

Abstract
A substantial proportion of tumors consist of genotypically distinct subpopulations of cancer cells. This intra-tumor genetic heterogeneity poses a significant challenge for the implementation of precision medicine. Single-cell genomics constitutes a powerful approach to resolve complex mixtures of cancer cells by tracing cell lineages and discovering cryptic genetic variations that would otherwise be obscured in tumor bulk analyses. Given the chemical alterations that result from formalin fixation, single-cell genomic approaches have largely remained limited to fresh/frozen specimens. Here we describe the development and validation of a robust and accurate methodology to perform whole-genome copy-number profiling of single nuclei obtained from formalin-fixed paraffin-embedded clinical tumor samples. We applied the single-cell sequencing approach described here to study the progression from in situ to invasive breast cancer, which revealed that ductal carcinomas in situ display intra-tumor genetic heterogeneity at diagnosis and that these lesions may progress to invasive breast cancer through a variety of evolutionary processes.
INTRODUCTION
The coexistence of genetically distinct tumor cells within a tumor, referred to as intra-tumor genetic heterogeneity (ITGH), is well documented in human cancers1–5. Whilst in some cancers ITGH is a widespread phenomenon1,6, in breast cancer, varying degrees of heterogeneity have been documented7,8. Sequencing studies have shed light on ITGH9,10, however, standard sequencing methods provide a compound measure of clonal complexity with subclonal frequencies of somatic alterations inferred statistically11,12. Single-cell genomic methods13–17 have been developed to provide orthogonal and complementary information to move beyond statistical inference and facilitate in-depth understanding of cancer clonal hierarchy and genetic heterogeneity18.
Single-cell genomics have thus far been limited to the analysis of fresh/frozen (i.e. fresh or rapidly frozen) tissues13,15. Fresh/frozen tumor specimen procurement is not part of the routine clinical and diagnostic practice in most institutions and for some tumor types (e.g. small tumors or tumors where near complete sampling is required for histopathology) fresh/frozen samples cannot be obtained. Hence, fresh/frozen specimens are only available for a subset of cancers, and they may not adequately represent the tumors from which they are derived. The vast majority of human tumor material is routinely formalin-fixed paraffin-embedded (FFPE) for diagnostic purposes. Molecular analysis of FFPE specimens, however, is challenging, given that formalin fixation introduces several types of artifacts, mainly caused by protein and nucleic acid cross-links19. Although whole-exome and targeted sequencing analyses have been successfully performed on DNA extracted from FFPE tumor bulk samples20,21, single-cell methods for genomic investigations of FFPE tissue samples have yet to be reported.
Here we describe and validate a robust approach to perform single-cell whole-genome copy number (CN) profiling from FFPE tissue (Fig. 1). We demonstrated that CN profiles of nuclei retrieved from FFPE and frozen samples of the same cancer cell lines and neoplastic lesions are equivalent. We validated the approach in clinical diagnostic specimens and gleaned insights into the progression from ductal carcinoma in situ (DCIS) to invasive breast cancer. This methodology makes it possible to apply single-cell sequencing to address biological and/or clinical questions that require FFPE samples.
Figure 1. Schematic representation of the formalin-fixed paraffin-embedded (FFPE) single-cell sequencing platform.
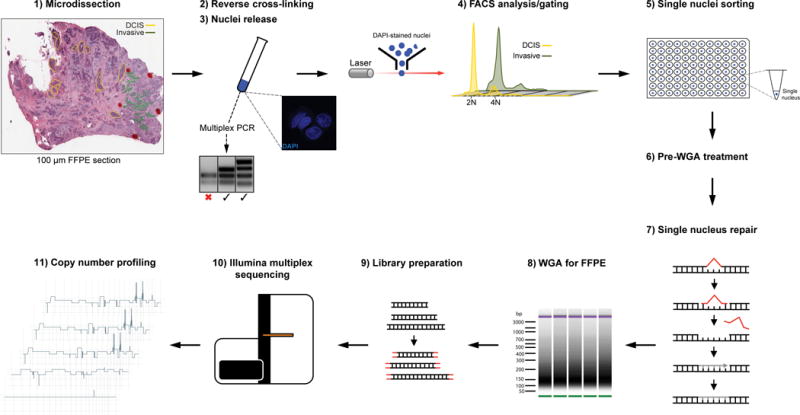
The procedure consists of 11 steps involving tissue microdissection, nuclei preparation and FACS analysis/sorting based on DAPI staining/DNA content (steps 1–5), single-nucleus DNA repair to correct for FFPE-induced DNA damage, whole-genome amplification (WGA), Illumina library preparation and multiplex sequencing (steps 6–10), and bioinformatics analysis (step 11). The multiplex PCR analysis is used to determine the quality of the DNA extracted from FFPE nuclei preparations, and is performed between steps 3 and 4.
RESULTS
FFPE single-cell whole-genome CN analysis methodology
Genome-wide CN profiling of single nuclei derived from fresh/frozen tissue involves the isolation of nuclei, followed by flow cytometry sorting based on DNA content to obtain single nuclei in individual wells of 96-well plates, whole genome amplification (WGA) and sequencing2,15. To develop a method for whole-genome-sequencing-based CN analysis of single cells derived from FFPE samples, we modified our protocol for frozen nuclei15 to include steps we deemed pertinent to the success of a method for the analysis of FFPE single cells. These steps are: (1) applying a molecular test to prioritize FFPE specimens likely to be amenable to single-cell analysis, (2) incorporating methods to ensure intact nuclei retrieval, and (3) treating isolated nuclei to repair damaged FFPE single-cell DNA (Fig. 1; Supplementary Figs. 1 and 2).
First, to select FFPE specimens we employed a multiplex-PCR22,23 assay to define the quality of DNA extracted from FFPE samples. This assay utilizes primer sets that amplify four genomic fragments (fragment sizes of 100, 200, 300 and 400 bp). Samples producing 300 bp and 400 bp fragments were deemed good in quality22,23 and the focus of this study. Tumor samples from eight individual patients in total were processed; of these, tumor samples from four patients yielded ≥300 bp multiplex-PCR fragments (Supplementary Figs. 1a and 1b). Single cells from these cases and from one case with 200 bp multiplex-PCR fragments were subjected to sequencing. One of the cases with 300 bp multiplex-PCR fragments displayed heavy immune-infiltrate (i.e. >80% of inflammatory cells within tumor clusters and in tumor stroma, a phenotype observed in <3% of breast cancers)24 and was not analyzed further (data not shown). Results from four cases are presented (Table 1). Second, to retrieve intact nuclei, FFPE blocks were sectioned into 100 μm–thick sections for microdissection5. The microdissected tissue was subjected to distinct treatment conditions (see Methods) including high-temperature (to remove protein and DNA cross-links), enzymatic processing (proteases to digest extracellular material), and mechanical stress (passage through a fine needle to ensure nuclei release). Third, to mitigate the formalin fixation artifacts22 (e.g. introduction of DNA adducts, gaps, nicks), single nuclei were treated with a cocktail of DNA repair enzymes (Methods)25, which we posited might enhance the performance of the WGA reaction by increasing the number of amplifiable DNA molecules/nuclei. Finally, for WGA, we utilized a degenerate oligonucleotide-priming PCR-based method (DOP-PCR) previously shown to provide robust and uniform amplification, suitable for CN analysis of single-cell2 and poor-quality fragmented26 DNA. DOP-PCR was chosen over other WGA methods such as Multiple Displacement Amplification27 or Multiple Annealing and Looping Based Amplification Cycles chemistry28, owing to its favorable performance with FFPE samples29,30.
Table 1.
Information of samples subjected to single-cell sequencing copy number profiling in this study.
| Case | Sample type | ER | PR | HER2 | Multiplex PCR result | Year FFPE block | Cancer ploidy |
|---|---|---|---|---|---|---|---|
| Case 1 | Triple-negative invasive ductal carcinoma | − | − | − | 4 bands (400 bp fragments) | 2008 | Triploid (~3N) |
| Case 2 | Synchronous DCIS/invasive carcinoma of mucinous type | + | + | + | 4 bands (400 bp fragments) | 2008 | Tetraploid (~4N) |
| Case 3 | Synchronous DCIS/invasive ductal carcinoma | + | + | − | 3 bands (300 bp fragments) | 2011 | Diploid (~2N) |
| Case 4 | Synchronous DCIS/invasive ductal carcinoma | + | + | + | 2 bands (200 bp fragments) | 2007 | Pseudo-Tetraploid (~3.7N) |
DCIS, ductal carcinoma in situ; ER, estrogen receptor; FFPE, formalin-fixed, paraffin-embedded; PR, progesterone receptor.
To test this approach, FFPE and frozen tissue sections from Case 1 (Fig. 2a), a triple-negative invasive ductal carcinoma, were reviewed by two pathologists (FCG, JSR-F), microdissected and processed as described for FFPE samples (Methods) and for frozen sections15 (Supplementary Table 1). Multiplex-PCR analysis of bulk FFPE DNA from Case 1 yielded PCR fragments of up to 400 bp (Supplementary Fig. 1a). Flow cytometry of FFPE nuclei based on DAPI staining yielded a ploidy profile similar to that of frozen nuclei with both exhibiting two distributions at ~2N (diploid) and ~3N (triploid) DNA content (Fig. 2b). Cytometric parameters such as forward/side scatter and DAPI signal were concordant between FFPE and frozen nuclei preparations, attesting to the quality of FFPE nuclei (Supplementary Fig. 2a). Furthermore, confocal imaging of FFPE and frozen nuclei revealed largely intact, DAPI-stained nuclei (Supplementary Fig. 1e). Single FFPE and frozen nuclei from each peak were sorted into 96-well plates for WGA.
