

Abstract
G-Protein-Coupled Receptors (GPCRs) are a large family of transmembrane receptors that play critical roles in normal cellular physiology and constitute a major pharmacological target for multiple indications, including analgesia, blood pressure regulation, and the treatment of psychiatric disease. Upon ligand binding, GPCRs catalyze the activation of intracellular G-proteins by stimulating the incorporation of guanosine triphosphate (GTP). Activated G-proteins then stimulate signaling pathways that elicit cellular responses. GPCR signaling can be monitored by measuring the incorporation of a radiolabeled and non-hydrolyzable form of GTP, [35S]guanosine-5'-O-(3-thio)triphosphate ([35S]GTPγS), into G-proteins. Unlike other methods that assess more downstream signaling processes, [35S]GTPγS binding measures a proximal event in GPCR signaling and, importantly, can distinguish agonists, antagonists, and inverse agonists. The present protocol outlines a sensitive and specific method for studying GPCR signaling using crude membrane preparations of an archetypal GPCR, the µ-opioid receptor (MOR1). Although alternative approaches to fractionate cells and tissues exist, many are cost-prohibitive, tedious, and/or require non-standard laboratory equipment. The present method provides a simple procedure that enriches functional crude membranes. After isolating MOR1, various pharmacological properties of its agonist, [D-Ala, N-MePhe, Gly-ol]-enkephalin (DAMGO), and antagonist, naloxone, were determined.
Keywords: Biochemistry, Issue 124, Cellular fractionation, membrane preparation, GPCR, agonist, antagonist, GTPγS binding, pharmacology, µ-opioid receptor
Introduction
G-Protein-Coupled Receptors (GPCRs) are a large family of cell-surface receptors responsible for a remarkable array of physiological processes, including analgesia, olfaction, and behavior1. GPCRs act by sensing specific external signals and subsequently stimulating intracellular signaling. They therefore mark a key junction between the external and internal environments of a cell. Due to the critical role GPCRs play in biology, they have become major targets for both basic research and drug discovery2,3.
Unlike other receptor families that bind discrete ligands, GPCRs can bind very different types of molecules. While one GPCR may interact with peptides, another may sense photons, small molecules, or ions1,4. While their ligands are diverse, GPCRs are unified in their overall architecture and function. Individual GPCRs are made up of seven α-helical transmembrane proteins with extracellular amino terminals and intracellular carboxyl terminals5,6. GPCRs are coupled to intracellular G-proteins—heterotrimeric protein complexes composed of α, β, and γ subunits—which mediate diverse signaling pathways7. The Gα subunit is a guanine nucleotide-binding protein that is inactive when bound to guanosine diphosphate (GDP) and active when bound to guanosine triphosphate (GTP)8,9. When GPCRs bind their ligands, they undergo a conformational change that permits Gα to dissociate from Gβγ, thereby allowing Gα to exchange GDP for GTP7. The receptor itself is phosphorylated at its carboxyl terminal by various serine/threonine kinases10,11 and internalized to attenuate receptor signaling12,13,14. Meanwhile, the activated Gα monomer and Gβγ dimer proceed to activate distinct signaling pathways7. There are several isoforms of each G-protein subunit, and each isoform targets particular downstream pathways and secondary messenger systems. The major Gα isoforms include Gs, Gq, Gi/o, and G12-13. Typically, individual GPCRs associate with a particular Gα isoform, thereby linking an external stimulus to a specific cellular response1.
Characterizing a GPCR-ligand interaction is critical to understanding the biology of the receptor. As GDP/GTP exchange is one of the earliest events that follows ligand binding, monitoring GTP binding can measure GPCR activation or inhibition. Assaying more downstream events in GPCR signaling is often not as quantitative or stoichiometric, may not distinguish full agonists from partial ones, and can require expensive reagents. Moreover, increased GTP binding to Gα proteins is an almost-universal event following GPCR activation, meaning that measuring GTP binding is a broadly applicable assay for monitoring the activity of most GPCRs. Measuring GTP binding is a simple and rapid approach to monitor GPCR signaling in cells overexpressing the receptor of interest or in native tissue. The present protocol details a functional GTP-binding assay using an archetypal GPCR, the µ-opioid receptor (MOR1), to quantitatively determine the activity of an agonist and antagonist on GPCR signaling.
