response against bacterial infection. Fish Shellfish Immunol 31: 517–523. doi: 10.1016/j.fsi.2011.05.029 [DOI] [PubMed] [Google Scholar]
- 8.Bao YB, Wang Q, Liu HM, Lin ZH (2011) A small HSP gene of bloody clam (Tegillarca granosa) involved in the immune response against vibrio parahaemolyticus and lipopolysaccharide. Fish Shellfish Immunol, 30: 729–733. doi: 10.1016/j.fsi.2010.12.002 [DOI] [PubMed] [Google Scholar]
- 9.Teaniniuraitemoana V, Lepretre M, Levy P, Vanaa V, Parrad S (2016) Effect of temperature, food availability, and estradiol injection on gametogenesis and gender in the pearl oyster Pinctada margaritifera. J Exp Zool A Ecol Genet Physiol, 325: 13–24. doi: 10.1002/jez.1992 [DOI] [PubMed] [Google Scholar]
- 10.Cai YY, Zhang Y, Wei RF. Overview of the bivalves (1995) Shanghai: Shanghai Science and Technology Press. [Google Scholar]
- 11.Larney C, Bailey TL, Koopman P (2014) Switching on sex: transcriptional regulation of the testis-determining gene Sry. Development 141: 2195–2205. doi: 10.1242/dev.107052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schepers G, Wilson M, Wilhelm D, Koopman P (2003) Sox8 is expressed during testis differentiation in mice and synergizes with SF1 to activate the Amh promoter in vitro. J Biol Chem 278: 28101–28108. doi: 10.1074/jbc.M304067200 [DOI] [PubMed] [Google Scholar]
- 13.Klattig J, Sierig R, Kruspe D, Besenbeck B, Englert C (2007) Wilms' tumor protein Wt1 is an activator of the anti-Müllerian hormone receptor gene Amhr2. Mol Cell Biol, 27: 4355–4364. doi: 10.1128/MCB.01780-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang T, Hou CC, She ZY, Yang WX (2013) The Sox gene family: function and regulation in testis determination and male fertility maintenance. Mol Biol Rep 40: 2187–2194. doi: 10.1007/s11033-012-2279-3 [DOI] [PubMed] [Google Scholar]
- 15.Sekido R, Bar I, Narváez V, Penny G, Lovell-Badge R (2004) Sox9 is up-regulated by the transient expression of SRY specifically in Sertoli cell precursors. Dev Biol 274: 271–279. doi: 10.1016/j.ydbio.2004.07.011 [DOI] [PubMed] [Google Scholar]
- 16.Barbara PD, Mejean C, Moniot B, Malcles MH, Berta P (2001) Steroidogenic factor-1 contributes to the cyclic-adenosine monophosphate down-regulation of human SRY gene expression. Biol Reprod 64: 775–783. [DOI] [PubMed] [Google Scholar]
- 17.Miyamoto Y, Taniguchi H, Hamel F, Silversides DW, Viger RS (2008) A GATA4/WT1 cooperation regulates transcription of genes required for mammalian sex determination and differentiation. BMC Mol Biol 9: 44 doi: 10.1186/1471-2199-9-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yueh-Chiang Hu, Okumura LM, Page DC (2013) Gata4 is required for formation of the genital ridge in Mice. PLoS Genet 9: e1003629 doi: 10.1371/journal.pgen.1003629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hossain A, Saunders GF (2001) The human sex-determining gene Sry is a direct target of WT1. J Biol Chem, 276: 16817–16823. doi: 10.1074/jbc.M009056200 [DOI] [PubMed] [Google Scholar]
- 20.Birk OS, Casiano DE, Wassif CA, Cogliati T, Zhao LP, Zhao Y, et al. (2000) The LIM homeobox gene Lhx9 is essential for mouse gonad formation. Nature 403: 909–913. doi: 10.1038/35002622 [DOI] [PubMed] [Google Scholar]
- 21.Kopp A (2012) Dmrt genes in the development and evolution of sexual dimorphism. Trends Genet 28: 175–184. doi: 10.1016/j.tig.2012.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Erdmanse Burtis KC (1993) The Drosophila doublesex proteins share a novel zinc finger related DNA binding domain. EMBO J 12: 527–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen MM, Hodgkin J (1988) mab-3, a gene required for sex-specific yolk protein expression and a male-specific lineage in C. elegans. Cell 54: 1019–1031. [DOI] [PubMed] [Google Scholar]
