

Abstract
Signaling pathways regulate gene expression programs via the modulation of the chromatin structure at different levels, such as by post-translational modifications (PTMs) of histone tails, the exchange of canonical histones with histone variants, and nucleosome eviction. Such regulation requires the binding of signal-sensitive transcription factors (TFs) that recruit chromatin-modifying enzymes at regulatory elements defined as enhancers. Understanding how signaling cascades regulate enhancer activity requires a comprehensive analysis of the binding of TFs, chromatin modifying enzymes, and the occupancy of specific histone marks and histone variants. Chromatin immunoprecipitation (ChIP) assays utilize highly specific antibodies to immunoprecipitate specific protein/DNA complexes. The subsequent analysis of the purified DNA allows for the identification the region occupied by the protein recognized by the antibody. This work describes a protocol to efficiently perform ChIP of histone proteins in a mature mouse T-cell line. The presented protocol allows for the performance of ChIP assays in a reasonable timeframe and with high reproducibility.
Keywords: Genetics, Issue 124, ChIP, H3K4me1, H3K4me3, H3K27ac, H3, histone marks, sonication, shearing, chromatin, regulation, transcription.
Introduction
Development, differentiation, and homeostasis depend upon specific gene expression programs that are established by signaling events that modulate chromatin structure and therefore determine if a specific gene is activated or repressed in a cell- and time-specific manner. During T-cell development, specific gene expression programs must be established to properly determine the maturation of T-cell precursors from the double-negative (DN) to the single-positive (SP) state, passing through several intermediate stages1. How the gene expression program is dynamically regulated during T-cell development has been broadly investigated in previous years by several laboratories2,3,4,5,6.
By post-translational modifications (PTMs) of histones, the exchange of canonical histones with histone variants, nucleosome eviction, and DNA methylation regulate the ON/OFF switch of genes. Several groups have investigated the genome-wide distribution of PTMs and histone variants to determine marks that are associated with different chromatin states at both proximal and distal regulatory regions7,8,9,10. Signaling cascades orchestrate dynamic chromatin regulation via the exchange of positive and negative chromatin-modifying enzymes (also known as chromatin modifiers) at specific enhancer elements. These chromatin modifiers regulate the chromatin structure and thus the transcriptional output by, for example, dynamic histone methylation and acetylation. This is the case in the Notch signaling pathway11,12,13,14.
Dynamic histone PTMs; exchanges with histone variants; and the dynamic occupancy of histones, transcription factors, and cofactors can be investigated by chromatin immunoprecipitation (ChIP) assays. Highly specific antibodies are used to purify specific DNA-protein complexes, and the purified DNA is analyzed by quantitative PCR (qPCR); deep-sequencing (ChIP-Seq); or, less frequently nowadays, hybridization to microarray (ChIP-ChIP).
ChIP assays are sometimes challenging due to complications with the lysis of the cells, shearing of the chromatin, and/or low specificity of the antibodies. Several strategies have been adopted to improve the protocol, as is the case for NEXSON15. The use of cooling water-bath sonicators avoids sample heating that might damage the epitopes present on the protein under investigation, but the energy required for the shearing of the samples gets dispersed into the water. Another improvement was made with the development of focused ultrasonication devices that prevent the dispersion of the energy. Therefore, focused ultrasonicator devices allow for improvements in cell lysis and chromatin shearing, eliminating operator-induced variations and significantly increasing the reproducibility.
This work describes a protocol (schematic overview in Figure 1) to efficiently perform the ChIP of histone proteins in a mature mouse T-cell line called E2-10HA16,17. T-cells are usually difficult to lyse, and the shearing of their chromatin has been revealed to be inefficient. In this protocol, which makes use of a cooling focused ultrasonicator, the number of cells, the lysis buffer, and the shearing setting were optimized for the E2-10HA mouse T-cell line. This protocol allows one to perform ChIP with high reproducibility and in a reasonable time. In fact, it requires approximately two days to shear the chromatin and to evaluate the quality of the shearing, and three days to perform the immunoprecipitation, reverse the crosslink, and purify the DNA.
Protocol
NOTE: All the buffers and the medium used in this protocol are listed in Table 1.
Days 1 and 2
1. Crosslinking the Cells
Transfer the mouse T-cells from a 6-well plate to a 50 mL tube and centrifuge for 5 min at 4 °C and ~271 x g. Resuspend the cell pellet in 30 mL of IMDM cell culture medium and count the cells using a Neubauer chamber.
Collect aliquots of 20 x 106 cells into new 50 mL tubes.