This protocol first outlines how to isolate crude membranes from cells overexpressing MOR1. Note that this protocol is not limited to overexpression systems and can be applied to many sources of membrane, including native tissue or preparations expressing multiple receptors and G proteins15. The protocol then details how to measure the binding of a radioactive GTP analog to these membranes in response to varying concentrations of [D-Ala, N-MePhe, Gly-ol]-enkephalin (DAMGO) or naloxone, a MOR1 agonist and antagonist, respectively. The GTP analog, [35S]guanosine-5'-O-(3-thio) triphosphate ([35S]GTPγS), is non-hydrolyzable. This property is critical because Gα subunits exhibit intrinsic GTPase activity7 and would eliminate the labeled gamma phosphate on a hydrolyzable GTP radiochemical. Membranes are then trapped onto glass fiber filters and washed, after which the radiolabeled GTP is quantified by liquid scintillation counting. Multiple pharmacological parameters can be derived to characterize the receptor-ligand interaction, including the half-maximal response (EC50) and Hill coefficient (nH) for agonists and the half-maximal inhibitory concentration (IC50) and equilibrium dissociation constant (Kb) for antagonists16,17,18.
Protocol
1. Expression of Recombinant HA-MOR1 in Cultured Cells
NOTE: Follow all cell culture protocols in a sterile laminar flow hood.
Sterilize the cell culture laminar flow hood with 70% ethanol and maintain sterile technique throughout the cell culture.
Prepare human embryonic kidney cells 293 (HEK293) cell culture medium, complete Dulbecco's-modified Eagle Medium (DMEM): DMEM, pH 7.4, supplemented with 2 mM L-glutamine, 1% penicillin/streptomycin, and 10% fetal bovine serum (FBS).
Plate 2.5 x 106 HEK293 cells onto a 10 cm tissue culture plate in 10 mL of complete DMEM and incubate at 37 °C and 5% CO2 until 70-80% confluency is reached (O/N). NOTE: To collect enough protein for a complete GTP-binding experiment, plate at least 3 x 10 cm plates of cells.
Transfect the cells with HA-MOR1 using a transfection reagent of choice and by following the manufacturer's guidelines. Incubate for 36-48 h. NOTE: Human MOR-1 cDNA (NCBI Reference Sequence: NM_000914.4) was a generous gift from Gavril Pasternak and was cloned into pCMV-HA.
2. Cell Fractionation and Membrane Collection
- Prepare the following lysis buffers:
- Buffer 1: 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.4; 1 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N',N'-tetraacetic acid (EGTA); 10% sucrose; protease inhibitor cocktail; and 1 mM dithiothreitol (DTT). Add the DTT fresh on the day of membrane isolation.
- Buffer 2: 10 mM HEPES, pH 7.4; 1 mM EGTA; 1 mM MgCl2; and 1 mM DTT. Add the DTT fresh on the day of membrane isolation.
Remove the transfected cells from the incubator (step 1.4), aspirate the medium, and rinse the cells with 5 mL of ice-cold Phosphate-buffered Saline (PBS). Aspirate the PBS and excess liquid from the plate.
Add 600 µL of Buffer 1 to the cells. Using a cell scraper, dislodge the cells from the plate surface and pipette the cell suspension into a 1.6 mL microcentrifuge tube.
Immediately snap-freeze the microcentrifuge tube in liquid nitrogen. Once the samples are frozen, thaw the lysates on ice or place them at -80 °C for long-term storage.
Repeat steps 2.3 and 2.4 with all plates of cells.
- Once the lysates have thawed, use a single-pulse micro-tube homogenizer to break open the cells. Pulse the homogenizer 3-5 times for 10 s, placing the tube back on ice for 30 s between pulses.
- Collect 20 µL as whole cell fraction.
Centrifuge the remaining sample for 10 min at 1,000 x g and 4°C. Collect the supernatant and place in a new 1.6 mL microcentrifuge tube on ice.
- Resuspend the pellet in 100 µL of Buffer 1 and rehomogenize with a pestle. Pulse the homogenizer 3-5 times for 5 s, placing the tube back on ice for 30 s between pulses.
- Centrifuge the sample a second time for 10 min at 1,000 x g and 4 °C. Combine the supernatant with the supernatant from step 2.7. NOTE: The remaining pellet contains nuclear and membrane proteins.
Centrifuge the combined supernatants for 20 min at 11,000 x g and 4 °C. Separate the supernatant (the cytosolic fraction) into a new 1.6 mL microcentrifuge tube.
Resuspend the pellet in 200 µL of Buffer 2. Homogenize the pellet by triturating it 3-5 times. Centrifuge the sample for 20 min at 21,100 x g and 4 °C. Aspirate the supernatant. Resuspend the pellet containing the crude membrane fraction in 50 µL of Buffer 2.
