

Abstract
The role of long noncoding RNAs (lncRNAs) in cancer is coming to the forefront due to growing interest in understanding their mechanistic functions during cancer development and progression. Despite this, the global epigenetic regulation of lncRNAs and repetitive sequences in cancer has not been well investigated, particularly in chronic lymphocytic leukemia (CLL). This study focuses on a unique approach: the immunoprecipitation-based capture of double-stranded, methylated DNA fragments using methyl-binding domain (MBD) proteins, followed by next-generation sequencing (MBD-seq). CLL patient samples belonging to two prognostic subgroups (5 IGVH mutated samples + 5 IGVH unmutated samples) were used in this study. Analysis revealed 5,800 hypermethylated and 12,570 hypomethylated CLL-specific differentially methylated genes (cllDMGs) compared to normal healthy controls. Importantly, these results identified several CLL-specific, differentially methylated lncRNAs, repetitive elements, and protein-coding genes with potential prognostic value. This work outlines a detailed protocol for an MBD-seq and bioinformatics pipeline developed for the comprehensive analysis of global methylation profiles in highly CpG-rich regions using CLL patient samples. Finally, a protein-coding gene and an lncRNA were validated using pyrosequencing, which is a highly quantitative method to analyze CpG methylation levels to further corroborate the findings from the MBD-seq protocol.
Keywords: Genetics, Issue 124, DNA methylation, methyl CpG DNA binding domain protein, differentially methylated regions, next-generation sequencing, protein coding and noncoding genes, chronic lymphocytic leukemia
Introduction
The use of next-generation sequencing techniques to analyze global DNA methylation profiles has been increasingly popular during recent years. Genome-wide methylation assays, including microarray- and non-microarray-based methods, were developed based on the following: the bisulfite conversion of genomic DNA, methylation-sensitive restriction enzyme digestions, and the immunoprecipitation of methylated DNA using methyl CpG-specific antibodies.
Aberrant DNA methylation is one of the hallmarks of leukemia and lymphomas, includingchronic lymphocytic leukemia (CLL). Earlier, several groups including ours characterized the DNA methylation profiles of different CLL prognostic subgroups and normal, healthy B-cell controls using the bisulfite conversion of genomic DNA, followed by micro-array-based methods or whole-genome sequencing1,2,3,4. The bisulfite conversion of genomic DNA leads to the deamination of unmodified cytosines to uracil, leaving the modified methylated cytosines in the genome. Once converted, the methylation status of the DNA can be determined by PCR amplification and sequencing using different quantitative or qualitative methods, such as micro-array-based or whole-genome bisulfite sequencing (WGBS). Although bisulfite conversion-based methods have many advantages and are widely used in different cancer types to analyze DNA methylation levels, there are a few drawbacks associated with this technique. WGBS sequencing allows single-base-pair resolution with lower amounts of DNA and is the best suitable option for analyzing a large number of samples. However, this method fails to differentiate the modifications between the 5 mC and 5 hmC levels in the genome5,6. Additionally, microarray-based methods do not offer complete coverage of the genome.
In a recent study from our laboratory7, immunoprecipitation based methods, rather than bisulfite conversion, were used to identify highly CpG-rich, differentially methylated regions on a global scale in CLL patients and normal healthy controls. Inmethyl-CpG-binding domain (MBD)next-generation sequencing (MBD-seq), the enrichment of double-stranded fragmented DNA depends upon the degree of CpG methylation. This method can overcome the drawbacks of the bisulfite conversion method and can also provide genome-wide coverage of CpG methylation in an unbiased and PCR-independent manner. Additionally, unlike bisulfite conversion-based microarray methods, MBD-seq can be used to analyze the methylation status of repetitive elements, such as long interspersed nuclear elements (LINEs), short interspersed nuclear elements (SINEs), long terminal repeats (LTRS), etc. However, compared to bisulfite conversion methods, an MBD-seq protocol requires a relatively large amount of input DNA. Also, the quality of the sequencing reads and the data depend on the specificity, affinity, and quality of the antibodies used.
The current study explains a detailed MBD-seq protocol to enrich methylated DNA for next-generation sequencing. It uses a commercial methylated DNA binding enrichment kit (listed in the Materials Table), as well as a computational pipeline to visualize and interpret methylation sequencing data to identify CLL-specific hyper- and hypomethylated regions compared to normal healthy controls. Basically, this method makes use of the ability of the MBD of the human MBD2 protein interaction with methylated CpGs to extract DNA enriched with methylated CpGs, and this is followed by the high-throughput sequencing of methylated DNA.