Figure 2. Establishment of a whole-genome copy-number profiling method for single nuclei derived from FFPE samples.
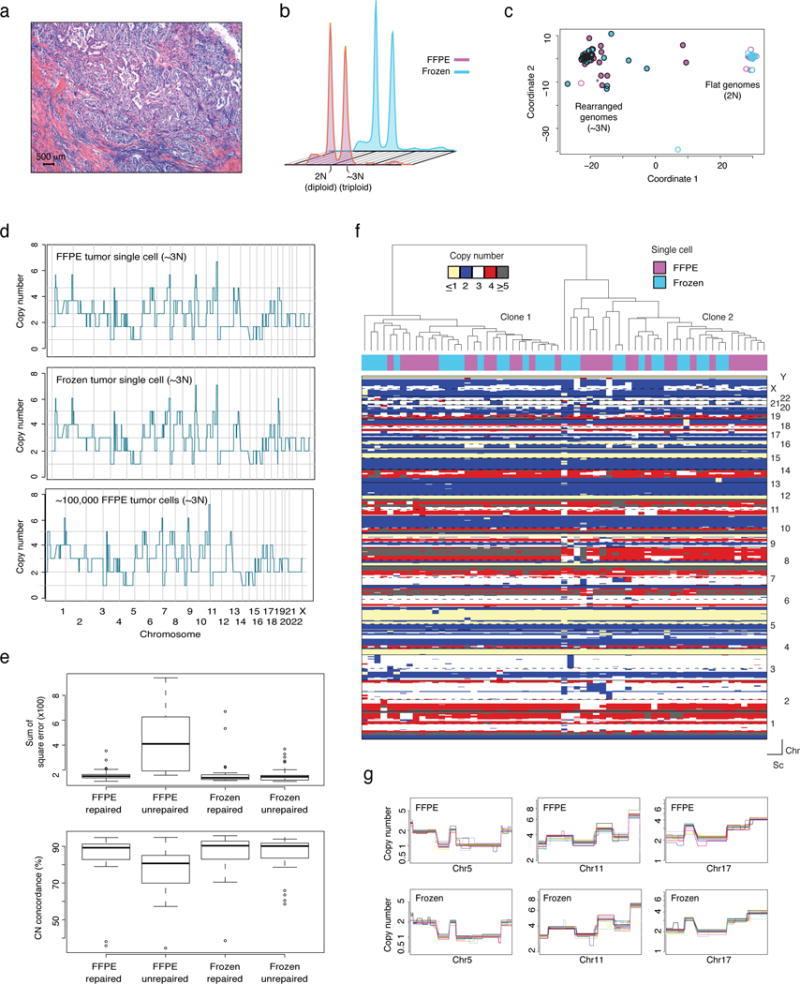
(a) Representative micrograph of the triple-negative invasive ductal carcinoma (Case 1) (hematoxylin-and-eosin; scale bar, 500 μm). (b) Ploidy flow cytometric profiles of nuclei extracted from FFPE/frozen tissue of Case 1 based on DAPI staining. (c) Multidimensional scaling of all FFPE and frozen nuclei sequenced. Each circle represents one cell (~3N FFPE (purple circles) and frozen (turquoise circles), n=36; 2N FFPE (purple circumference) and frozen (turquoise circumference), n=24). (d) Representative examples of genome-wide single-nucleus copy number (CN) profiles from FFPE (top) and frozen (middle) single nuclei derived from the ~3N (tumor) distribution. Bulk CN profile of 100,000 FFPE tumor cells is also shown (bottom). CN values of the bulk were rounded to the nearest integer. (e) Data bin-to-bin variance (expressed as the Sum of Square error - top panel) and percent concordance with bulk CN profiles (bottom panel) of FFPE repaired (n=36), FFPE unrepaired (n=24), frozen repaired (n=24) and frozen unrepaired (n=36) nuclei. Data are mean ± s.d. (f) Hierarchical clustering analysis of CN data from FFPE (n=33) and frozen (n=30) single cells from Case 1, using Manhattan distance and Ward’s method. (g) Close-up views of representative clonal CN alterations on chromosomes 5, 11 and 17 identified in FFPE (top) and frozen (bottom) from a minimum of 10 randomly selected single cells. Each colored line represents an individual single nucleus. Chr: chromosome, Sc: single cell.
We next sought to define the impact of the DNA repair step on FFPE nuclei, to compare repaired DNA from FFPE nuclei to a “gold standard” (i.e. DNA from frozen nuclei), and to test whether the repair step would introduce artifacts (i.e. comparison of repaired/unrepaired frozen nuclei). Repaired and unrepaired sets of FFPE and frozen nuclei (from 2N and 3N distributions) were sequenced (Supplementary Table 1). Amplified FFPE and frozen nuclei DNA were processed for multiplex sequencing as previously described15. Ct values (threshold cycle) of real-time WGA reactions (Methods) from the different conditions were used to approximate the relative abundance of amplifiable DNA molecules in frozen versus FFPE in repair/no-repair conditions. This qualitative analysis revealed that the relative amount of template available for WGA was enhanced significantly by single-nucleus DNA repair (p<0.0001, Mann-Whitney U test; Supplementary Fig. 1g).
Multi-dimensional scaling of all single nuclei sequenced revealed two clusters. One cluster contained ‘flat’ (i.e. devoid of gene copy number alterations) profiles (all derived from 2N peak and presumably representing intra-tumor stromal and inflammatory cells) from FFPE and frozen samples while the other cluster contained rearranged cancer CN profiles (frozen and FFPE nuclei, all derived from the 3N peak, Fig. 2c), which were indicative of the cells’ neoplastic origin. Consistent with this, bulk sequencing of DNA extracted from a population of 100,000 nuclei from 2N and 3N peaks yielded similar CN profiles (Fig. 2d and Supplementary Figs. 3a and b). Unrepaired FFPE single nuclei displayed substantial bin-to-bin variability in normalized read count data and an over abundance of CN segments, likely artifactual (Supplementary Figs. 3d and e). These artifacts were not observed in the matched frozen and repaired FFPE datasets (Fig. 2e). A systematic comparison of bin-to-bin variability (Fig. 2e and Supplementary Fig. 3e) and the concordance between CNAs detected in unrepaired FFPE, repaired FFPE and frozen single nuclei profiles and tumor bulk profiles (Fig. 2d and Supplementary Fig. 3b) revealed that single nucleus DNA repair rendered the profiles of FFPE single nuclei comparable to those obtained from frozen single nuclei and bulk tumor samples. The CNAs detected in FFPE repaired and frozen nuclei revealed high, significant correlations with the corresponding tumor bulk profiles (Spearman’s correlation r2=0.9058983 for FFPE repaired and 0.9116344 for frozen unrepaired, p-values <2.2e−16). These analyses corroborate the notion that FFPE single nucleus repair significantly improves the quality of the data. Importantly, no significant differences between unrepaired and repaired frozen nuclei were observed (Fig. 2e), suggesting that artifacts potentially introduced by the single nucleus DNA repair step are negligible.
Hierarchical clustering of CN profiles of all 3N FFPE (repaired) and frozen (unrepaired) nuclei revealed that all harbored a highly rearranged genome and that FFPE and frozen single nuclei displayed concordant profiles (Fig. 2f). Clonal alterations identified via bulk profiling were consistently present in both FFPE and frozen nuclei sequenced (Fig. 2g). Two clonally related subpopulations differing by a CN gain on 8q21.3-q23.1 were detected in both FFPE repaired and frozen nuclei (Fig. 2f), and evident in the CN profiles from the tumor bulk sequencing in the form of a non-integer subclonal CN alteration (CNA, Supplementary Fig. 3c). Lastly, an analysis of 2N single normal nuclei data from FFPE (repaired) and frozen (unrepaired) tissue showed that the percentage of the genome that deviates from CN=2 (i.e. hypothetical ground truth) was negligible and not statistically different (average 0.63% and 0.38% for FFPE and frozen, respectively). Taken together, these results support the robustness and accuracy of the approach in retrieving genome-wide CN profiles from single FFPE nuclei.
Validation using breast cancer cell lines
Despite the high concordance between the CN profiles of FFPE and frozen nuclei from Case 1 and their concordance with profiles of tumor bulk, one could argue that the genetic variation observed between individual nuclei may represent artifacts. Therefore, to investigate single-cell CN variation in a controlled setting, FFPE and frozen blocks of two commonly studied triploid breast cancer cell lines, MCF-7 and CAMA-1, were prepared (Methods). Flow cytometric analysis based on DAPI staining, forward and side scatter (Supplementary Fig. 2b and Supplementary Fig. 4a) was used to determine single-nucleus event rates in each preparation and were subsequently used to prepare 1:1 and 4:1 mixes (MCF-7:CAMA-1). The mixes were then used to make FFPE and frozen blocks (Methods). Blocks were processed as described for Case 1 and for both conditions, 96 cells per mix were sorted (cytometric parameters from both conditions were qualitatively similar, Supplementary Fig. 2), amplified (only FFPE nuclei were repaired; Supplementary Fig. 1d) and sequenced. In addition, bulk DNA from each cell line was sequenced.