Immediately proceed to the GTPγS binding experiments (section 3) or snap-freeze in liquid nitrogen and store at -80 °C.
3. [35S]GTPγS Binding
NOTE: Use standard radiochemical safety protocol when handling [35S]GTPγS and when conducing [35S]GTPγS binding experiments. Wear protective gloves and a lab coat at all times. Check the packaging material for leaks or cracks. Dispose of waste and excess reagents according to institutional protocols.
Prepare Incomplete Binding Buffer (IBB) by mixing 50 mM HEPES, pH 7.4; 5 mM MgCl2; 100 mM NaCl; and 1 mM ethylenediaminetetraacetic acid (EDTA) in distilled water.
Dilute stock [35S]GTPγS to 50 nM in 10 mM Tris-HCl, pH 7.6, and 10 mM DTT. Make 500 µL aliquots and store them at -80 °C. Use only a freshly thawed aliquot immediately before initiating the experiment. Thaw the aliquots on ice.
Prepare nonradio-labeled GTPγS by dissolving 1 mg of GTPγS powder in 177.62 µL of pure water for a stock concentration of 10 mM. Make 10-µL aliquots and store them at -20 °C.
Prepare complete binding buffer (CBB) by supplementing IBB to include a final concentration of 1 mM DTT, 0.1% (wt/vol) bovine serum albumin (BSA), 10 µM GDP, and 0.1 nM [35S]GTPγS. NOTE: The CBB now contains radiolabeled GTP. Take appropriate safety precautions while working with radioactive solutions.
Thaw the membrane fraction from step 2.11 on ice.
- Quantify the protein concentration of the membrane fraction via a Bradford protein assay using a spectrophotometer at 595 nm.
- Prepare a dilution series of BSA protein standards with Buffer 2, at final concentrations of 0, 250, 500, 750, 1,500, 2,500, and 5,000 µg/mL.
- Add 2 µL of each standard or membrane to 1 mL of Bradford reagent in a cuvette. Mix thoroughly by vortexing.
- Adjust the spectrophotometer to a wavelength of 595 nm and blank using the cuvette with 0 µg/mL protein.
- Wait 5 min and read each of the standards and each of the samples at a 595 nm wavelength.
- Plot the absorbance of the standards versus the concentration. Using the Beer-Lambert Law (A = εcl, where ε = extinction coefficient, l = length of cuvette, and c = concentration), calculate the concentration of the membrane samples.
Dilute the membrane fractions to a concentration of 100 µg protein/mL in IBB. NOTE: One 10 cm dish of HEK293 cells typically yields between 1 & 3 mg protein/mL.
- Prepare the following experimental conditions in individual 1.6 mL microcentrifuge tubes: a ligand dilution series, a basal activity condition to measure basal GTP binding, and a nonspecific binding condition to measure nonspecific GTP binding.
- For the ligand dilution series, prepare a dilution series of the ligand of interest in a final volume of 100 µL of CBB. Prepare the ligand series at 2x the desired final concentrations. Space the ligand concentrations to adequately cover a range of responses.
- To assay the effect of DAMGO on GTP binding via MOR1, prepare DAMGO dilutions of 2 mM, 200 µM, 20 µM, 2 µM, 200 nM, 20 nM, 2 nM, 200 pM, 20 pM, and 2 pM in CBB (final volume: 100 µL). NOTE: For example, if the ligand of interest has an expected EC50 value of 10 µM, prepare ligand dilutions of 200 nM, 2 µM, 20 µM, 200 µM, and 2 mM. These five conditions cover a wide range of concentrations and are 2x the concentration desired for the assay.
- For basal binding, prepare a tube with 100 µL of CBB only.
- For nonspecific binding, prepare a tube with 99 µL of CBB supplemented with 1 µL of 2 mM nonradio-labeled GTPγS.
Add 100 µL of the diluted membrane solution (step 3.7) to each experimental condition.
Incubate the membranes with the various experimental conditions in 1.6 mL microcentrifuge tubes for 30 min at 25 °C in a thermomixer or on an orbital shaker.
4. Membrane Filtration
Prepare wash buffer by combining 50 mM Tris-HCl, pH 7.4; 5 mM MgCl2; and 50 mM NaCl in distilled water.
Presoak the glass fiber filters in water for 10 min. NOTE: Filters have a pore size of 1 µm, a diameter of 2.1 cm, and a thickness of 675 µm.
Remove the samples from the thermomixer or orbital shaker (step 3.10). Briefly pulse-spin the samples for 5 s to collect each sample at the bottom of the tube.