Protocol
The ethical approval for collecting the CLL samples is from 2007-05-21, with the following registration number: EPN Gbg dnr 239/07. All CLL patients were diagnosed according to recently revised criteria8, and the samples were collected at the time of diagnosis. The patients in the study were included from different hematology departments in the western part of Sweden after written consent had been obtained. Only CLL peripheral blood mononuclear cell (PBMC) samples with a tumor percentage of leukemic cells ≥70% were selected in this study.
1. Preparations
Isolate PBMCs for DNA extractions from CLL and normal, healthy peripheral blood samples using a commercial isolation kit (see the Materials Table), according to the manufacturer's instructions.
Autoclave 1.5 mL tubes. Thaw all reagents in the methylated DNA binding kit (see the Materials Table).
Use DNase-free water to prepare 10 mL of 1x bead wash buffer from the 5x stock wash buffer provided by the kit to wash beads and dilute the MBD protein provided by the kit.
Obtain or prepare the following items in advance: 3 M sodium acetate, absolute ethanol, and 70% ethanol for DNA precipitation (Materials Table).
2. Genomic DNA Extraction and Sonication
Use a commercially available DNA extraction kit (Materials Table) to isolate genomic DNA from patient and normal PBMC samples according to the manufacturer's protocol. Quantify the genomic DNA using a spectrophotometer at 260 nm. NOTE: The DNA extraction column capacity is 5-6 million cells, maximum; hence, it is important to use more than one column for samples with more than 5 million cells. Elute the DNA twice in equal volumes totaling 100 µL of 10 mM Tris EDTA (TE) buffer. The MBD-biotin protein used in the methyl miner kit for MBD sequencing will not bind to single-stranded DNA. Thus, it is very important to keep the eluted DNA either frozen or at 4 °C to preserve its double-stranded nature.
Dilute 5 µg of genomic DNA from each sample to a total of 200 µL using TE buffer (pH 8), giving a final concentration of 25 ng/µL.
Perform sonication in specially designed tubes using a sonicator (Materials Table) for a total of 30 cycles (30 s on and 30 s off per cycle; after every 5 cycles, briefly spin the tubes to collect the samples to the bottom). NOTE: This will produce fragmented DNA ranging between 150 bp and 300 bp, which is optimal for this protocol.
Check the sonication range for all samples before proceeding to the next step of MBD sequencing by running 1 µL of the fragmented DNA samples and a DNA size ladder on a commercially available, pre-cast 2% agarose gel using DNA electrophoresis imaging equipment. Visualize the DNA using a standard UV transilluminator.
3. Bead Preparation Prior to Binding with MBD-biotin Protein
Resuspend the magnetic streptavidin beads from the stock tube provided by the kit, gently pipetting up and down to obtain a homogenous suspension. Do not vortex the beads or allow them to dry.
Place 50 µL of beads (for each 5 µg of fragmented DNA sample) in separate, clean, and labelled 1.5 mL tubes. Add 50 µL of 1x bead wash buffer to reach the final volume of 100 µL. NOTE: When using small 0.5 mL polymerase chain reaction (PCR) stripes for washing beads, around 150-200 µL of wash buffer per tube is used, as mentioned in the kit protocol. However, in the case of 1.5 mL tubes, add at least 250 µL to 300 µL of wash buffer per tube for the bead wash.
Place the tubes on a magnetic stand for one minute to allow all the magnetic beads to concentrate on the inner wall of the tube facing the magnet. Remove the liquid, without touching the beads, using a 200 µL pipette.
Remove the tubes from the magnetic stand, add 250 µL of 1x bead wash buffer, and mix the beads gently with a pipette.
Repeat steps 3.3 and 3.4 at least 4-5 times for all samples and finally resuspend in 250 µL of 1x bead wash buffer. Keep them on ice.
4. Binding the MBD-biotin Protein to the Washed Beads
Add 35 µL of MBD protein (7 µL for 1 µg of DNA sample) to separate tubes and bring the total volume up to 250 µL using 1x bead wash buffer.
Add 250 µL of diluted MBD protein to the 250 µL of washed beads and leave them on end-to-end rotation at room temperature for 1 h.
- After mixing the beads and protein for 1 h, wash the MBD protein biotin bound with magnetic streptavidin beads.