Hierarchical clustering revealed that MCF-7 and CAMA-1 nuclei clustered according to their specific CN profiles irrespective of the preparation procedure (i.e. FFPE and frozen, Supplementary Fig. 4b). In addition, no differences between repaired FFPE, frozen nuclei and bulk data of each cell line in terms of bin-bin variance and copy number concordance were observed (Supplementary Figs. 4b–d, Supplementary Table 1), attesting to the equivalence of FFPE and frozen single nuclei sequencing methods. The numerically lower concordances between MCF-7 profiles (FFPE and frozen single nuclei) than CAMA-1 profiles stems from the higher genetic heterogeneity observed for this cell line; an observation supported by comparing single-cell to bulk data (Supplementary Figs. 4b and e).
The observed MCF-7:CAMA-1 ratios (i.e. 50% and 20% CAMA-1 cells in repaired FFPE samples and 46% and 14% CAMA-1 cells in frozen samples) as defined by the CN profiles were consistent with the theoretical mixing ratios (i.e. 50% and 20% of CAMA-1, respectively), providing evidence that with 96 cells sequenced, one can confidently detect a subclone comprising 20% of the tumor bulk (Supplementary Fig. 4f). Furthermore, down-sampling analysis of the single nuclei from 96 to 48 (in both 4:1 mixes) revealed that the clone comprising 20% of the mix could be detected accurately using 48 single nuclei (Supplementary Table 1).
Analysis of synchronous ductal carcinoma in situ (DCIS) and invasive disease
To illustrate the potential of this single-cell CN analysis method to address biologically relevant questions, we used it to study breast cancer progression and selected for analysis two cases of synchronously diagnosed DCIS and invasive breast cancer (Figs. 3 and 4a). Areas of DCIS and invasive cancer (Figs. 3a and 4a) from FFPE and frozen blocks were separately microdissected5 and nuclei prepared as described above. A fraction of the nuclei was used for bulk tumor sequencing.
Figure 3. Validation of the whole-genome CN method for FFPE-derived single nuclei using samples from Case 2, a synchronous DCIS and invasive breast cancer.
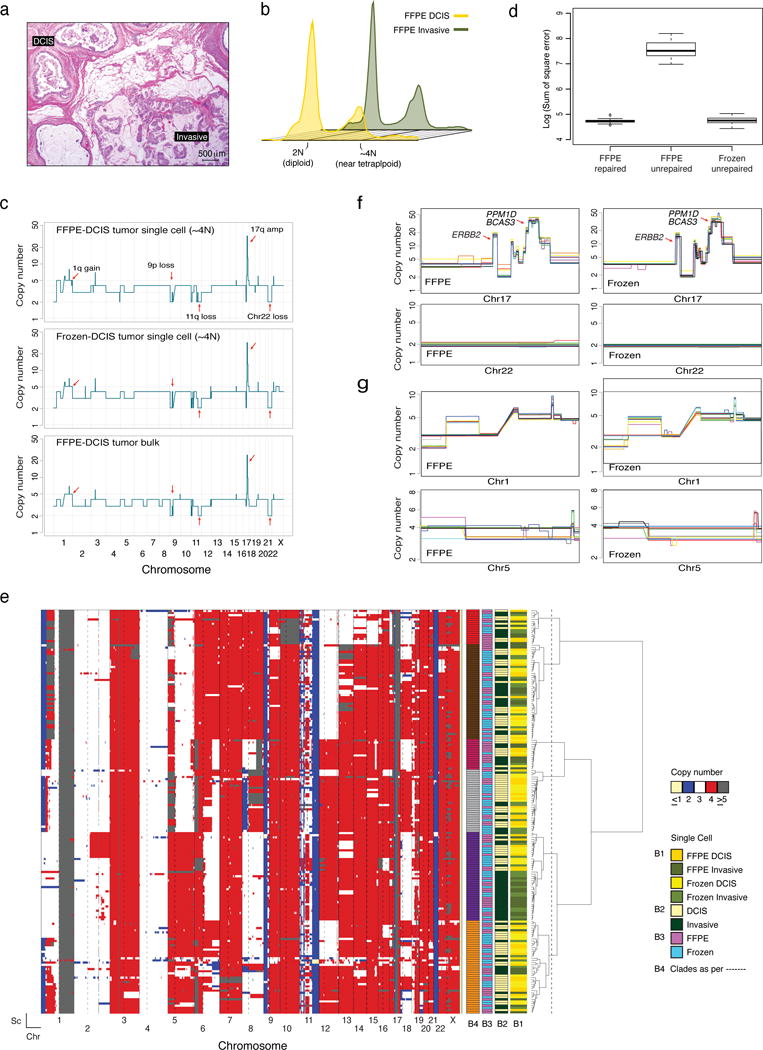
(a) Representative micrograph of an estrogen receptor-positive/ERBB2 (HER2)-negative invasive breast cancer (mucinous type) with synchronous DCIS (hematoxylin-and-eosin; scale bar, 500 μm). (b) Ploidy flow cytometric profiles of nuclei extracted from DCIS/invasive and FFPE/frozen tissue of Case 2. (c) Representative examples of genome-wide single-cell copy number (CN) profiles from FFPE (top) and frozen (middle) single nuclei derived from the ~4N (tumor) distribution, and the bulk CN profile of the FFPE DCIS component (bottom). CN values of the bulk were rounded to the nearest integer. Red arrows indicate clonal CN events. (d) Bin-to-bin variance expressed as Sum of Square error of FFPE repaired (n=12), FFPE unrepaired (n=12) and frozen unrepaired (n=12) single cells (Methods). (e) Hierarchical clustering analysis of FFPE (n=120) and frozen (n=120) single cells from Case 2, using Manhattan distance and Ward’s method. Reference bars under the dendrogram indicate tumor component of origin (B1 and B2), type of tissue of origin (B3) and the clades (B4) defined by the conservative and arbitrary cut of the dendrogram (dotted line). The heatmap was constructed as described in the Methods. (f) Zoomed views of representative clonal CN alterations on chromosome 17 and chromosome 22 identified in FFPE (left) and frozen (right) single cells. (g) Zoomed views of representative subclonal CN alterations identified on chromosome 1 and chromosome 5 identified all FFPE (left) and frozen (right) nuclei sequenced. In the overlay images of (f) and (g) each colored line represents an individual single nucleus and a minimum of 10 randomly selected profiles are presented. Chr: chromosome, Sc: single cell.
Figure 4. Sequencing of FFPE single nuclei from samples of case 3 and phylogeny reconstruction of progression from DCIS to invasive breast cancer.
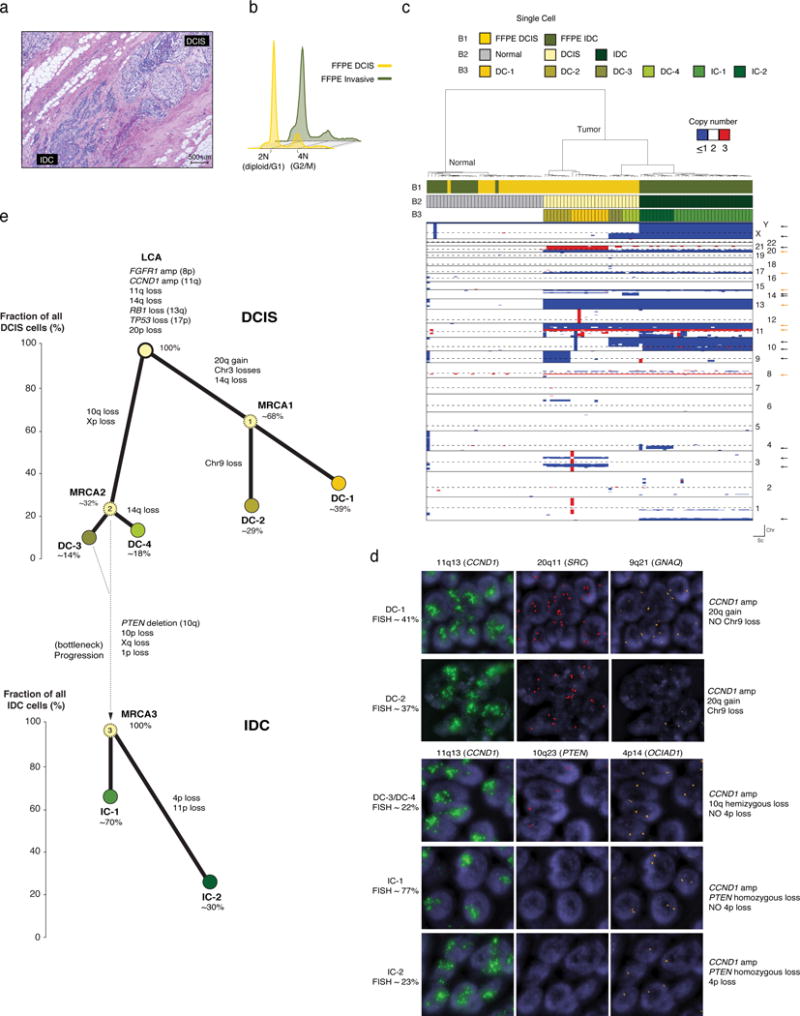
(a) Representative micrograph of a diploid estrogen receptor-positive ERBB2 (HER2)-negative invasive ductal carcinoma (IDC) with synchronous DCIS (hematoxylin-and-eosin; scale bar, 500 μm). (b) Ploidy cytometry profiles of Case 3 FFPE DCIS and invasive (IDC) components. (c) Hierarchical clustering analysis of FFPE single nuclei (n=96) from Case 3, using Manhattan distance and Ward’s method. Reference bars: Bar 1 (B1), sample of origin from which the single cells were isolated (i.e. FFPE DCIS or FFPE IDC); Bar 2 (B2), classification of cells into normal, DCIS or IDC; Bar 3 (B3), subclonal populations identified in DCIS and IDC. Orange and black arrows (right side of heatmap) indicate clonal and subclonal alterations, respectively. Chr: chromosome, Sc: single cell. (d) Representative micrographs of fluorescence in situ hybridization analysis with probes for specific loci indicated in the Figure, confirming the existence of selected subclones present in the DCIS (DC-1 and DC-3) and IDC (IC-1 and IC-2) identified by single-cell sequencing (63× magnification). Percentages of the distinct clones as defined on the basis of quantification of FISH results as described in the Methods and Supplementary Table 2. (e) Phylogenetic trees of Case 3 based on single-cell CN data. Nodes in the trees correspond to observed subclones in DCIS (DC-1, DC-2, DC-3 and DC-4) and IDC (IC-1 and IC-2), which are color-coded to match the clusters in the dendrogram in panel c. The inferred most recent common ancestors (MRCAs) of subclones are indicated with a light-yellow circle with dashed outline. Values within circles indicate percentage of cells inferred as carrying distinct clonal/subclonal alterations. Connectors (thick black lines) are annotated with subclone specific CN alterations. The percentage of tumor cells (cancer cell fraction) harboring any given set of defining alterations is indicated for each subclone on the Y-axis of each DCIS and invasive phylogeny (i.e. the Y-axis coordinate of the top of each circle indicating a clone refers to its cancer cell fraction). For each phylogeny, branches were drawn to connect the clones for illustration purposes.