Remove the lid of the vacuum filtration apparatus. Lay the presoaked filters onto the vacuum ports of the apparatus. Re-secure the apparatus lid to form a vacuum seal. Turn on the vacuum.
Pipette 195 µL of each 200 µL experimental condition onto filters to minimize error from adsorption to the walls of the tube and/or from pipetting.
Wash the filters three times with 1 mL of ice-cold wash buffer.
5. Liquid Scintillation Counting
Place 5 mL scintillation counting vials in a counting rack. Add 5 mL of scintillation fluid to each vial.
Turn off the vacuum (step 4.4). Remove the lid of the vacuum filtration apparatus. Using tweezers, pick up the filters from the vacuum ports of the filtration apparatus and drop each filter into an individual 5 mL scintillation vial.
Prepare one vial with 195 µL of CBB to determine the maximal signal.
Cap each vial securely. Incubate the vials on an orbital shaker at 25 °C for 10 min.
Turn on the scintillation counter. Program the scintillation counter to measure 35S isotope emission for 5 min per sample using the standard associated scintillation program.
Press "Start" to take a count.
When the counting is done, dispose the counted vials as hazardous waste.
6. Data Analysis
- Enter the data into statistics and analysis software.
- Subtract the non-specific binding from each of the other measurements to determine the specific binding. NOTE: The degree of nonspecific binding is determined by the sample incubated with non-radiolabeled GTPγS (step 3.8.3).
- Open the statistics and analysis software and select an XY dataset entry.
- Enter the specific binding data from the [35S]GTPγS-binding experiments into a data table in the statistics and analysis software. Enter the agonist concentrations, as molar concentrations, in the "X" column. Enter the associated 35S counts as "Y" values.
- Transform the data.
- Convert the X values to their respective logarithm by clicking "Analyze." Select "Transform" from the built-in analyses.
- Within the "Transform" dialog box, select "Transform X values using" and select the function "X=log(X)." Choose to create a new graph of the results.
- Click "OK."
- Normalize the Y-values.
- With the transformed results displayed, click "Analyze." Select "Normalize" from the built-in analyses.
- Within the "Normalize" dialog box, select the radio button to define 0% as the "smallest value in each data set" and 100% as the "largest value in each data set." Select the box to present the results as percentages. Select the box to create a new graph of the results.
- Click "OK."
- Fit a nonlinear regression curves to the plotted data. Perform a nonlinear regression analysis.
- With the normalized results displayed, click "Analyze." From the built-in analyses, select "Nonlinear regression (curve fit)."
- If investigating an agonist, select "log(agonist) vs. normalized response - variable slope" from the dialog box.
- If investigating an antagonist, "select log(antagonist) vs. normalized response - variable slope" from the dialog box.
- Click "OK."
Review the graph and results to ensure that the fit and data are in reasonable agreement. Ensure that the regression matches the general pattern of the plotted data and that the residuals are not so large that they render the dose-response curve flat. NOTE: The software will summarize the results of the nonlinear regression and details the concentration that elicits the half-maximal response (EC50), Hill coefficient (nH), and half-maximal inhibitory concentration (IC50).
- Derive agonist/antagonist parameter potency.
- Derive the agonist equilibrium dissociation constants (Kb) from either of the following experiments16,17,18
- Assess the shift in the dose-response of an agonist in competition with a fixed concentration of antagonist. Here, EC50' = EC50 (1 + [antagonist]/antagonistKb), where EC50' is the right-shifted EC50.
- Assess [35S]GTPγS binding with varying concentrations of antagonist in competition with a fixed concentration of agonist. Here, Kb = IC50/ (2 +([agonist]/agonist EC50)n)1/n - 1, where n is the Hill coefficient of the agonist.
Depending upon the variability of the data, repeat the experiment (steps 3.8-5.6) for each ligand approximately 3-5 times and average the results using a statistics and analysis software.