- Place the tubes on the magnetic stand for 1 min and remove the liquid without touching the beads using a pipette. Add 250 µL of 1x bead wash buffer and place the tubes on a rotation mixer for 5 min at room temperature.
Repeat step 4.3.1 two more times and finally resuspend the washed MBD-biotin beads in 200 µL of 1x bead wash buffer, making the beads ready for methylated DNA capture. NOTE: Washing 2-3 times in this manner removes all the background unbound MBD protein beads and improves the efficient binding of MBD protein-coupled beads with fragmented genomic DNA.
5. Binding MBD-biotin Beads with Fragmented Genomic DNA
In a clean 1.5 mL DNase-free tube, add 100 µL of 5x bead wash buffer and 180 µL of the fragmented genomic DNA (step 2.3). Bring the final volume to 500 µL using DNase free water.
Add 380 µL of DNase-free water to the remaining 20 µL of the fragmented genomic DNA and freeze them. Use these samples as input DNA controls and precipitate them later along with the final eluted methylated DNA samples (see step 7.1).
Place the tubes containing washed MBD-biotin beads (step 4.4) on a magnetic stand for 1 min and remove the liquid without disturbing the beads. Add 500 µL of fragmented genomic DNA diluted in bead wash buffer.
Seal all the tubes tightly with paraffin film and leave them overnight at 4 °C on an end-to-end rotation stand at 8-10 rpm. NOTE: The DNA and biotin-bead binding reaction can be done for 1 h at room temperature, but leaving it at 4 °C overnight can improve the recovery of the final methylated DNA.
6. Removing the Unbound DNA and Eluting the Methylated DNA from the Beads
After the DNA and MBD-bead binding reaction, place the tubes on the magnetic rack for 1 min to concentrate all the beads on the inner wall of the tube.
Remove the supernatant liquid with a pipette without touching the beads and save this unbound DNA sample fraction on ice.
Add 200 µL of 1x bead wash buffer to the beads and place the tubes on the rotating stand for 3 min at room temperature. Place the tubes on the magnetic stand and remove the liquid. Repeat the wash another two times to remove the residual unbound DNA.
After the final wash, add 200 µL of high-salt elution buffer (2,000 mM NaCl), provided in the kit to elute the DNA.
Place the tubes on the rotating stand for 15 min at room temperature. Place them on the magnetic stand for 1 min and use a pipette to carefully transfer the supernatant to a new, clean 1.5 mL tube.
Add 200 µL of high-salt elution buffer and repeat the elution by rotating the tubes at room temperature for 15 min. Add the second elute to the same tube containing the 200 µL of the first elute. NOTE: The final 400 µL of total eluted DNA is now ready for ethanol precipitation to extract the purified DNA suitable for next-generation sequencing.
7. Ethanol Precipitation and the Enrichment of Methylated DNA
Add 1 µL of glycogen (20 µg/µL; included in the kit); 40 µL of 3 M sodium acetate, pH 5.2; and 800 µL of ice-cold absolute ethanol to 400 µL of eluted DNA and also to the 400 µL of input DNA samples prepared in step 5.2.
Mix the tubes well by vortexing and incubate them at -80 °C overnight.
Centrifuge the tubes at 12,000 x g (maximum speed) and 4 °C. Discard the supernatant carefully, without disturbing the pellet, add 500 µL of 70% ethanol, and vortex the tubes.
Centrifuge again at maximum speed for 15 min at 4 °C and remove the supernatant by carefully using a pipette.Centrifuge the tubes at maximum speed for 1 min at room temperature and remove the residual ethanol completely using a pipette tip.
Air-dry the pellet for 5 min at room temperature. Add 10 µL of DNase-free water to the DNA pellet. Proceed to the methylated DNA quantification (step 7.6), MBD sequencing (step 7.7), and analysis (sections 8 and 9).
Quantify the final recovered methylated DNA samples using a commercially available fluorometric quantitation kit (see the Materials Table), according the manufacturer's instructions.
Note: Using this protocol, 30-50 ng of final DNA was recovered from each sample. DNA samples are ready to send on dry ice for downstream library construction and high-throughput MBD sequencing
Perform DNA library constructionand high-throughput MBD sequencing using a commercial platform (Materials Table), as described in reference9. NOTE: For the initial quality control of the final methylated DNA, perform library preparations using 50-bp pair-end sequencing for all samples. Process the raw data for post-sequencing quality control, as elaborated in step 8.1, and further process it using bioinformatics approaches and statistical methods, as described below (steps 8.2-9.6).