Polyploid Case 2
Flow cytometry data of both components from FFPE and frozen tissues displayed equal quality (Supplementary Fig. 2c). Both DCIS and invasive samples exhibited two DNA content distributions at 2N (diploid) and ~4N (near-tetraploid) (Fig. 3b and Supplementary Fig. 5a). Sequencing of 2N frozen DCIS nuclei revealed all to be genomically normal (Supplementary Fig. 5b), while the ~4N nuclei displayed rearranged genomes (Fig. 3c We thus focused on ~4N cancer nuclei from the cancer sample.
Similar to what was performed for Case 1, single nuclei from the ~4N distribution were analyzed in three conditions: (1) FFPE repaired, (2) FFPE unrepaired, and (3) frozen unrepaired. Consistent with the results of Case 1, unrepaired FFPE nuclei yielded artifactual results, (i.e. non-integer CN states; Supplementary Figs. 6a and 6b), whereas the introduction of the single nucleus DNA repair step resulted in the detection of CNAs largely concordant with those detected in frozen nuclei and matching tumor bulk profiles (Figs. 3c and 3d, Supplementary Fig. 6c). Spearman’s correlations between CNAs detected in FFPE and frozen nuclei and matching tumor bulks were significant (p-values < 2.2e−16) and similar to one another (r2=0.7627261 for FFPE and r2=0.7802684 for frozen). Correlations were numerically but not statistically significantly lower than those of Case 1, probably owing to the profound ITGH in Case 2 (Fig. 3e).
Hierarchical clustering of all nuclei sequenced (60 DCIS FFPE, 60 invasive FFPE, 60 DCIS frozen and 60 invasive frozen, Supplementary Table 1) illustrated the robustness and accuracy of the data with all single nuclei displaying, among other clonal events, gain of 1q and losses of 11q and 22 (Figs. 3c, 3e, 3f). Focal amplifications on 17q encompassing the ERBB2 (17q12-q21.2), PPM1D and BCAS3 (17q22-q23.2) loci were also observed in all nuclei sequenced (Figs. 3c and 3e). Subclonal events such as losses of 1p and 8p and alterations on chromosomes 5 and 8 were found in the profiles from DCIS and from the invasive tumor samples from both FFPE and frozen tissue (Figs. 3e and 3g). Taken together, the presented data, along with the fact that FFPE and frozen single cells clustered together observed in the heatmap (Fig. 3e), further support the robustness of CN profiling of nuclei retrieved from FFPE samples.
We next defined subpopulations (i.e. clades) based on conservatively estimated cut-points of the dendrogram (dashed line in Fig. 3e, Methods). Six distinct but highly related subpopulations were identified (Fig. 3e). For all six clades, the constituting nuclei were derived from both the in situ and invasive components. This implies that either invasion was unrelated to the CNAs or the ability to invade was acquired early in disease development (i.e. in situ disease) followed by genome instability and the development of multiple genetically heterogeneous DCIS subclones that in parallel progressed to invasive disease (Supplementary Fig. 7a).
Diploid Case 3
Flow cytometry of DCIS and invasive components displayed diploid (2N) profiles (Fig. 4b). 48 FFPE repaired and frozen diploid nuclei from both DCIS and invasive disease were analyzed (Supplementary Table 1). FFPE repaired single nuclei gave qualitatively similar CN profiles to frozen nuclei and were concordant with the CN profiles derived from tumor bulk whole-exome sequencing (WES, Supplementary Figs. 8a–c). Hierarchical clustering of all nuclei revealed the presence of two major groups, one with genomically normal cells and another with rearranged, clonally related cancer cells from the DCIS and invasive components (Fig. 4c). Clonal alterations identified in analysis of the matched frozen bulk tumor tissue such as loss of 13q (RB1) and 17p (TP53) as well as focal amplifications on 8p11.2-p12 (FGFR1) and 11q13.3-q13.4 (CCND1), were detected in all single FFPE and frozen nuclei (Fig. 4c, orange arrows, and Supplementary Fig. 8c). Subclonal events included 20p–20q gain (20p11.21–q13.33), loss of chromosome 9, and segmental losses at 3p21.31–p12.3 and 3q21.2–q24 (Fig. 4c, black arrows). These and other subclonal alterations defined several distinct clusters, which were restricted to either the DCIS (DC-1, DC-2, DC-3, DC-4) or the invasive component (IC-1, IC-2; Fig. 4c and Supplementary Fig. 8d). These clusters were found to be robust, given the low (<10−15) probability of each subcluster of cells within a cluster sharing a set of breakpoints by chance (Methods). Based on the identified subclones, some of which were validated at similar clonal frequencies by fluorescence in situ hybridization (FISH, Fig. 4d, Supplementary Table 2 and Supplementary Fig. 9), we sought to define the likely evolutionary process that occurred during disease progression (Fig. 4e). The reconstructed phylogeny of Case 3 suggests a scenario where ITGH occurred early in disease development and that progression from DCIS to invasive carcinoma might have occurred via clonal selection (Fig. 4e and Supplementary Fig. 7b). Interestingly, this putative evolutionary bottleneck was associated with a homozygous deletion at the PTEN locus (Fig. 4d and Supplementary Figs. 8b and d), an observation validated by FISH with PTEN specific probes (Fig. 4d, Supplementary Fig. 9).
We next asked whether the clonal selection process deduced from the single-cell reconstructed phylogeny of Case 3 could be also evidenced in the mutational space extracted from WES of the tumor bulk. Indeed, WES data analysis from microdissected DCIS and invasive components (Methods) also indicate progression from in situ to invasive disease resulting in clonal shifts with a reduction in clonal diversity in the latter (Supplementary Fig. 10).
Single-cell CN information from suboptimal FFPE samples
While we focused our analysis on FFPE samples yielding 300 bp and above in the multiplex-PCR assay, we asked whether it is also possible to retrieve accurate CN information from suboptimal (i.e. overly damaged) FFPE samples. Thus, we analyzed an additional Case (i.e. Case 4, Fig. 5a), which yielded 200 bp fragments in the multiplex PCR assay, even after single nucleus DNA repair (Methods and Supplementary Fig. 1a). This analysis revealed that clonal alterations, identified in bulk WES and matched single frozen nuclei data, were found in all single FFPE repaired nuclei sequenced (Figs. 5b and c), and in the case of the clonal ERBB2 (i.e. HER2) amplification, confirmed by FISH analysis (Fig. 5d). The overall pattern of CN alterations of FFPE repaired and frozen single nuclei was similar, however frozen nuclei preferentially clustered together (Fig. 5e). Whether this was due to geographic heterogeneity or the overly damaged nature of the sample is unclear and requires analysis of more overly damaged FFPE samples. Nonetheless, our findings suggest that even in relatively degraded samples, our method is capable of capturing clonal alterations at the single nucleus resolution.
Figure 5. Whole-genome single-nucleus copy-number information for case 4, a suboptimal, overly damaged FFPE sample.