- 24.Kobayashi T, Matsuda M, Kajiura-Kobayashi H, Suzuki A, Saito N, Nakamoto M, et al. (2004) Two DM domain genes, DMY and DMRT1, involved in testicular differentiation and development in the medaka, Oryzias latipes. Dev Dynam 231: 518–526. [DOI] [PubMed] [Google Scholar]
- 25.Yoshimoto S, Okada E, Umemoto H, Tamura K, Uno Y, Nishida-Umehara C, et al. (2008) A W-linked DM-domain gene, DM-W, participates in primary ovary development in Xenopus laevis. Proc Natl Acad Sci USA 105: 2469–2474. doi: 10.1073/pnas.0712244105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shetty S, Kirby P, Zarkower D, Graves JAM (2002) DMRT1 in a ratite bird: evidence for a role in sex determination and discovery of a putative regulatory element. Cytogenet Genome Res 99: 245–251. doi: 71600 [DOI] [PubMed] [Google Scholar]
- 27.Zhao L, Svingen T, Ng ET, Koopman P (2015) Female-to-male sex reversal in mice caused by transgenic overexpression of Dmrt1. Development 142: 1083–1088. doi: 10.1242/dev.122184 [DOI] [PubMed] [Google Scholar]
- 28.Naillat F, Yan WY, Karjalainen R, Liakhovitskaia A, Samoylenko A, Xu Q, et al. (2015) Identification of the genes regulated by Wnt-4, a critical signal for commitment of the ovary. Exp Cell Res 332: 163–178. doi: 10.1016/j.yexcr.2015.01.010 [DOI] [PubMed] [Google Scholar]
- 29.Maatouk DM, DiNapoli L, Alvers A, Parker KL, Taketo MM, Capel B, et al. (2008) Stabilization of beta-catenin in XY gonads causes male-to-female sex-reversal[J]. Hum Mol Genet 17: 2949–2955. doi: 10.1093/hmg/ddn193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lavery R, Chassot AA, Pauper E, Gregoire EP, Klopfenstein M, de Rooij DG, et al. (2012) Testicular differentiation occurs in absence of R-spondin1 and Sox9 in mouse sex reversals. PLoS Genet 8: e1003170 doi: 10.1371/journal.pgen.1003170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pisarska MD, Barlow G, Kuo FT (2011) Minireview: roles of the forkhead transcription factor Foxl2 in granulosa cell biology and pathology. Endocrinology 152: 1199–1208. doi: 10.1210/en.2010-1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Swain A, Narvaez S, Burgoyne P, Camerino G, Lovell-Badge R (1998) Dax1 antagonizes Sry action in mammalian sex determination. Nature 391: 761–767. doi: 10.1038/35799 [DOI] [PubMed] [Google Scholar]
- 33.Gailey DA, Billeter JC, Liu JH, Bauzon F, Allendorfer JB, Goodwin SF, et al. (2006) Functional conservation of the fruitless male sex-determination gene across 250 Myr of insect evolution. Mol Biol Evol 23: 633–643. doi: 10.1093/molbev/msj070 [DOI] [PubMed] [Google Scholar]
- 34.Sun X, Yang HW, Sturgill D, Oliver B, Rabinow L, Samson ML, et al. (2015) Sxl-dependent, tra/tra2-independent alternative splicing of the Drosophila melanogaster X-linked gene found in neurons. G3 (Bethesda) 5: 2865–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hargitai B, Kutnyánszky V, Blauwkamp TA, Steták A, Csankovszki G, et al. (2009) Xol-1, the master sex-switch gene in C. elegans, is a transcriptional target of the terminal sex-determining factor TRA-1. Development 136: 3881–3887. doi: 10.1242/dev.034637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davis TL, Meyer BJ (1997) SDC-3 coordinates the assembly of a dosage compensation complex on the nematode X chromosome. Development 124: 1019–1031. [DOI] [PubMed] [Google Scholar]