8. Bioinformatics Analysis Method 1: Identifying CLL-associated Differentially Methylated Regions (cllDMRs)
Clean the obtained 49-bp reads (FASTQ format) for adaptors using available quality-control tools, such as Trimmomatic10 or Cutadapt. Crosscheck the quality of the trimmed reads using the FastQC toolkit: java -jar trimmomatic.jar SE SAMPLE_uncleaned.fastq SAMPLE.fastq ILLUMINACLIP:adapters.fasta:2:30:10
- Align the cleaned FASTQ files with short-read genome aligner Bowtie against a reference genome11. Specify the parameters as below:
- Allow up to two mismatches (Bowtie parameter: -v 2).
- Limit the reporting to the six best alignments per read (Bowtie parameter: -m 6) to control for multi-mapping reads. bowtie -v 2 -a -m 6 HG19_INDEX -S SAMPLE.fastq > SAMPLE.sam NOTE: Convert the SAM files generated for individual samples after alignment into BAM using SAMtools. samtools view -bS -o SAMPLE.bam SAMPLE.sam
Use the model-based analysis of ChIP-Seq (MACS) peak caller on aligned samples (BAM) to predict enriched/methylated regions from the CLL subgroups12. NOTE: Comparison I: Use the Input sample as the control group and both the Normal and CLL patient samples as treatment groups. This step is to collect all negative peaks (peaks enriched in Input/background). Comparison II: Use the Normal sample group as the control group and the CLL patient sample group as the treatment group. Obtained positive peaks are CLL hyper-methylated regions, and negative peaks are CLL hypomethylated regions over normal or differentially methylated regions (DMRs). macs14 -t SAMPLE_TREATMENT.bam -c SAMPLE_CONTROL.bam --format BAM -g hs
Remove Comparison I background peaks from differentially methylated regions (DMRs) using BEDtools13. bedtools subtract -a <CLL_enriched_regions> -b <Input_enriched_regions>
- Predict the percentage of repeat elements (SINE-Alu, LINE, etc.) enriched in DMRs.
- Use fastacmd to extract the FASTA sequence of DMRs by the chromosomal coordinates from reference genome HG19. fastacmd -d HG19_genome.fa -s Chromosome -L Start,End -l 50000 > DMRs.fasta
- Use the RepeatMasker commandline tool to predict the percentage of repeat elements present in teh FASTA sequence of DMRs. RepeatMasker -gc -gccalc -s -species human -html DMRs.fasta
Annotate the predicted DMRs using Homer "annotatePeaks.pl" with the available Ensembl [PMC4919035] or Gencode [PMID 22955987] transcript annotation (protein-coding and noncoding transcripts). annotatePeaks.pl DMRs.bed <GENOME> -gtf <Ensembl or Gencode GTF> NOTE: This provides information on the distribution of peaks over different genic regions (i.e., promoter, exon, intron, 3' UTRs, and 5' UTRs) and intergenic regions. Transcripts or genes associated with DMRs are called differentially methylated genes (DMGs).
Use the enrichment tool with the updated or current functional database to find functions enriched by CLL DMGs (only protein-coding genes)14. NOTE: Gene Set Clustering based on Functional Annotation (GeneSCF)14 is a real-time-based tool for functional enrichment analysis that uses updated KEGG and Gene Ontology as a reference database.
9. Bioinformatics Analysis Method 2: Identifying CLL-associated Significantly Differentially Methylated Regions (cll sigDMR's)
Follow steps 8.1-8.5 from method I (section 8) and use read-count-based differential enrichment analysis by utilizing the available tools, as elaborated in steps 9.2 - 9.6, to add one more level of statistics to the analysis pipeline.
Quantify the number of reads mapped to individual peaks or DMRs in Normal and CLL patient samples using "featureCounts" from the "Subread" package15. subread/bin/featureCounts -Q 30 -F SAF -a DMRs.SAF -o DMRs_counts.table SAMPLE_TREATMENT.bam SAMPLE_CONTROL.bam NOTE: A mapping quality filter can be introduced to avoid quantifying bad-quality reads (e.g.,-Q 30). For this step, prepare SAF files for the obtained DMRs. For more information on SAF file format, please use this link http://bioinf.wehi.edu.au/featureCounts/.