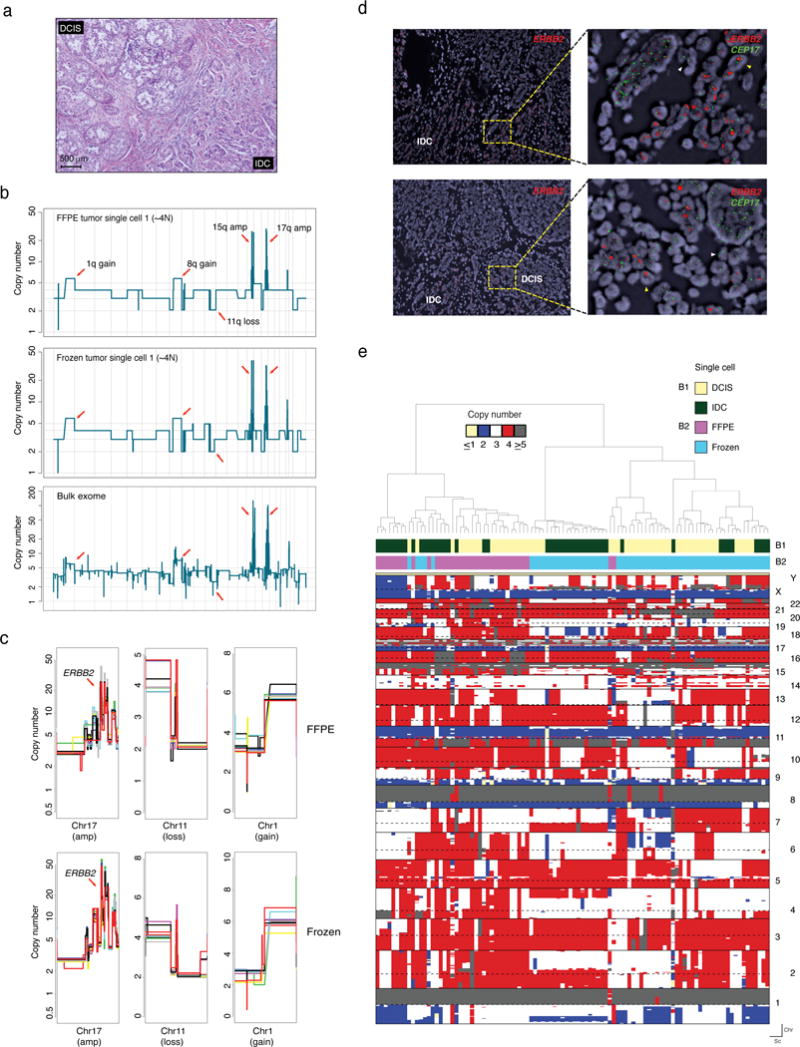
(a) Representative micrograph of an estrogen receptor-positive/ERBB2 (HER2)-positive invasive ductal carcinoma (IDC) with DCIS. (b) Representative CN plots of single FFPE and frozen nuclei compared to the tumor bulk CN profile derived from whole exome sequencing data. (c) Zoomed in views of representative clonal alterations identified in single FFPE and frozen nuclei sequenced. Each colored line represents an individual single nucleus with a minimum of 10 randomly selected profiles presented. (d) Fluorescence in situ hybridization with probes for ERRB2 (HER2) highlighting the presence of clonal ERBB2 gene amplification, consistent with the single nuclei sequencing results. (e) Hierarchal clustering analysis of all single nuclei (FFPE, n=48 and frozen, n=48) sequenced from Case 4. Reference bars on the top indicate the tumor component of origin (B1) and type of tissue of origin (B2). Hierarchical clustering heatmap was constructed using CN profiles of individual nuclei using Manhattan and Ward as the distance function and clustering method, respectively (Methods). Chr: chromosome, Sc: single cell.
DISCUSSION
Here we describe the first method for the genome-wide CN profiling of single nuclei using routinely processed FFPE clinical samples. Aspects germane to the success of this methodology were (1) selection of FFPE samples based on objective assessment of DNA quality/size, (2) the implementation of a variety of treatments to retrieve intact nuclei, and (3) the introduction of a single nucleus DNA repair step. The single-cell CN information obtained from FFPE samples using this approach was highly concordant with those obtained from matched frozen samples and the corresponding bulk CN profiles. The consistent detection of clonal and subclonal CN events in FFPE and matched frozen cells highly supports the sensitivity and reproducibility of the method.
Although our methodology allows CN determination of single-cell FFPE genomes, the WGA strategy employed carries an intrinsic limitation. The poor coverage breadth associated with DOP-PCR renders our approach unsuitable for applications such as single-cell WES. However, WGA methods that allow for high coverage of single-cell genomes are currently not compatible with amplifying FFPE DNA17. Hence, the detection of nucleotide substitutions, small insertions and deletions or rearrangements in FFPE single cells, alone or in combination with CN profiling, will require further development. Another limitation of the method described here is that it performs optimally in FFPE samples with good quality DNA, which may account for approximately 50% of the samples depending on the fixation protocols employed. The multiplex-PCR method described as part of the sample work-up, however, provides an upfront assessment of the samples that are likely to yield optimal results. The technical failure rate in samples with PCR fragments >300 bp is likely minimal, given that all samples analyzed in this study that passed the quality control PCR yielded high quality single-cell CN data and that FFPE breast cancer tumor bulk samples with even lower fragment sizes have been successfully employed for the analysis of CNAs23.
Nonetheless, the methodology developed here can potentially unlock pathology archives by providing access to a large repository of material for single-cell genomics, which could be employed for detailed studies of cancer evolution and the chronology of somatic genetic events in cancer development and progression, in particular in the progression from pre-invasive lesions (e.g. DCIS, lobular carcinoma in situ) to invasive breast cancer and from primary breast cancers to metastatic lesions where only limited and FFPE tumor material is available. This could facilitate biomarker discovery in the form of indices of ITGH for prognostication and prediction, identification of clonal vs. subclonal genetic alterations, and co-occurrence or mutual exclusivity of specific somatic subclonal genetic alterations within the cancer cell populations of a tumor via retrospective analysis of FFPE archived, clinically annotated samples obtained from clinical trials or available in pathology departments. The method also has the potential to inform the underlying genetics and biology of cancer as illustrated by the results from matched DCIS and invasive samples. Our study provides evidence at single-cell resolution to support different mechanisms for the progression from DCIS to invasive breast cancer (Supplementary Fig. 7), suggesting that progression from in situ to invasive disease is complex and variable depending on the patient. In some cases the ability to invade may be an intrinsic characteristic acquired early in the development of the DCIS, whereas in others it may be the result of genetic selection of an invasive clone. It should be noted, however, that synchronous DCIS and invasive breast cancer samples were analyzed here; further studies to define the progression of pure DCIS (i.e. diagnosed in the absence of invasive disease and treated surgically with or without radiation therapy) to invasive breast cancer are warranted. Given that in FFPE samples, histologic features can be optimally obtained, this method will allow for detailed genotypic-phenotypic analyses through the observation and dissection of phenotypically distinct components within FFPE tumor tissues followed by single-cell genomic analyses, as illustrated by the DCIS and invasive breast cancer examples described in this study.
Taken together, our results demonstrate the reproducibility and accuracy of the single-cell FFPE CN profiling method described here, and illustrate its potential to unravel ITGH in cancers, dissect the genetics of histologically/phenotypically distinct cancer components and, trace their evolutionary history.
ONLINE METHODS
Case selection and sample labeling
FFPE and frozen blocks from Case 1 (2008) were purchased from the UMass Cancer Center Tissue and Tumor Bank, University of Massachusetts. FFPE and frozen blocks from the remaining samples analyzed, including cases 2, 3 and 4, which comprise synchronous DCIS and invasive cancer, were retrieved from the Memorial Sloan Kettering Cancer Center (MSKCC) pathology archives (Table 1). All cases were reviewed by JSR-F and FCG who classified the tumors following the World Health Organization (WHO) criteria31 and identified DCIS and invasive components for subsequent microdissection. Estrogen receptor (ER), progesterone receptor (PR) and ERBB2 (i.e. HER2) status was assessed as previously described32, following the American Society of Clinical Oncology (ASCO)/College of American Pathologists (CAP) guidelines33,34. This study was approved by the MSKCC institutional review board (IRB Number: WA0174-13), and informed consent was obtained where appropriate according to the protocol approved.
Sample preparation, microdissection and single nuclei preparation
Up to three 100 μm-thick sections were cut onto positively-charged microscope slides. Sections were washed three times with 1 ml Xylene for 5 min to remove the paraffin, rehydrated in sequential 5 min ethanol immersions (2× 100%, followed by 1× 95%, 70%, 50% and 30% ethanol), stained with nuclear fast red (NFR, Sigma-Aldrich) and dehydrated by reverse sequential ethanol washes. The areas of interest were microdissected as previously described5, washed three times at 90 °C for 60 min with 1 ml of Tris-EDTA pH 9.0 (IHC antigen retrieval reagent, Enzo) or 5 mM EDTA buffer pH 8 (Sigma-Aldrich) to facilitate the removal of the cross-links (i.e. reverse cross-linking) present in the FFPE tissues. Samples were cooled at room temperature (RT) and washed four times (3× 1.2 mL wash and 1× 800 μL final wash) with Phosphate Buffered Saline (PBS) supplemented with 0.5 mM CaCl2 to remove EDTA. The tissues were digested for 16 h at 37°C in 1 ml of an enzymatic cocktail containing 1 mg/ml of Collagenase/Dispase (Roche) and 100 units/ml of Hyaluronidase (Calbiochem) in PBS/0.5 mM CaCl2. Next, 400 μl NST buffer (146 mM NaCl, 10 mM Tris base at pH 7.8, 1 mM CaCl2, 21 mM MgCl2, 0.05% BSA, 0.2% Nonidet P-40) was added to the samples and centrifuged for seven minutes at 5000 rpm. The pellets were resuspended in 800 μl of NST-DAPI (DAPI; 10 μg/mL), 0.1% DNase-free RNase A and 10% fetal bovine serum (LifeTechnologies), partially disaggregated by pipetting and passed through a 25 G needle at least 30 times. Nuclei suspensions were washed an additional three times with 800 μl of NST-DAPI, then filtered twice through a 35 μm-strainer mesh and collected into a Polystyrene round-bottom FACS tube. Samples were rested on wet ice for immediate sorting or supplemented with 10% DMSO for cryopreservation at −80°C. Frozen samples were processed in a similar manner with modifications due to the fragile nature of fresh/frozen nuclei. The 100 μm-thick sections were maintained on ice when possible, all washes (including NFR solution and washes during staining) were done using PBS/0.5 mM CaCl2, hydration/dehydration immersions were carried out using ethanol dilutions with PBS/0.5 mM CaCl2.