Representative Results
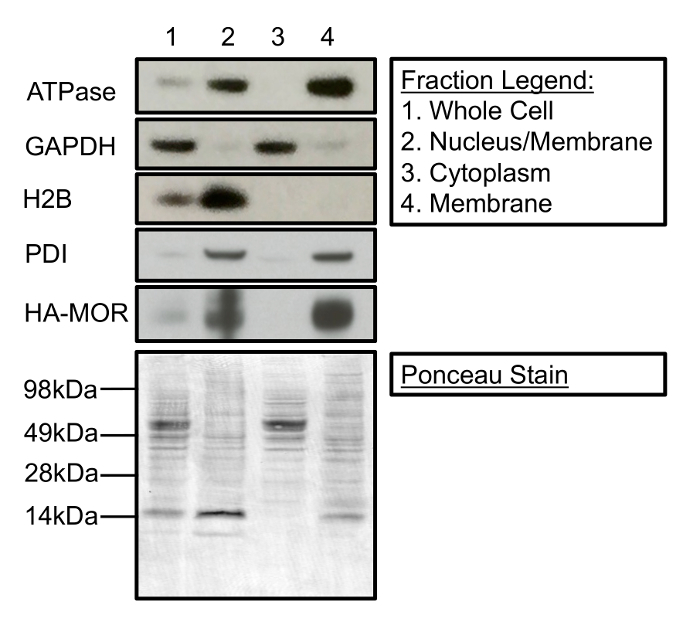
Cell fractionation can be used to isolate and enrich membrane-associated proteins from cytosolic and nuclear proteins. Figure 1 is a Western blot demonstrating the contents of the three primary fractions that can be collected during the subcellular fractionation process. Specifically, Figure 1 shows that fractionation cleanly separates membrane proteins (i.e. Na+/K+ ATPase, protein disulfide isomerase (PDI), and HA-MOR1) from histone H2B and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), nuclear proteins, and cytosolic proteins, respectively. Additionally, Figure 1 demonstrates the enrichment of proteins (lane 1 compared to lane 4) in their respective subcellular fractions as a result of fractionation. A representative Ponceau stain demonstrates equal protein loading in each fraction. It is important to note that this fractionation protocol does not distinguish between different cellular membranes. PDI is generally localized to the endoplasmic reticulum (ER), whereas Na+/K+ATPase is predominantly at the plasma membrane. However, both proteins are present in the final crude membrane fraction (Figure 1, lane 4). Additionally, while this protocol robustly separates nuclear proteins from the cytosolic and final membrane fraction (Figure 1, lanes 2-4), the fraction enriched for nuclear proteins contains some membrane proteins (Figure 1, lane 2), possibly from the ER.
Multiple pharmacological parameters can be derived to characterize a GPCR-ligand interaction via GTP-binding experiments (Table 1). For example, the half-maximal response (EC50) and Hill coefficient (nH) of an agonist can be derived by monitoring GTP binding in response to varying doses of the agonist. Figure 3A demonstrates dose-responsive GTP binding to MOR1 after DAMGO treatment. When the data is fit to a four-parameter nonlinear regression, the fit describes a receptor-ligand interaction with an EC50 of 185 ± 23 nM and a Hill coefficient of 0.46 ± 0.06. The shallow GTP-binding curve observed after DAMGO treatment suggests negative cooperativity between DAMGO and MOR1. This method can also identify and describe the pharmacology of an antagonist. As Figure 3A and B illustrates, naloxone is an MOR1 antagonist. The agonist potency (Kb) of naloxone, 97 ±20 nM, was determined by varying the concentration of naloxone in competition with a fixed concentration of DAMGO (Figure 3A). Naloxone exhibited a Hill coefficient of 0.88 ±0.06, suggesting independent binding between naloxone and MOR1. If the action of a ligand is unknown, this assay can discriminate between an agonist, antagonist, and inverse agonist. If the ligand is an agonist, there would be an increase in GTP binding, as in Figure 3A, following DAMGO application. If the ligand is an inverse agonist, there would be diminished binding of GTP relative to basal binding. If the ligand is an antagonist, there would be no effect upon treatment with ligand alone. If applied concomitantly with an agonist, an antagonist would inhibit the ability of the agonist to stimulate GTP binding. Figure 3A illustrates the antagonist activity of naloxone against the agonist DAMGO.
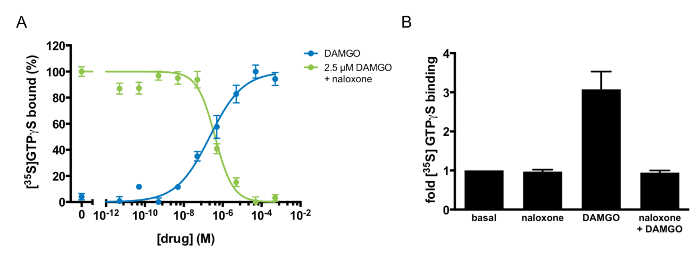
Figure 1: Cellular Fractionation Separates Membrane-associated, Nuclear, and Cytosolic proteins. (Top) Lane 1 represents the protein present in the whole cell. Lane 2 contains nuclear and membrane-associated proteins separated during the first centrifugation steps. Lane 3 is the cytosolic fraction separated following 20 min of centrifugation at 11,000 x g. Lane 4 contains a crude membrane fraction suitable for [35S]GTPγS-binding experiments. (Bottom) Ponceau stain of a Western membrane demonstrates protein loading for each cellular fraction. The following antibodies were used for immunoblotting: mouse anti-Na+/K+-ATPase (1:1,000), mouse anti-GAPDH (1:5,000), mouse anti-H2B (1:2,500), rabbit anti-PDI (1:1,000), and rat anti-HA (1:2,000). Please click here to view a larger version of this figure.