Use a RAW read-count table containing the number of reads for the individual peaks in Normal and CLL patient sample groups in edgeR as the input16. NOTE: Compare normal versus CLL patient sample groups to find sigDMRs. For differential enrichment analysis, follow the guide from edgeR for detailed step-by-step instructions (https://bioconductor.org/packages/release/bioc/vignettes/edgeR/inst/doc/edgeRUsersGuide.pdf).
Filter the sigDMRs using the false discovery rate (FDR) and log-fold change predicted by edgeR16.
Annotate the predicted sigDMR's using Homer "annotatePeaks.pl" with the available Ensembl or Gencode transcript annotation (protein-coding and noncoding transcripts). annotatePeaks.pl sigDMRs.bed HG19_genome.fa -gtf <Ensembl or Gencode GTF> -CpG NOTE: This provides information on the distribution of peaks over different genic regions (i.e., promoter, exon, intron, 3' UTRs, and 5' UTRs) and intergenic regions. Transcripts or genes associated with sigDMRs are called sigDMGs.
Use an enrichment tool with the updated or current functional database to find functions enriched by CLL sigDMGs (only protein-coding genes)14. NOTE: GeneSCF, one of the real-time tools for functional enrichment analysis, uses KEGG and Gene Ontology as reference databases.
Representative Results
MBD-seq was recently performed on CLL patients and matched, normal, healthy controls to identify CLL-specific differentially hyper- and hypomethylated genes7. The experimental and bioinformatic pipeline used for analyzing the data generated from CLL and normal healthy samples are shown in Figure 1A and 1B. These analyses identified several CLL-specific differentially methylated regions (cllDMRs), which were significantly hyper/hypomethylated from IGHV-mutated and IGHV-unmutated samples compared to control samples, with a p-value <0.00001. Figure 