Nuclear DNA extraction and Multiplex-PCR assay
DNA from nuclei preparations was prepared using the PureLink™ DNA Mini Kit (LifeTechnologies). Fifty nanograms of DNA were subjected to template restoration using PreCR Repair Mix (NEB) in a reaction containing 1× ThermoPol Buffer, 100 μM dNTPs, 1× NAD+, 1 μl of PreCR mix and H2O to 50 μl, incubated 20 min at 37°C. The restored DNA was concentrated using the DNA Clean & Concentrator (Zymo Research) and identical amounts of repaired and unrepaired bulk DNA were subjected to Multiplex-PCR as previously described23 with minor modifications. The PCR reaction was performed with four primer sets that produce 100, 200, 300 and 400 bp fragments from non-overlapping target sites in the GAPDH gene (chr12) in 25 μl with final concentrations of 0.133 μM of each of the eight primers (Supplementary Table 3) and 1× AmpliTaq® Gold 360 DNA Master Mix. Multiplex-PCR was conducted as follows: 5 min 95°C, 37 cycles each of 15 sec 95°C, 15 sec 55°C and 30 sec 72°C, followed by 5 min 72°. Fifteen microliters of PCR product was loaded on a 2% TAE agarose gel and electrophoresed. Samples (unrepaired) were classified based on the largest of four possible PCR products detected. Unrepaired and repaired DNA samples were run side-by-side for comparison purposes (Supplementary Fig. 1).
DNA fluorescence in situ hybridization analysis
FISH analysis for Case 3 was performed using four 3-color probe panels on sequential sections of two frozen tissue blocks. Probe panel-1 consisted of bacterial artificial chromosome (BAC) clones spanning CCND1/11q13 (RP11-300I6, RP11-804L21; labeled with Green dUTP), SRC/20q11 (RP11-151C5, RP11-451G10; labeled with Red dUTP) and GNAQ/9q21 (RP11-747P3, RP11-574G7; labeled with Orange dUTP). Probe panel-2 consisted of BAC clones spanning CCND1/11q13 (as above), PTEN/10q23 (RP11-165M8, RP11-765C10; labeled with Red dUTP) and OCIAD1/4p14 (RP11-36B15; labeled with Orange dUTP). Probe panel-3 consisted of BAC clones spanning CCND1/11q13 (as above), PTEN/10q23 (as above) and the chromosome 10 centromeric region (CEN) (p10RP8; labeled with Orange dUTP). Probe panel-4 consisted of BAC clones spanning CCND1/11q13 (as above), OCIAD1/4p14 (as above) and chromosome 4 CEN (RP11-365H22, RP11-779E21; labeled with Red dUTP). FISH analysis for Case 4 was performed using a two-color ERBB2–centromeric chromosome 17 (CEP17) probe. The probe mix consisted of BAC clones containing the ERBB2 gene (RP11-94L15, RP11-62N23 and CTD-3211L18; labeled with Red dUTP) and a centromeric repeat plasmid for Chromosome 17 (clone p17H8p; labeled with Green dUTP). Probe labeling, tissue processing, hybridization, post-hybridization washing, and fluorescence detection were performed according to standard lab procedures35. Slides were scanned using a Zeiss Axioplan 2i epifluorescence microscope equipped with a CCD camera (CV-M4+CL, JAI) controlled by Isis 5.5.9 (MetaSystems Group Inc.). Each probe panel was hybridized on a separate unstained slide and the corresponding H&E slide/image was used to identify regions of DCIS or IDC. The entire section was first scanned under 63× objective to assess signal pattern. Normal breast epithelium and stromal elements served as the controls (i.e. internal controls: normal tissues adjacent to the lesions; external controls: three distinct normal breast tissue samples) to assess quality of hybridization and nuclear truncation (i.e. nuclei present only in part in the histologic section). At least five images/representative region were captured and a minimum of 50 discrete nuclei scored per region (each image was a compressed stack of 12 z-section images taken at 0.5 μm intervals). For Case 3, the distinct probe panels were used to validate and map the location of the distinct DCIS and IDC subclones previously identified by single-cell sequencing. For assessment of ITGH in Case 3 and ERBB2 amplification in Case 4, we followed the ASCO/CAP guidelines33.
Cell lines culture conditions
MCF-7 and CAMA-1 cells were purchased from the American Type Culture Collection (ATCC), authenticated and tested for mycoplasma contamination as previously described36,37, and maintained in 5% CO2-humidified atmosphere at 37°C in DMEM supplemented with 10% FBS, 2 mM L-Glutamine and 1% pen/strep.
Confocal imaging of FFPE and frozen nuclei
Nuclei derived from FFPE and frozen tissue from Cases 1 (tumor) and 2 (tumor and normal) were mounted with ProLong® Gold Antifade Reagent with DAPI (LifeTechnologies). Fluorescence stacked images (40–70 stacks/image) were acquired using a Leica TCS SP5-II Upright microscope (Leica Microsystems). Maximum Intensity Projection images from Supplementary Fig. 1e were generated using Fiji (ImageJ 2.0.0-rc-43-1.50e).
Formalin-fixed paraffin-embedded (FFPE) and frozen cell line pellet block preparation
The content of one confluent T-75 flask each line (i.e. MCF-7 and CAMA-1) was split in two identical aliquots and pelleted by centrifugation 5 min at 800 rpm. For FFPE blocks, the pellet was incubated for 1 h in 10% neutral buffered formalin at RT, then resuspended in 100μl warm HistoGel (ThermoScientific) and placed on ice for solidification. Solidified HistoGel pellets were then processed following the same routine protocol employed for surgical pathology specimens at MSKCC. For frozen blocks, the pellet was resuspended in O.C.T (Tissue-Tek) and placed on dry ice for solidification. Nuclei suspensions were prepared exactly as described above for tumor-derived tissue samples. To prepare accurate mixes (1:1 and 4:1, MCF-7:CAMA-1), cytometric analyses were performed for both FFPE and frozen nuclei preparations of each cell line. The number of total events per second and the percentage of single-cell events for FFPE MCF-7, FFPE CAMA-1, frozen MCF-7 and frozen CAMA-1 were determined and used to compute the fraction of single-cell events per second in each nuclei preparation, which were employed to prepare the mixes for sorting.
Flow cytometry analysis and sorting of single nuclei
Sorting of single nuclei was performed using a FACSAria II SORP (BD Biosciences). The DAPI signal was detected by a 355-nm UV laser (450/50 bandpass filter). Gains were set for the UV-photomultiplier based on the DNA content equivalent to human diploid lymphoblast cells. Prior to FACS, a cytometric analysis of ploidy (i.e. DNA content) distributions within each tumor sample was performed and compared to that of a diploid control sample (derived from a lymphoblastoid cell line of a healthy individual) to accurately determine the diploid peak position within the tumor and establish FACS collection gates. Single nuclei were determined by doublet discrimination as described by Wersto et al.38. Single cells were deposited into individual wells of a 96-well plate containing 10 μl of 1× Single Cell Lysis & Fragmentation Buffer and Proteinase K (Sigma-Aldrich).
Single-nucleus DNA repair and whole-genome amplification
Immediately after sorting, single nuclei retrieved from microdissected FFPE samples were incubated at 50°C for 2 h and stored at −80°C until further use. Given that the nuclear DNA of cases 3 and 4 was found to be fragmented (multiplex-PCR assay, Supplementary Fig. 1), the non-enzymatic random fragmentation prior to OmniPlex library generation was omitted. For cases 1 and 2, and the cancer cell lines, whose DNA was found to be of high quality (multiplex-PCR assay; Supplementary Fig. 1), a 2-min heating period at 99 °C (i.e., fragmentation to ~400 bp) was included. Single-nucleus DNA repair was performed prior to WGA by adding 1.3 μl of 10× ThermoPol Reaction Buffer, 0.26 μl of 50× dNTPs/NAD+ mix (5 mM/25 mM), 1.18 μl of H2O and 0.26 μl of PreCR mix (NEB) to the 10 μl nuclear lysis reaction and incubated for 30 min at 37°C. Repaired DNA was then directly combined with 2.6 μl 1× Single Cell Library Preparation Buffer and 1.3 μl Library Stabilization Solution, mixed thoroughly and placed in a thermocycler at 95°C for 2 min. The sample was then cooled to 4°C, briefly span down and supplemented with 1.3 μl of Library Preparation Enzyme, mixed thoroughly, placed in a thermocycler and incubated as follows: 16°C for 20 min, 24°C for 20 min, 37°C for 20 min, 75°C for 5 min and cooled to 4°C. These OmniPlex libraries were then combined with 44.3 μl of H2O, 7.5 μl of 10× Amplification Master Mix, mixed thoroughly and placed in a thermocycler, heating at 94°C for 3 min followed by 28 (Cases 1 and 2) or 30 (Cases 3 and 4) cycles of 94°C for 30 sec and 65°C for 5 min. Unrepaired DNA samples extracted from FFPE nuclei were subjected to identical cycling conditions. WGAs were assessed on a 1.5% agarose gel to confirm amplification and positive samples were processed further. Successful WGA reactions yielded ~50 μl of material at >100 ng/μl and had a WGA product spread between 100 and 800 bp. Frozen nuclei WGAs were performed following the standard protocol15.