- 37.Streit A, Li WQ, Robertson B, Schein J, Kamal IH, Marra M, et al. (1999) Homologs of the Caenorhabditis elegans masculinizing gene her-1 in C. briggsae and the filarial parasite brugia malayi. Genetics 152: 1573–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.HamaokaBY Dann CE, Geisbrecht BV Leahy DJ (2004) Crystal structure of Caenorhabditis elegans Her-1 and characterization of the interaction between Her-1 and Tra-2A. Proc Natl Acad Sci U S A 101: 11673–11678. doi: 10.1073/pnas.0402559101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Doniacht, Hodgkin J (1984) A sex-determining gene, fem-1, required for both male and hermaphrodite development in Caenorhabditis elegans. Dev Biol 106(1): 223–235. [DOI] [PubMed] [Google Scholar]
- 40.ChinSangID Spence AM (1996) Caenorhabditis elegans sex-determining protein Fem-2 is a protein phosphatase that promotes male development and interacts directly with Fem-3. Genes Dev 10: 2314–2325. [DOI] [PubMed] [Google Scholar]
- 41.Gempe T, Beye M (2011) Function and evolution of sex determination mechanisms, genes and pathways in insects. Bioessays 33: 52–60. doi: 10.1002/bies.201000043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li HL, Zhang ZF, Bi Y, Yang DD, Zhang LT, et al. (2014) Expression characteristics of beta-catenin in Scallop Chlamys farreri gonads and its role as a potential upstream gene of Dax1 through canonical Wnt signalling pathway regulating the spermatogenesis. PLoS One 9: e115917 doi: 10.1371/journal.pone.0115917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Naimi A, Martinez AS, Specq ML, Diss B, Mathieu M, et al. (2009) Molecular cloning and gene expression of Cg-Foxl2 during the development and the adult gametogenetic cycle in the oyster Crassostrea gigas. Comp Biochem Physiol B Biochem Mol Biol 154: 134–142. doi: 10.1016/j.cbpb.2009.05.011 [DOI] [PubMed] [Google Scholar]
- 44.Santerre C, Sourdaine P, Adeline B, Martinez AS (2014) Cg-SoxE and Cg-beta-catenin, two new potential actors of the sex-determining pathway in a hermaphrodite lophotrochozoan, the Pacific oyster Crassostrea gigas. Comp Biochem Physiol A Mol Integr Physiol 167: 68–76. doi: 10.1016/j.cbpa.2013.09.018 [DOI] [PubMed] [Google Scholar]
- 45.Yu FF, Wang MF, Zhou L, Gui JF, Yu XY (2011) Molecular cloning and expression characterization of DMRT2 in akoya pearl oysters, Pinctada Martensii. J Shellfish Res 30: 247–254. [Google Scholar]
- 46.SEQC/MAQC-III Consortium (2014) A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium. Nat Biotechnol 32: 903–914. doi: 10.1038/nbt.2957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Teaniniuraitemoana V, Huvet A, Levy P, Klopp C, Lhuillier E (2014) Gonad transcriptome analysis of pearl oyster Pinctada margaritifera: identification of potential sex differentiation and sex determining genes. BMC Genomics 15: 491 doi: 10.1186/1471-2164-15-491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tong Y, Zhang Y, Huang JM, Xiao S, Zhang YH (2015) Transcriptomics analysis of Crassostrea hongkongensis for the discovery of reproduction-related genes. PLoS One 10: e0134280 doi: 10.1371/journal.pone.0134280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Y, Zhang L, Sun Y, Ma X, Wang J (2016) Transcriptome sequencing and comparative analysis of ovary and testis identifies potential key sex-related genes and pathways in scallop Patinopecten yessoensis. Mar Biotechnol (NY) 1: 453–465. [DOI] [PubMed] [Google Scholar]