Add formaldehyde (FMA) to a 1% final concentration directly to the cell culture medium and incubate at room temperature for 10 min. Caution: FMA is toxic if inhaled, so work under a fume hood and wear proper protective equipment. Also, please handle FMA waste according to the rules of the host institution.
Add a 1/8-volume of 1 M glycine, pH 7.0, and incubate at room temperature for 5 min.
Pellet the cells by centrifugation for 5 min at 4 °C and ~271 x g and wash with 10 mL of phosphate-buffered saline (PBS).
Wash the cell pellet with 1 mL of PBS and transfer to 1.5 mL tubes.
2. Cell Lysis and Chromatin Shearing
Resuspend the cell pellet in 1 mL of SDS lysis buffer and incubate on ice for 10 min.
Transfer each sample to a sonication tube and perform shearing using a focused ultrasonicator according to the settings indicated in Table 2.
After shearing, transfer the samples to 1.5 mL tubes and centrifuge for 10 min at 4 °C and ~18,000 x g.
Transfer the supernatant to new tubes.
Collect 50 µL to determine the shearing efficiency according to step 3 and snap-freeze the sheared lysate in liquid nitrogen. Store the lysate in single-use aliquots at -80 °C.
3. Determination of Shearing Efficiency
Add 50 µL of elution buffer to the 50 µL of each sheared lysate and incubate at 65 °C overnight, with shaking.
Add 100 µL of TE buffer and 4 µL of 10 mg/mL RNase A (0.2 µg/µL final concentration). Mix and incubate for 2 h at 37 °C, with shaking.
Add 2 µL of 20 mg/mL proteinase K (0.2 µg/µL final concentration) to each sample and incubate for 2 h at 55 °C, with shaking.
Transfer each sample to a gel tube suitable for phenol/chloroform/isoamyl alcohol extraction; handle according to the manufacturer's instructions.
- Add 200 µL of phenol/chloroform/isoamyl alcohol (25:24:1) and shake the tubes. Centrifuge for 5 min at 24 °C and ~16,000 x g and transfer the upper phase to new tubes. Caution: Phenol, chloroform, and isoamyl alcohol are toxic if inhaled and are corrosive; work under a fume hood and wear proper personal protective equipment. Also, handle phenol, chloroform, and isoamyl alcohol waste according to the rules of the host institution.
- Repeat once.
- Purify the DNA by using purification columns according to the manufacturer's instruction, with the following modifications:
- Wash the membrane twice. After the last wash, leave the tube open for 2 min at room temperature to evaporate the residual ethanol.
- For the elution, add 50 µL of H2O, incubate at room temperature for 1 min, spin the tube for 1 s to properly wet the membrane, and incubate at room temperature for 1 min before eluting the DNA by centrifugation for 1 min at 24 °C and ~17,900 x g.
Quantify the purified DNA on a fluorometer using a high-sensitivity kit according to the manufacturer's instructions.
Analyze approximately 500 ng of the purified DNA on a 1.8% agarose gel and approximately 1 ng on an electrophoresis system using a high-sensitivity kit. NOTE: The expected size of the DNA fragments is between 200 and 500 bp.
Days 3 and 4
4. Immunoprecipitation
Dilute 1 volume of sheared lysate with 5 volumes of dilution buffer.
Preclear with 30 µL/mL protein A agarose beads (prepared according to Appendix A) for 30 min while rotating in the cold room.
Centrifuge at 4 °C for 5 min at ~750 x g.
Collect 10% of the input and store it at 4 °C.
Aliquot the desired amount of lysate into new tubes (see Table 3 for details). Add the desired antibodies to each tube (see Table 3 for details) and rotate overnight at 4 °C.
Add 40 µL of protein A beads and incubate for 1 h at 4 °C while rotating.
Wash the beads with 1 mL of low-salt buffer, high-salt buffer, LiCl buffer, and TE buffer (for each wash, incubate for 5 min at 4 °C while rotating and centrifuge for 3 min at 4 °C and ~950 x g). See Table 3 for details.
Add 110 µL of elution buffer and incubate for 15 min at room temperature, vortexing every 2 min.
Centrifuge for 3 min at 4 °C and ~950 x g and transfer 100 µL of supernatant to a new 1.5-mL tube.
Repeat steps 4.7-4.8 with 100 µL of elution buffer and combine the eluates. Add elution buffer up to 200 µL to input the samples.
Incubate all samples overnight at 65 °C, with shaking.
Day 5
5. DNA Purification
Add 200 µL of TE buffer to each eluate and 8 µL of 10 mg/mL RNase A (0.2 µg/µL final concentration). Mix and incubate for 2 h at 37 °C, with shaking.