For real-time WGA, the amplification reactions were supplemented with 0.1× SYBR® Green (Sigma-Aldrich) and 1 μl ROX® reference dye (LifeTechnologies) and ran in a StepOne Plus Real-Time PCR System (Applied Biosystems). Comparisons were performed using Mann-Whitney U test (GraphPad Prism 6).
Illumina library preparation, library pooling and multiplex sequencing
Five hundred nanograms of WGA products were acoustically sonicated using the Covaris E210 focus acoustics system (Covaris): Duty Cycle: 10%, Intensity: 4, Cycle/Bust: 200 and Time: 80 sec. The sonicated WGA was end-repaired, dA-tailed and barcoded (indices listed in Supplementary Table 3) following standard Illumina protocols. The FFPE and frozen indexed/barcoded libraries were pooled, PCR-enriched (primers listed in Supplementary Table 3 and QCed prior to sequencing on a HiSeq instrument (paired-end 76, 101 or single-read 76 Illumina sequencing runs).
Bioinformatics analysis of single-cell sequencing data
Single-cell sequencing data were processed and analyzed as previously described2,14,15. In brief, sequence reads were mapped using Bowtie39, PCR duplicates removed, and indexed using SAMtools40. Uniquely mapping reads were counted for each bin and normalized for GC-bias using lowess smoothing. Normalized read count data were then segmented using circular binary segmentation (CBS)41. For CN determination we employed our previously published approach based on least squares fitting14. Briefly, for single-cell data the CN at any position (bin) along the genome must be an integer value. In the case of a normal female diploid genome, the value of the CN in the majority of bins is expected to be 2, thus, the best-fit multiplier is 2. For rearranged cancer cells where CN of genome segments is unknown, the normalized bin count data is iteratively multiplied by 81 values ranging from 1.5 to 5.5 (0.05 increments). Next, a “quantal error” is computed for each multiplier, which is the sum of the squared difference between the multiplied segmented profile values and their nearest integer rounded counterparts. The multiplier that minimizes the “quantal error” is deemed as the best fit and used to estimate CN. Given that the ploidy of the tumor cells for each case was pre-estimated based on flow cytometry data, the thresholds of the multiplier were further constrained within the range determined by flow cytometry. All genome-wide CN profiles illustrated in the Figures were constructed using 20k data resolution14. Given the coverage we sequenced at, the analysis allows for accurate determination of CN states between 0–7 at a resolution of roughly 700 kbs. It also allows the capture of higher CN states in the form of amplifications. It should be noted however that for amplifications, the CN at a particular state in a collection of single cells is observed as a tight distribution of CN values.
Code availability
The code employed for the single-cell sequencing data analysis is available in Baslan et al.15
Bin-to-bin variance calculation
Noise in the read count data (i.e. bin-to-bin variance) was calculated by taking the summation of the square of the differences between the values of the normalized read count and the segmented data for all 5k bins14. The data were plotted in box plot format for each experimental group to illustrate subgroup variance.
Percent genome match analysis between single cells and corresponding bulk samples
Concordance of the CN at a particular bin between the single-cell and the corresponding bulk data was taken as a relative measure of accurate CN calling. For this, the percentage of matching bins, across all 5k bins, was calculated for each single cell and plotted in the form of box plot for each experimental condition. This was calculated for datasets that were rounded to the nearest integer following the least square-fitting algorithm CN estimation for the single-cell and bulk data. This represents a lower bound for the accuracy measure since regions that display subclonality in the bulk, and hence are not at integer CN states, are rounded to the nearest integer and therefore are expected to be discordant with the values at a subset of the single-cell data.
Calculation of number of single cells to sequence
To estimate a suitable number of single cells to sequence binomial statistics was used to calculate the power to observe a particular genetic alteration in single-cell data in 10%, 15% and 20% of the single cells. With the requirement of a particular alteration being found in a minimum of three single cells, it was determined that sequencing 48 single cells gives sufficient power to observe subclonal alterations at the aforementioned percentages.
False positive estimates
False positive estimation was calculated using non-rearranged single-cell data. Estimation was performed by assuming that for these cells any deviation from CN=2 in the autosomal bins represents a false positive. The percentage of bins that show this deviation was calculated for all cells and the average was computed. These calculations represent an upper bound on the false positive estimates as CNAs detected in single cells may actually represent somatic CNAs.
Coverage uniformity and GC-content bias
We used Ginkgo29 (http://qb.cshl.edu/ginkgo) to compute and plot the coverage uniformity (Lorenz curve28) and the GC-bias of repaired and unrepaired single-nuclei.
Hierarchical clustering and multi-dimensional scaling
Hierarchical clustering heatmaps were constructed using CN profiles of individual cells at 5K resolution20 using Manhattan and Ward as the distance function and clustering method, respectively. For multi-dimensional scaling (MDS), classical MDS was implemented using Euclidean distances with k=2 dimensions.
Statistical analysis of clustering
To estimate the probability of observing a cluster of cells sharing a group of breakpoints we estimated the false-positive breakpoint rate, p, from the normal-like diploid FFPE cell population, assuming as an upper bound that all events in this population are false positive calls. To estimate the probability of observing Nc or more cells out of N cells sharing B breakpoints we computed Σ (i=Nc−1)^N (N¦i) p^Bi (1−p^B)^(N−i). For the statistical analysis comparing FFPE and frozen normal cells we used the Mann-Whitney U test.
Low-pass whole genome sequencing and analysis
Libraries were prepared and analyzed as described in Baslan et al.14.
Whole-exome sequencing of Case 3 and bioinformatics analysis
Case 3 WES was performed on two sets of samples (plus matching normal) for each component, namely DCIS-1/IDC-1 and DCIS-2/IDC-2. The whole-exome capture for DCIS-1 and IDC-1 was performed using the NimbleGen SeqCap EZ Human Exome Library v2.0 (Roche), following the manufacturer’s protocol. The whole-exome enrichment for DCIS-2 and IDC-2 was performed with SureSelect Human All Exon v4 (Agilent Technologies), following the manufacturer’s protocol. The exon-enriched libraries were subjected to 2× 76 bp paired-end sequencing on an Illumina HiSeq2000 instrument. The median depths of coverage were 138× (range 125–150×, DCIS-1/IDC-1) and 203× (range 93–217×, DCIS-2/IDC-2) (Supplementary Table 4). Bioinformatics analysis was conducted exactly as previously described4. CN alterations were inferred from MPS data using FACETS42 and genes with altered CN were determined adopting the methods described in Curtis et al.43. Gene amplification, homozygous deletion and loss of heterozygosity events were visually reviewed using plots of raw Log2 and allele ratios.
The cancer cell fraction (CCF) of each mutation was inferred using the number of reads supporting the reference and the alternate alleles and the segmented log ratio from WES as input for ABSOLUTE (v1.0.6)11. Solutions from ABSOLUTE were manually reviewed as described11,44. A mutation was classified as clonal if its probability of being clonal was >50% or if the lower bound of the 95% confidence interval of its CCF was >90%, as previously described45.
Phylogeny reconstruction
CN and mutation based phylogenetic trees were made using the R package Phangorn46, with the maximum parsimony optimality criterion, as previously described45. The levels for the mutational tree were binary, based on the presence or absence of the mutations within each sample. For the CN data the levels of alteration were categorized as homozygous deletion, loss, unchanged, gain and amplification. The Bioconductor package CGHregions47 was used for dimension reduction of the CN alteration matrix, in order minimize the weight of large alterations, and the resulting matrix was used as input for Phangorn.
Representation of the evolutionary path based on single-cell data from Case 3 was constructed manually using the sub-clonal information inferred from the breakpoint probability analysis, identified clonal alterations, and the calculated relative abundance of each identified sub-clone, essentially as previously described48.
Statistical analysis
Comparisons of Ct values to define the template DNA available in WGA nuclei using real-time SYBR Green reactions were performed using the Mann-Whitney U test (GraphPad Prism 6). Spearman’s correlations were employed to define the correlations between single cell and bulk CN profiles.