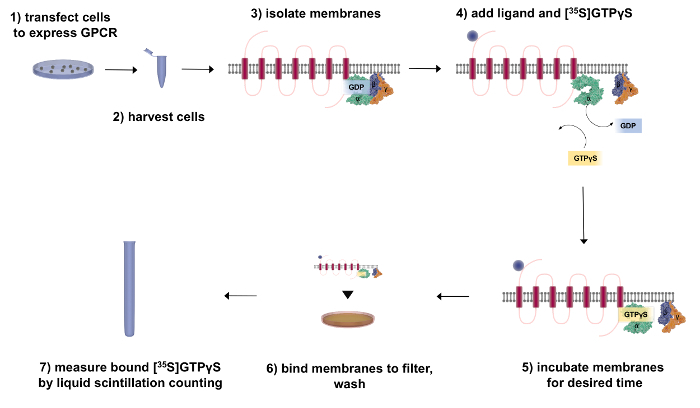
Figure 2: Flow Chart Outlining the Fractionation and [35S]GTPγS-binding Procedure. First, transfect the cells to express the GPCR of interest. After 48 h, harvest and fractionate the cells to isolate the receptor (red) and associated G-proteins (green = Gα, purple = Gβ, and orange = Gγ). To perform GTP-binding experiments, add [35S]GTPγS and incubate the membranes with the ligand of interest. To measure G-protein activity, bind the membranes to a filter to wash away any unbound radiochemical and then quantify the bound [35S]GTPγS by liquid scintillation counting. Please click here to view a larger version of this figure.
Figure 3: Agonism and Antagonism at µ-opioid Receptors Defined by [35S]GTPγS binding. (A) [35S]GTPγS dose-response curve to DAMGO alone (blue) or naloxone in competition with 2.5 µM DAMGO (green). [35S]GTPγS binding was normalized to the maximal stimulation in each experiment and was expressed as a percentage. The points shown are the mean of triplicate determinations and are expressed as the mean ±S.E.M. (B) Antagonism of DAMGO-stimulated [35S]GTPγSbinding by naloxone. [35S]GTPγSbinding was quantified after the addition of naloxone alone (100 µM), DAMGO alone (10 µM), or DAMGO (10 µM) in competition with naloxone (100 µM). The results are expressed as the mean ±S.E.M. of three independent experiments. Please click here to view a larger version of this figure.
| Ligand | EC50 or IC50 (nM) | nH | Kb (nM) |
| DAMGO | 185 ±23 | 0.46 ±0.06 | |
| Naloxone | 420 ±87 | 0.88 ±0.06 | 97 ±20 |
Table 1: Pharmacological Parameters of DAMGO and Naloxone Activity at µ-opioid Receptors. The half-maximal response (EC50) and Hill coefficient (nH) for DAMGO were derived from [35S]GTPγS binding in response to varying doses of DAMGO. The half-maximal inhibitory concentration (IC50) and equilibrium dissociation constant (Kb) for naloxone were determined from the effect of competition between naloxone and 2.5 µM DAMGO on [35S]GTPγS binding. The results are expressed as the mean ±S.E.M. of three independent experiments.
Discussion
The present protocol describes two separate but complementary methods: a simple approach to fractionate cells and tissues into broad but distinct compartments and a means to investigate GPCR signaling by measuring [35S]GTPγS binding.
Efficient cellular fractionation has a wide range of applications, ranging from the extraction and enrichment of proteins, to the assessment of the subcellular localization of proteins, to the study of receptor pharmacology. Although alternative approaches to fractionate cells and tissues exist, the protocol presented here is comparatively cheaper, faster, and simpler. Unlike more established methods, however, this method is unable to cleanly separate plasma membrane-associated proteins from other membranous compartments, such as endosomes or the ER. It must also be noted that while the above protocol uses a transient overexpression system to generate cell membranes containing the GPCR of interest, this protocol is compatible with stable overexpression systems, as well as with animal or human tissue19.