2A shows all the cllDMRs obtained from both normal B-cell and normal PMBC comparisons. All the cllDMRs were mapped to different classes of protein-coding and noncoding genes, as shown in Figure 2B. Importantly, in this analysis, the comparisons were performed independently between the CLL patient samples, with two different normal controls, sorted B-cell and PMBCs. Interestingly, the analysis resulted in a large overlap of common CLL-specific differentially methylated genes (cllDMGs; 851 hypermethylated and 2,061 hypomethylated) when compared to normal B-cell control and normal PBMC control samples (Figure 2C), suggesting that these cllDMGs can be possible CLL signature genes with significant roles in disease pathogenesis. To strengthen the analyzed data, several CpG sites located in one hypermethylated protein coding gene, SKOR1, and one hypomethylated long noncoding lncRNA, AC012065.7, were validated using a pyrosequencing method, as described in earlier publications7,17,18 (Figure 3A and B). Figure 4 shows sheared DNA after the sonication protocol. The fragmented DNA ranges between 150 and 300 bp, making it ideal for sequencing purposes.
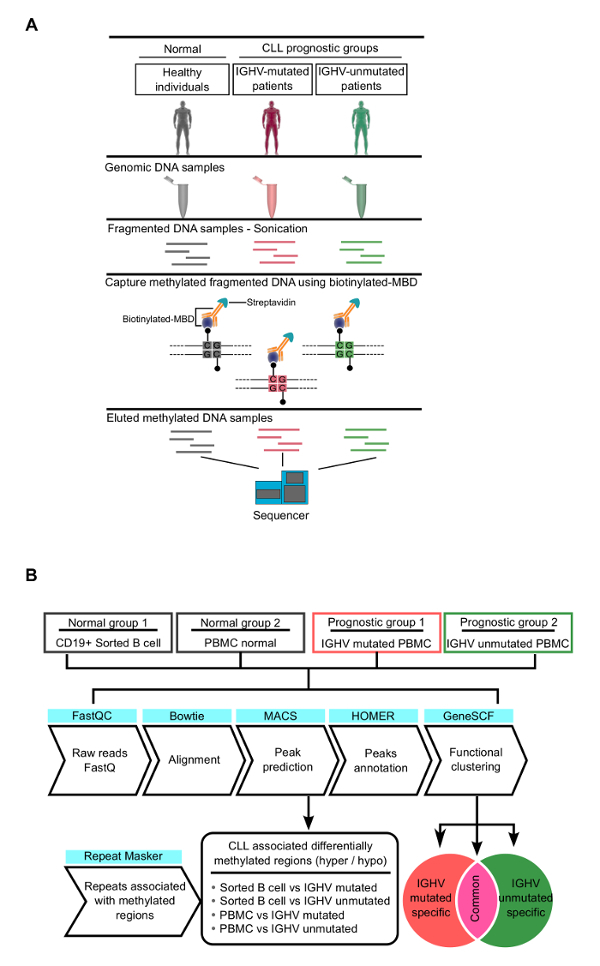
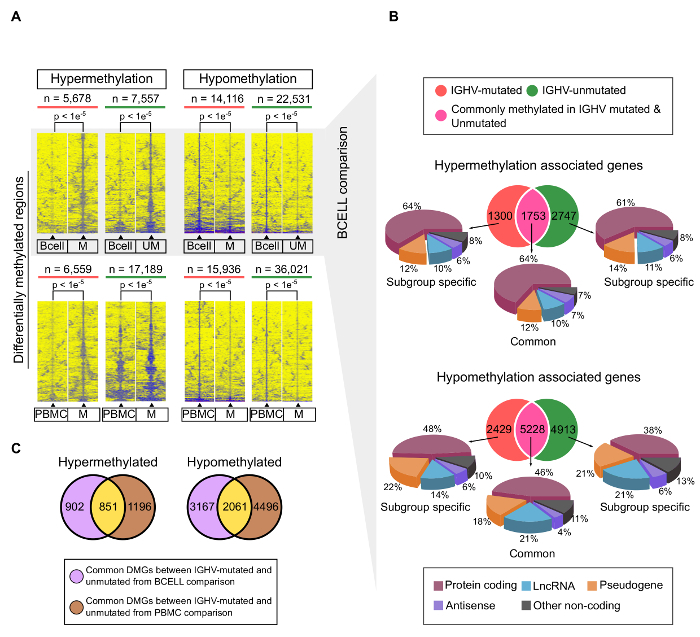
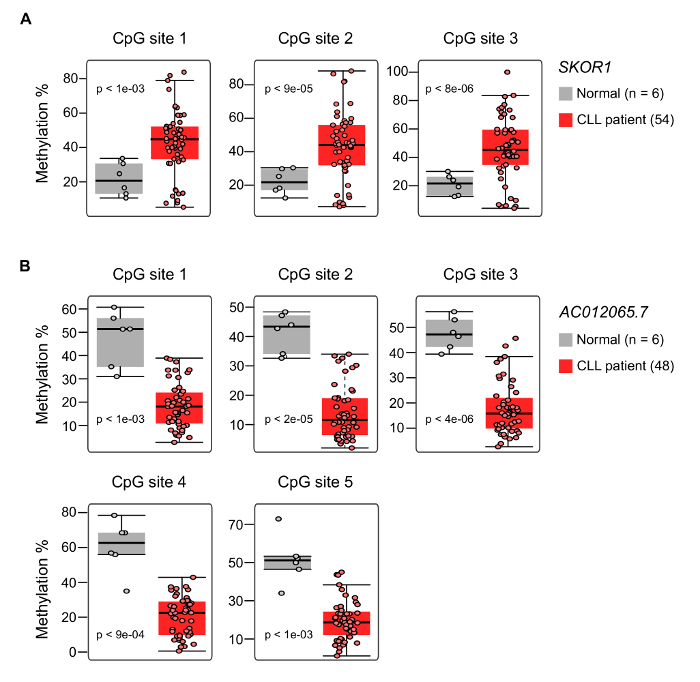
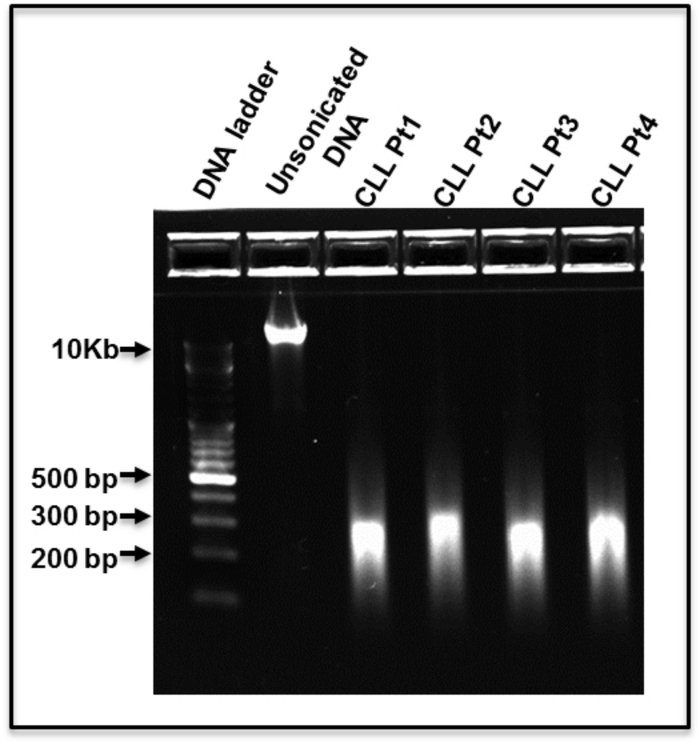
Figure 1: Overview of the Work Flow Used in this Study. This figure, obtained from our recent publication7, shows the design of the overall analysis used to identify the differentially methylated regions (DMRs) in chronic lymphocytic leukemia (CLL) patient samples. (a) Experimental design of the MBD-based enrichment of methylated DNA. (b) The bioinformatics analysis pipeline used to identify CLL-specific differentially methylated regions (cllDMRs). Please click here to view a larger version of this figure.