- 50.Teaniniuraitemoana V, Huvet A, Levy P, Gaertner-Mazouni N, Gueguen Y (2015) Molecular signatures discriminating the male and the female sexual pathways in the pearl oyster Pinctada margaritifera. PLoS One 10: e0122819 doi: 10.1371/journal.pone.0122819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghiselli F, Milani L, Chang PL, Hedgecock D, Davis JP (2012) De Novo assembly of the manila clam Ruditapes philippinarum transcriptome provides new insights into expression bias, mitochondrial doubly uniparental inheritance and sex determination. Mol Biol Evol 29: 771–786. doi: 10.1093/molbev/msr248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bettinazzi S, Plazzi F, Passamonti M (2016) The complete female- and male-transmitted mitochondrial genome of Meretrix lamarckii. PLoS One 11: e0153631 doi: 10.1371/journal.pone.0153631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Milani L, Ghiselli F, Iannello M, Passamonti M (2014) Evidence for somatic transcription of male-transmitted mitochondrial genome in the DUI species Ruditapes philippinarum (Bivalvia: Veneridae). Curr Genet 60: 163–173. doi: 10.1007/s00294-014-0420-7 [DOI] [PubMed] [Google Scholar]
- 54.Huang XC, Rong J, Liu Y, Zhang MH, Wan Y (2013) The complete maternally and paternally inherited mitochondrial genomes of the endangered freshwater mussel Solenaia carinatus (bivalvia: unionidae) and implications for unionidae taxonomy. PLoS One 8: e84352 doi: 10.1371/journal.pone.0084352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smietanka B, Wenne R, Burzynski A (2016) Complete male mitochondrial genomes of European Mytilus edulis mussels. Mitochondrial DNA 27: 1634–1635. doi: 10.3109/19401736.2014.958704 [DOI] [PubMed] [Google Scholar]
- 56.Lu RM, Lin ZH, Zhang YP, Chai XL, Dong YH, Xiao GQ, et al. (2008) Comparison on the karyotypes of Scapharca subcrenata, Tegillarca granosa and Estellarca olivacea. Journal of Shanghai Fisheries University 17: 625–629. [Google Scholar]
- 57.Pertea G, Huang XQ, Liang F, Antonescu V, Sultana R, et al. (2003) TIGR gene indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics 19: 651–652. [DOI] [PubMed] [Google Scholar]
- 58.Iseli C, Jongeneel CV, Bucher P (1999) ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc Int Conf Intell Syst Mol Biol 99: 138–148. [PubMed] [Google Scholar]
- 59.Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M, et al. (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21: 3674–3676. doi: 10.1093/bioinformatics/bti610 [DOI] [PubMed] [Google Scholar]
- 60.Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, et al. (2006) WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34(Web Server issue): W293–W297. doi: 10.1093/nar/gkl031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hansen KD, Wu Z, Irizarry RA, Leek JT (2011) Sequencing technology does not eliminate biological variability. Nat Biotech pp: 572–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schulze SK, Kanwar R, Gölzenleuchter M, Therneau TM, Beutler AS (2012) SERE: Single-parameter quality control and sample comparison for RNA-Seq. BMC genomics 13: 524 doi: 10.1186/1471-2164-13-524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biology 11: R106 doi: 10.1186/gb-2010-11-10-r106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Q, Lin ZH, Bao YB, Huo LH, Gu HL (2012) Clone and analysis of hemoblobin gene (Tg-HbIIA) and immune expression research in Tegillarca granosa. Oceanologia Et Limnologia Sinica 43: 88–94. [Google Scholar]
- 65.Li CH, He JJ, Su XR, Li TW (2011) A manganese superoxide dismutase in blood clam Tegillarca granosa: Molecular cloning, tissue distribution and expression analysis[J]. Comp Biochem Physiol B Biochem Mol Biol 159: 64–70. doi: 10.1016/j.cbpb.2011.02.003 [DOI] [PubMed] [Google Scholar]