Add 4 µL of 20 mg/mL proteinase K (0.2 µg/µL final concentration) to each eluate and incubate for 2 h at 55 °C, with shaking.
Transfer each sample to a gel tube suitable for phenol/chloroform/isoamyl alcohol extraction; handle according to the manufacturer's instructions.
- Add 400 µL of phenol/chloroform/isoamyl alcohol (25:24:1) and shake the tubes. Centrifuge for 5 min at 24 °C and ~16,000 x g. Transfer the upper phase to new tubes. Caution: Phenol, chloroform, and isoamyl alcohol are toxic if inhaled and are corrosive; work under a fume hood and wear proper personal protective equipment. Also, handle phenol, chloroform, and isoamyl alcohol waste according to the rules of the host institution.
- Repeat once.
Purify the DNA using purification columns according to the manufacturer's instructions, with the following modifications:
Wash the membrane twice. After the last wash, leave the tubes open for 2 min at room temperature to evaporate the residual ethanol.
For the elution, add 50 µL of H2O, incubate at room temperature for 1 min, spin the tubes for 1 s to properly wet the membrane, and incubate at room temperature for 1 min before eluting the DNA by centrifugation for 1 min at 24 °C and ~17,900 x g.
Representative Results
Mature murine T-cells were subjected to ChIP analysis to investigate the enrichment of the histone mark mono-methylation of lysine 4 on histone 3 (H3K4me1), tri-methylation of lysine 4 on histone 3 (H3K4me3), and acetylation of lysine 27 on histone 3 (H3K27ac), as well as the nucleosome occupancy, as revealed by panH3 ChIP. The shearing quality was evaluated by analysis on a 1.8% agarose gel (Figure 2A) and on an electrophoresis system (Figure 2B). H3K4me1 and H3K4me3 are generally used to identify enhancer sites and promoters, respectively (Figure 3A). In fact, H3K4me3 is prominent but not exclusively enriched at promoters5,8,14. A characteristic of active enhancers is to be highly enriched for H3K4me1 and H3K27ac and poorly enriched for H3K4me3 (Figure 3A, upper panel), whereas inactive enhancers present low levels of H3K4me3 and H3K27ac but high levels of H3K4me1 (Figure 3A, lower panel). As shown in Figure 3B, the Glyceraldehyde 3-phosphate dehydrogenase (Gapdh) gene is representative of the analysis of active gene promoters (Gapdh promoter). In fact, it shows high levels of H3K4me3 and H3K27ac