Figure 2: Hypermethylated and Hypomethylated cllDMRs and cllDMGs of CLL Patient Samples Compared to Normal, Healthy, Sorted B Cells7. (a) All the chronic lymphocytic leukemia (CLL)-associated differentially methylated regions with significant p-values (< 0.00001) obtained from comparing the IGHV-mutated and -unmutated CLL samples to normal sorted B cells (upper panel) and normal PBMC samples (lower panel). The enrichments shown in the heatmap were within a ± 3-kb window from differentially methylated region (DMRs). (b) Venn diagram showing the overlap of CLL-specific differentially methylated genes (cllDMGs; hypermethylated and hypomethylated) between IGHV-mutated and IGHV-unmutated groups. The pie chart represents the percentage of genes belonging to different classification, such as protein-coding, long noncoding RNA (lncRNA), pseudogenes, antisense, and other noncoding RNAs. (c) Venn diagram showing the overlap of common differentially methylated genes (IGHV-mutated and IGHV-unmutated prognostic groups) between B-cell and PBMC comparisons. The left panel of the Venn diagram shows the overlap for hypermethylated genes and the right panel for hypomethylated genes. Please click here to view a larger version of this figure.
Figure 3: Validation of the DNA Methylation Status for Individual CpG Sites on the Selected Significantly Differentially Methylated Target Genes. Pyrosquencing data for the quantification of the percentage of DNA methylation for two selected genes using an independent chronic lymphocytic leukemia (CLL) sample cohort containing 54 samples and 6 normal, healthy, age-matched B-cell samples.(A) The boxplots represent the percentage of methylation levels for 3 individual CpG sites of the hypermethylated SKOR1 gene using pyrosequencing. (B) The boxplot shows the degree of methylation for 5 individual CpG sites of hypomethylated AC012065.7 gene using pyrosequencing. The boxes indicate the interquartile range (25-75%), while the inner horizontal line indicates the median value. The whiskers represent the minimum and maximum values. The p-value corresponding to the degree of differential methylation of CLL samples over normal B cells are shown in the boxplots for each individual CpG site. Please click here to view a larger version of this figure.
Figure 4: Shearing of Genomic DNA. Representative results of four chronic lymphocytic (CLL) DNA samples after sonication, along with one CLL sample of before (0 min) sonication was run in a 2% pre-cast agarose gel, stained and visualized under UV light. The DNA size ladder is indicated on lane 1. Please click here to view a larger version of this figure.
Discussion
MBD-seq is a cost-effective, immunoprecipitation-based technique that can be used to study methylation patterns with complete genome-wide coverage. Both MeDIP seq (methylated DNA immunoprecipitation followed by sequencing) and MBD-seq result in the enrichment of CpG-rich methylated DNA. However, MBD-seq shows more affinity towards binding to highly CpG-rich regions when compared to MeDIP seq19. Using a methyl-binding enrichment kit, one can fractionate the DNA into high-CpG- and low-CpG-rich regions by eluting DNA with high-salt and low-salt buffers, respectively. In this investigation, only a single fraction elution was performed to capture highly enriched CpG-rich regions covering most of the CpG islands.
MBD-seq can be a powerful alternative to WGBS, which is commonly used in CLL and other leukemia investigations. Even though MBD-seq requires a relatively higher amounts of input DNA compared to the bisulfite conversion-based methods, it allows for the investigation of global methylation changes that are specific only for 5 mC modifications, without any PCR amplification bias created after the conversion. Thus, MBD-seq is an ideal method to address cllDMGs, which could be potential epigenetic-based CLL signature genes with prognostic value.