- 66.Benson G. Tandem repeats finder: a program to analyze DNA sequences (1999) Nucleic Acids Res 27: 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. (2010) The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Research 20: 1297–1303. doi: 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pannetier M, Fabre S, Batista F, Kocer A, Renault L, Jolivet G, et al. (2006) Foxl2 activates P450 aromatase gene transcription: towards a better characterization of the early steps of mammalian ovarian development. J Mol Endocrinol 36: 399–413. doi: 10.1677/jme.1.01947 [DOI] [PubMed] [Google Scholar]
- 69.Liu XL, Zhang ZF, Shao MY, Liu JG, Muhammad F (2012) Sexually dimorphic expression of foxl2 during gametogenesis in scallop Chlamys farreri, conserved with vertebrates. Dev Genes Evol 222: 279–286. doi: 10.1007/s00427-012-0410-z [DOI] [PubMed] [Google Scholar]
- 70.Santerre C, Sourdaine P, Martinez AS (2012) Expression of a natural antisense transcript of Cg-Foxl2 during the gonadic differentiation of the oyster Crassostrea gigas: first demonstration in the gonads of a lophotrochozoa species. Sex Dev 6: 210–221. doi: 10.1159/000338085 [DOI] [PubMed] [Google Scholar]
- 71.Santerre C, Sourdaine P, Adeline B, Martinez AS (2014) Cg-SoxE and Cg-β-catenin, two new potential actors of the sex-determining pathway in a hermaphrodite lophotrochozoan, the Pacific oyster Crassostrea gigas. Comp Biochem Physiol A Mol Integr Physiol 167: 68–76. doi: 10.1016/j.cbpa.2013.09.018 [DOI] [PubMed] [Google Scholar]
- 72.Li H, Zhang Z, Bi Y, Yang D, Zhang L, Liu J, et al. (2014) Expression characteristics of β-catenin in scallop Chlamys farreri gonads and its role as a potential upstream gene of Dax1 through canonical Wnt signalling pathway regulating the spermatogenesis. PLoS One 9: e115917 doi: 10.1371/journal.pone.0115917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee SH, Um SJ, Kim EJ (2016) CBX8 antagonizes the effect of Sirtinol on premature senescence through the AKT-RB-E2F1 pathway in K562 leukemia cells. Biochem Biophys Res Commun 469: 884–890. doi: 10.1016/j.bbrc.2015.12.070 [DOI] [PubMed] [Google Scholar]
- 74.Tang J, Wang G, Zhang M, Li FY, Sang Y, Wang B, et al. (2014) Paradoxical role of CBX8 in proliferation and metastasis of colorectal cancer. Oncotarget 5: 10778–10790. doi: 10.18632/oncotarget.2502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li J, Li Y, Cao Y, Yuan M, Gao Z, Guo X, et al. (2014) Polycomb chromobox 7 (Cbx7) modulates activation-induced CD4+ T cell apoptosis. Arch Biochem Biophys 564: 184–188. doi: 10.1016/j.abb.2014.10.004 [DOI] [PubMed] [Google Scholar]
- 76.Baumann C, De La Fuente R (2011) Role of polycomb group protein cbx2/m33 in meiosis onset and maintenance of chromosome stability in the Mammalian germline. Genes (Basel) 2: 59–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Katoh-Fukui Y, Tsuchiya R, Shiroishi T, Nakahara Y, Hashimoto N, Noguchi K,et al. (1998) Male-to-female sex reversal in M33 mutant mice. Nature 393: 688–692. doi: 10.1038/31482 [DOI] [PubMed] [Google Scholar]
- 78.Katoh-Fukui Y, Miyabayashi K, Komatsu T, Owaki A, Baba T, Shima Y, et al. (2012) Cbx2, a polycomb group gene, is required for Sry gene expression in mice. Endocrinology 153: 913–924. doi: 10.1210/en.2011-1055 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All transcriptome files of Tegillarca granosa are available from the SRA database (accession number SRR5512703, SRR5515063).