Supplementary Material
Acknowledgments
We thank the M. Wigler lab (Cold Spring Harbor Laboratory, CSHL) for kindly providing access to necessary equipment, and M. Schatz, T. Garvin and R. Aboukhalil (CSHL) for their assistance with Ginkgo, an interactive, online platform for the analysis of CNAs from single cells. We thank the CSHL Flow Cytometry Shared Resources, which is supported in part by the National Cancer Institute Cancer Center Shared Grant award number CA045508. We thank S. Turcan and J. Taranda for critically reviewing the manuscript. S. Piscuoglio was funded in part by a Susan G. Komen Postdoctoral Fellowship Grant (PDF14298348). T. Baslan is supported by the MSKCC Single Cell Sequencing Initiative and the William and Joyce O’Neil Research Fund. J.S. Reis-Filho is funded in part by Breast Cancer Research Foundation. This study was funded in part by a Susan G. Komen Investigator-Initiated Research Grant (IIR13265578). Research reported in this publication was supported in part by the Cancer Center Support Grant of the National Institutes of Health/National Cancer Institute under award number P30CA008748. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
AUTHOR CONTRIBUTIONS
J.S.R.-F., B.W. and J.B.H. conceived and supervised the study. L.G.M. developed the single-cell FFPE methodology, designed and conducted the experiments. TB and JK conducted the bioinformatics and statistical analyses of the single-cell data. K.B. and C.K.Y.N. performed the bioinformatics of WES data. M.S. performed single-cell data preprocessing for use in the Ginkgo platform. S.P. prepared the cell lines FFPE/frozen blocks. L.S. prepared the sequencing libraries of FFPE/frozen cell lines and performed confocal microscopy of sorted nuclei. K.C. and G.N. conducted the FISH experiments, which were analyzed by K.C., G.N. and F.C.G. T.A.K. and H.Y.W. provided with the tumor samples. J.S.R.-F. and F.C.G. reviewed and microdissected the histological samples. P.M. and L.R. performed flow-cytometric analysis and sorting. T.B. prepared the LP-WGS libraries. H.C., A.S. and A.d.C.P. prepared the WES libraries. L.G.M., T.B., J.K., B.W., J.B.H. and J.S.R.-F. analyzed, discussed and interpreted the data and wrote the manuscript. All authors reviewed and approved the manuscript for submission.
COMPETING FINANCIAL INTERESTS STATEMENT
The authors have no conflicts of interest to declare.
Accession codes
Sequence Read Archive (SRA), accession SRP008292.
Data availability statement
Single-cell, low-pass whole-genome and whole-exome sequencing data have been deposited in the Sequence Read Archive (SRA) under accession SRP008292.
References
- 1.Gerlinger M, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Navin N, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aparicio S, Caldas C. The implications of clonal genome evolution for cancer medicine. N Engl J Med. 2013;368:842–851. doi: 10.1056/NEJMra1204892. [DOI] [PubMed] [Google Scholar]
- 4.Ng CK, et al. Intra-tumor genetic heterogeneity and alternative driver genetic alterations in breast cancers with heterogeneous HER2 gene amplification. Genome Biol. 2015;16:107. doi: 10.1186/s13059-015-0657-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hernandez L, et al. Genomic and mutational profiling of ductal carcinomas in situ and matched adjacent invasive breast cancers reveals intra-tumour genetic heterogeneity and clonal selection. J Pathol. 2012;227:42–52. doi: 10.1002/path.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Bruin EC, et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science. 2014;346:251–256. doi: 10.1126/science.1253462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yates LR, et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med. 2015;21:751–759. doi: 10.1038/nm.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ng CK, Schultheis AM, Bidard FC, Weigelt B, Reis-Filho JS. Breast cancer genomics from microarrays to massively parallel sequencing: paradigms and new insights. J Natl Cancer Inst. 2015;107 doi: 10.1093/jnci/djv015. pii: djv015. [DOI] [PubMed] [Google Scholar]
- 9.Ding L, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nik-Zainal S, et al. The life history of 21 breast cancers. Cell. 2012;149:994–1007. doi: 10.1016/j.cell.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carter SL, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roth A, et al. PyClone: statistical inference of clonal population structure in cancer. Nat Methods. 2014;11:396–398. doi: 10.1038/nmeth.2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512:155–160. doi: 10.1038/nature13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baslan T, et al. Optimizing sparse sequencing of single cells for highly multiplex copy number profiling. Genome Res. 2015;25:714–724. doi: 10.1101/gr.188060.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baslan T, et al. Genome-wide copy number analysis of single cells. Nat Protoc. 2012;7:1024–1041. doi: 10.1038/nprot.2012.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leung ML, Wang Y, Waters J, Navin NE. SNES: single nucleus exome sequencing. Genome Biol. 2015;16:55. doi: 10.1186/s13059-015-0616-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Voet T, et al. Single-cell paired-end genome sequencing reveals structural variation per cell cycle. Nucleic Acids Res. 2013;41:6119–6138. doi: 10.1093/nar/gkt345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zardavas D, Irrthum A, Swanton C, Piccart M. Clinical management of breast cancer heterogeneity. Nat Rev Clin Oncol. 2015;12:381–394. doi: 10.1038/nrclinonc.2015.73. [DOI] [PubMed] [Google Scholar]
- 19.Gilbert MT, et al. The isolation of nucleic acids from fixed, paraffin-embedded tissues-which methods are useful when? PLoS One. 2007;2:e537. doi: 10.1371/journal.pone.0000537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng Z, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med. 2014;20:1479–1484. doi: 10.1038/nm.3729. [DOI] [PubMed] [Google Scholar]
- 21.Van Allen EM, et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat Med. 2014;20:682–688. doi: 10.1038/nm.3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greer CE, Peterson SL, Kiviat NB, Manos MM. PCR amplification from paraffin-embedded tissues. Effects of fixative and fixation time. Am J Clin Pathol. 1991;95:117–124. doi: 10.1093/ajcp/95.2.117. [DOI] [PubMed] [Google Scholar]
- 23.van Beers EH, et al. A multiplex PCR predictor for aCGH success of FFPE samples. Br J Cancer. 2006;94:333–337. doi: 10.1038/sj.bjc.6602889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adams S, et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J Clin Oncol. 2014;32:2959–2966. doi: 10.1200/JCO.2013.55.0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hosein AN, et al. Evaluating the repair of DNA derived from formalin-fixed paraffin-embedded tissues prior to genomic profiling by SNP-CGH analysis. Lab Invest. 2013;93:701–710. doi: 10.1038/labinvest.2013.54. [DOI] [PubMed] [Google Scholar]
- 26.Bosso M, Al-Mulla F. Whole genome amplification of DNA extracted from FFPE tissues. Methods Mol Biol. 2011;724:161–180. doi: 10.1007/978-1-61779-055-3_11. [DOI] [PubMed] [Google Scholar]
- 27.Dean FB, et al. Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci U S A. 2002;99:5261–5266. doi: 10.1073/pnas.082089499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zong C, Lu S, Chapman AR, Xie XS. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science. 2012;338:1622–1626. doi: 10.1126/science.1229164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garvin T, et al. Interactive analysis and assessment of single-cell copy-number variations. Nat Methods. 2015;12:1058–1060. doi: 10.1038/nmeth.3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deleye L, et al. Whole genome amplification with SurePlex results in better copy number alteration detection using sequencing data compared to the MALBAC method. Sci Rep. 2015;5:11711. doi: 10.1038/srep11711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lakhani SR, Ellis IO, Schnitt SJ, Tan PH, van de Vijver MJ. WHO Classification of Breast Tumors. IARC; Lyon: 2012. [Google Scholar]
- 32.Sakr RA, et al. PI3K pathway activation in high-grade ductal carcinoma in situ–implications for progression to invasive breast carcinoma. Clin Cancer Res. 2014;20:2326–2337. doi: 10.1158/1078-0432.CCR-13-2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wolff AC, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol. 2013;31:3997–4013. doi: 10.1200/JCO.2013.50.9984. [DOI] [PubMed] [Google Scholar]
- 34.Hammond ME, et al. American Society of Clinical Oncology/College Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol. 2010;28:2784–2795. doi: 10.1200/JCO.2009.25.6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Piscuoglio S, et al. Uterine adenosarcomas are mesenchymal neoplasms. J Pathol. 2016;238:381–388. doi: 10.1002/path.4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weigelt B, Warne PH, Lambros MB, Reis-Filho JS, Downward J. PI3K pathway dependencies in endometrioid endometrial cancer cell lines. Clin Cancer Res. 2013;19:3533–3544. doi: 10.1158/1078-0432.CCR-12-3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weinreb I, et al. Hotspot activating PRKD1 somatic mutations in polymorphous low-grade adenocarcinomas of the salivary glands. Nat Genet. 2014;46:1166–1169. doi: 10.1038/ng.3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wersto RP, et al. Doublet discrimination in DNA cell-cycle analysis. Cytometry. 2001;46:296–306. doi: 10.1002/cyto.1171. [DOI] [PubMed] [Google Scholar]
- 39.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Venkatraman ES, Olshen AB. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics. 2007;23:657–663. doi: 10.1093/bioinformatics/btl646. [DOI] [PubMed] [Google Scholar]
- 42.Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016 doi: 10.1093/nar/gkw520. pii: gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Curtis C, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Landau DA, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152:714–726. doi: 10.1016/j.cell.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schultheis AM, et al. Massively Parallel Sequencing-Based Clonality Analysis of Synchronous Endometrioid Endometrial and Ovarian Carcinomas. J Natl Cancer Inst. 2016;108 doi: 10.1093/jnci/djv427. djv427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schliep KP. Phangorn: phylogenetic analysis in R. Bioinformatics. 2011;27:592–593. doi: 10.1093/bioinformatics/btq706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van de Wiel MA, van Wieringen WN. CGHregions: Dimension Reduction for Array CGH Data with Minimal Information Loss. Cancer Inform. 2007;3:55–63. [PMC free article] [PubMed] [Google Scholar]
- 48.Krasnitz A, Sun G, Andrews P, Wigler M. Target inference from collections of genomic intervals. Proc Natl Acad Sci U S A. 2013;110:E2271–2278. doi: 10.1073/pnas.1306909110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.