Sample preparation is critical to maximize fraction yield and purity. It is particularly important to ensure complete homogenization and to minimize the transition times between centrifugation steps. Using a pellet homogenizer/pestle to completely disrupt the cell membrane significantly increases the crude membrane yield in the final cell fraction. If the membrane yield is too low, the two simplest solutions would be to scale up the initial cell culture and/or to homogenize the cell pellet a few more times. If individual fractions show a high degree of impurity, reduce the transition times between centrifugation and separation. Do not allow the samples to sit following centrifugation, as proteins may diffuse into the sample and reduce fraction purity.
Filtration-based [35S]GTPγS binding is a rapid and quantitative method to measure the activation of G-proteins using the associated receptor. Measuring GTP binding allows for a more quantitative measurement of GPCR activity than is generally possible when monitoring downstream processes. However, there are two critical limitations to the method outlined here. Most importantly, filtration-based [35S]GTPγS quantitation is not equally feasible with all Gα isoforms20,21. In general, this method is restricted to Gi/o-coupled GPCRs like MOR122. Gs- and Gq-coupled receptors tend to be less sensitive. This is likely due to the combination of the lower abundance of Gs/Gq and their relatively slower nucleotide exchange rate, making it difficult to distinguish real [35S]GTPγS binding above background. This limitation has been addressed elsewhere using G-protein antibody capture techniques20. Some groups have found that eliminating GDP from the reaction improves the signal-to-noise ratio for Gs- or Gq-coupled receptors23. The second limitation is the membrane sensitivity to lipophilic agonists or surfactants. [35S]GTPγS assays require functional receptor-G-protein complexes, and the addition of lipophilic molecules to the reaction system may disrupt the structure of the membranes or these complexes. If this is a potential concern, it may be advantageous to test whether the reagent of interest disrupts [35S]GTPγS binding in an already well-characterized system, such as in DAMGO-mediated MOR1 stimulation. Other conditions to consider optimizing include the binding buffer pH, the concentrations of Mg2+ and NaCl, the protein concentration, and the incubation time23,24.
This protocol focused on a well-characterized GPCR, the MOR1, and well-characterized drugs, DAMGO (agonist) and naloxone (antagonist), that act on MOR1. However, one of the advantages of this technique is that it can be used to characterize unknown ligands as agonists, antagonists, or inverse agonists, depending upon how the ligand modulates GTP binding. When designing experiments, it is critical to use a range of concentrations of the ligand of interest and to be mindful of the range of outcomes: agonists will cause an increase in GTP binding over baseline, inverse agonists will cause a decrease in GTP binding compared to baseline, and antagonists will have little to no effect when added in isolation on baseline GTP binding.
In summary, this protocol describes a technique for performing subcellular fractionation, collecting crude membrane preparations, and investigating GPCR activation by measuring [35S]GTPγS binding. These techniques can be readily adapted to a variety of cell culture and tissue models to study the pharmacology of one of the most important families of receptors.
Disclosures
The authors declare no competing interests.
Acknowledgments
This work was supported by National Institutes of Health grant DA-000266 and the Medical Scientist Training Program T32 grant (C.V., N.W.Z., and P.C.S.). The authors would also like to acknowledge somersault18:24 (somersault1824.com) for the Library of Science & Medical Illustrations.