The crucial factors for performing this method are the quality and the sonication range of the fragmented DNA used prior to the binding reaction. All samples showed shorter fragment ranges, between 150 and 300 bp (Figure 4), resulting in good-quality sequencing results, with around 25-33 million unique reads for each sample mapping to the genome after data filtering and cleaning.
Finally, the methylation status of hyper- and hypomethylated cllDMGs has been validated for few genes using a pyrosequencing method in an independent sample cohort. This method gives the percentage of DNA methylation, depending on the ratio of C-to-T at individual CpG sites based on the amount of C and T incorporated during the sequence extension. The hypermethylated cllDMGs showed high percentages of methylation in CLL samples compared to the normal samples. Likewise, the hypomethylated cllDMGs showed the opposite. Pyrosequencing data for two cllDMGs is shown in Figure 3.
Compared to array-based methods, this method allows for the wider examination of sequences across the genome, including previously annotated regions spanning protein-coding regions and non-annotated sequences spanning repetitive elements and lncRNAs. Thus, this approach on CLL patients identified several novel cllDMGs spanning lncRNAs and repetitive elements. Since these investigations have not been performed previously in CLL patient samples, this study serves as a valuable resource for identifying CLL-associated differential methylated regions in different prognostic subgroups. These cllDMGs will serve as novel biomarkers and targets for epigenetic drug therapy.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This study was supported by the Swedish Research Council, the Swedish Cancer Society, the Knut and Alice Wallenberg Foundation (KAW), and FoU VästraGötalandsregionen.
References
- Kanduri M, et al. Differential genome-wide array-based methylation profiles in prognostic subsets of chronic lymphocytic leukemia. Blood. 2010;115(2):296–305. doi: 10.1182/blood-2009-07-232868. [DOI] [PubMed] [Google Scholar]
- Cahill N, et al. 450K-array analysis of chronic lymphocytic leukemia cells reveals global DNA methylation to be relatively stable over time and similar in resting and proliferative compartments. Leukemia. 2013;27(1):150–158. doi: 10.1038/leu.2012.245. [DOI] [PubMed] [Google Scholar]
- Kanduri M, et al. Distinct transcriptional control in major immunogenetic subsets of chronic lymphocytic leukemia exhibiting subset-biased global DNA methylation profiles. Epigenetics. 2012;7(12):1435–1442. doi: 10.4161/epi.22901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulis M, et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet. 2012;44(11):1236–1242. doi: 10.1038/ng.2443. [DOI] [PubMed] [Google Scholar]
- Booth MJ, et al. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science. 2012;336(6083):934–937. doi: 10.1126/science.1220671. [DOI] [PubMed] [Google Scholar]
- Yu M, et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012;149(6):1368–1380. doi: 10.1016/j.cell.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subhash S, Andersson PO, Kosalai ST, Kanduri C, Kanduri M. Global DNA methylation profiling reveals new insights into epigenetically deregulated protein coding and long noncoding RNAs in CLL. Clin Epigenetics. 2016;8:106. doi: 10.1186/s13148-016-0274-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallek M, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111(12):5446–5456. doi: 10.1182/blood-2007-06-093906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Meyer T, et al. Quality evaluation of methyl binding domain based kits for enrichment DNA-methylation sequencing. PLoS One. 2013;8(3):e59068. doi: 10.1371/journal.pone.0059068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9(9):R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subhash S, Kanduri C. GeneSCF: a real-time based functional enrichment tool with support for multiple organisms. BMC Bioinformatics. 2016;17(1):365. doi: 10.1186/s12859-016-1250-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinelli S, et al. ANGPT2 promoter methylation is strongly associated with gene expression and prognosis in chronic lymphocytic leukemia. Epigenetics. 2013;8(7):720–729. doi: 10.4161/epi.24947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopparapu PK, et al. Epigenetic silencing of miR-26A1 in chronic lymphocytic leukemia and mantle cell lymphoma: Impact on EZH2 expression. Epigenetics. 2016;11(5):335–343. doi: 10.1080/15592294.2016.1164375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, et al. Evaluation of affinity-based genome-wide DNA methylation data: effects of CpG density, amplification bias, and copy number variation. Genome Res. 2010;20(12):1719–1729. doi: 10.1101/gr.110601.110. [DOI] [PMC free article] [PubMed] [Google Scholar]