References
- Kobilka BK. G protein coupled receptor structure and activation. Biochim. Biophys. Acta. 2007;1768(4):794–807. doi: 10.1016/j.bbamem.2006.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubig RR, Siderovski DP. Regulators of G-protein signalling as new central nervous system drug targets. Nat. Rev. Drug Discov 1. 2002. pp. 187–197. [DOI] [PubMed]
- Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat. Rev. Drug Discov. 2006;5(12):993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- Buck L, Axel R. A novel multigene family may encode odorant receptors: a molecular basis for odor recognition. Cell. 1991;65(1):175–187. doi: 10.1016/0092-8674(91)90418-x. [DOI] [PubMed] [Google Scholar]
- Kobilka BK, et al. Science. 4827. Vol. 238. New York, NY: 1987. Cloning, sequencing, and expression of the gene coding for the human platelet alpha 2-adrenergic receptor; pp. 650–656. [DOI] [PubMed] [Google Scholar]
- Palczewski K, et al. Science. 5480. Vol. 289. New York, NY: 2000. Crystal structure of rhodopsin: A G protein-coupled receptor; pp. 739–745. [DOI] [PubMed] [Google Scholar]
- Neer EJ. Heterotrimeric c proteins: organizers of transmembrane signals. Cell. 1995;80(2):249–257. doi: 10.1016/0092-8674(95)90407-7. [DOI] [PubMed] [Google Scholar]
- Londos C, Salomon Y. 5'-Guanylylimidodiphosphate, a Potent Activator of Adenylate Cyclase Systems in Eukaryotic Cells. Proc. Natl. Acad. Sci. USA. 1974;71(8):3087–3090. doi: 10.1073/pnas.71.8.3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross EM, Gilman AG. Resolution of Some Components of Adenylate-Cyclase Necessary for Catalytic Activity. J. Biol. Chem. 1977;252(20):6966–6969. [PubMed] [Google Scholar]
- Stadel JM, Nambi P, Shorr R, Sawyer DF, Caron MG, Lefkowitz RJ. Catecholamine-Induced Desensitization of Turkey Erythrocyte Adenylate-Cyclase Is Associated with Phosphorylation of the Beta-Adrenergic-Receptor. Proc. Natl. Acad. Sci. USA. 1983;80(11):3173–3177. doi: 10.1073/pnas.80.11.3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilden U, Kuhn H. Light-Dependent Phosphorylation of Rhodopsin - Number of Phosphorylation Sites. Biochemistry. 1982;21(12):3014–3022. doi: 10.1021/bi00541a032. [DOI] [PubMed] [Google Scholar]
- Lohse M, Benovic J, Codina J, Caron M, Lefkowitz R. Science. 4962. Vol. 248. New York, NY: 1990. beta-Arrestin: a protein that regulates beta-adrenergic receptor function; pp. 1547–1550. [DOI] [PubMed] [Google Scholar]
- Leftowitz RJ, Shenoy SK. Science. 5721. Vol. 308. New York, NY: 2005. Transduction of receptor signals by beta-arrestins; pp. 512–517. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ. Historical review: a brief history and personal retrospective of seven-transmembrane receptors. Trends Pharmacol. Sci. 2004;25(8):413–422. doi: 10.1016/j.tips.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Salah-Uddin H, Thomas DR, et al. Pharmacological assessment of m1 muscarinic acetylcholine receptor-gq/11 protein coupling in membranes prepared from postmortem human brain tissue. J. Pharm. Exp. Ther. 2008;325(3):869–874. doi: 10.1124/jpet.108.137968. [DOI] [PubMed] [Google Scholar]
- Yung-Chi C, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22(23):3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Lazareno S, Birdsall N. Estimation of competitive antagonist affinity from functional inhibition curves using the Gaddum, Schild and Cheng-Prusoíf equations. Br. J. Pharmacol. 1993;109(4):1110–1119. doi: 10.1111/j.1476-5381.1993.tb13737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maréchal E. Chemogenomics and Chemical Genetics. Springer; 2011. Chapter 5, Measuring Bioactivity: KI, IC50 and EC50; pp. 55–65. [Google Scholar]
- Salah-Uddin H, Thomas DR, et al. Pharmacological Assessment of M1 Muscarinic Acetylcholine Receptor-Gq/11 Protein Coupling in Membranes Prepared from Postmortem Human Brain Tissue. J. Pharm. Exp. Ther. 2008;325(3):869–874. doi: 10.1124/jpet.108.137968. [DOI] [PubMed] [Google Scholar]
- Milligan G. Principles: extending the utility of [35S]GTP gamma S binding assays. Trends. Pharmacol. Sci. 2003;24(2):87–90. doi: 10.1016/s0165-6147(02)00027-5. [DOI] [PubMed] [Google Scholar]
- Harrison C, Traynor JR. The [35S]GTPgammaS binding assay: approaches and applications in pharmacology. Life Sci. 2003;74(4):489–508. doi: 10.1016/j.lfs.2003.07.005. [DOI] [PubMed] [Google Scholar]
- Traynor JR, Nahorski SR. Modulation by mu-opioid agonists of guanosine-5'-O-(3-[35S]thio)triphosphate binding to membranes from human neuroblastoma SH-SY5Y cells. Mol. Pharmacol. 1995;47(4):848–854. doi: 10.1016/S0026-895X(25)08634-1. [DOI] [PubMed] [Google Scholar]
- Sittampalam GS, et al., editors. GTPγS Binding Assays. Assay Guidance Manual. 2004.
- Strange PG. Use of the GTPγS ([35S]GTPγS and Eu-GTPγS) binding assay for analysis of ligand potency and efficacy at G protein-coupled receptors. Br. J. Pharmacol. 2010;161(6):1238–1249. doi: 10.1111/j.1476-5381.2010.00963.x. [DOI] [PMC free article] [PubMed] [Google Scholar]