and low levels of H3K4me1. In Figure 3B, Deltex1 (Dtx1) is representative of inactive enhancers (Dtx1 enhancer), as it is highly enriched for H3K4me1 and poorly enriched for H3K4me3 and H3K27ac. Gene desert is representative of a region lacking coding genes and is usually sued as a negative control for active chromatin marks. The observation that H3K4me1, a well-accepted enhancer mark4,5,7,14,18, is not enriched at Gene desert suggests that this region lacks enhancers.
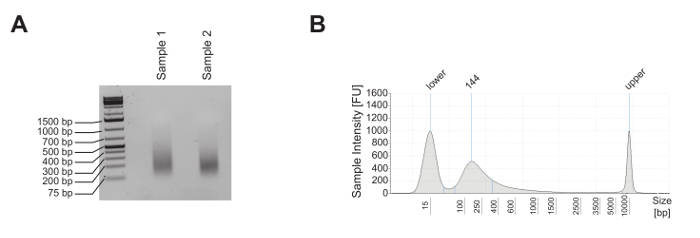
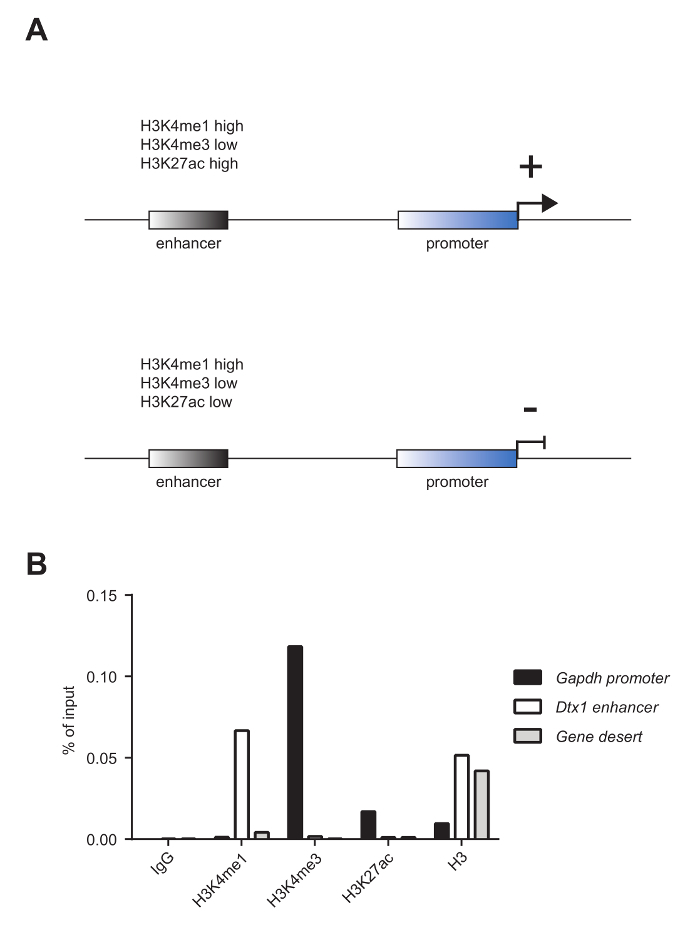
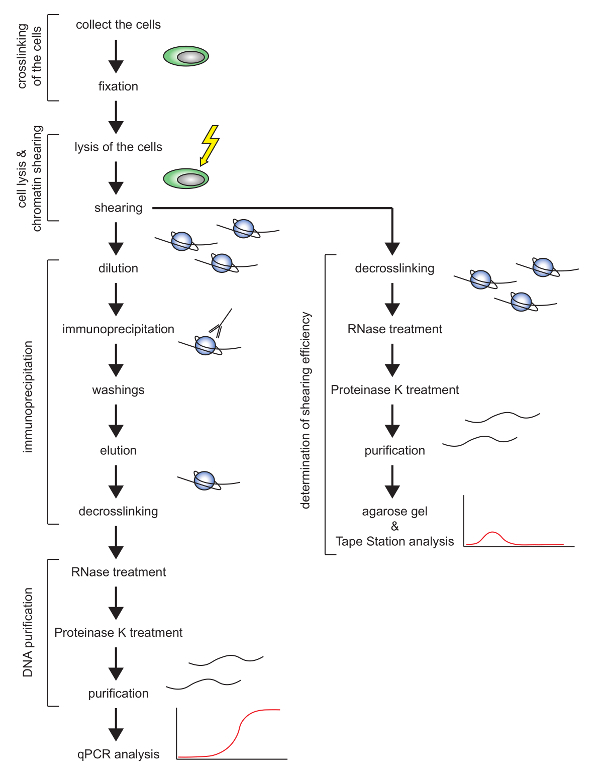
Figure 1: Schematic overview of the ChIP protocol presented. Please click here to view a larger version of this figure.
Figure 2: Shearing quality control of the mature mouse T-cell line. (A) Two different aliquots of mature mouse T-cell line E2-10HA were sheared, and approximately 500 ng of purified DNA were analyzed on a 1.8% agarose gel to evaluate their shearing quality. (B) 1 ng of purified DNA from sample 2 was analyzed by electrophoresis to evaluate its shearing quality. Please click here to view a larger version of this figure.
Figure 3: Chromatin marks that characterize enhancers and promoters. (A) Active enhancers (upper panel) are characterized by high H3K4me1, low H3K4me3, and high H3K27ac, whereas inactive enhancers (lower panel) present high H3K4me1, low H3K4me3, and low H3K27ac. (B) The samples presented in Figure 2A were pooled together and used for ChIP analysis versus histone marks H3K4me1, H3K4me3, and H3K27ac and histone occupancy, as revealed by panH3 ChIP. This analysis shows that the promoter of the housekeeping gene Glyceraldehyde 3-phosphate dehydrogenase (Gapdh promoter) is highly enriched for H3K4me3 and H3K27ac and lowly enriched for H3K4me1. On the other hand, the enhancer of the inactive gene Deltex1 (Dtx1 enhancer) is highly enriched for H3K4me1 but poorly enriched for H3K4me3 and H3K27ac. ChIP using a panH3 antibody was used to investigate nucleosome occupancy. Gene desert was used as a negative control. One representative experiment is shown. Please click here to view a larger version of this figure.
| Name of the buffer | Reagent | Final concentration |
| Dilution buffer | Sodium dodecyl sulfate (SDS) | 0.01% (w/v) |
| Triton X-100 | 1.1% (v/v) | |
| Ethylenediaminetetraacetic acid (EDTA) pH 8.0 | 1.2 mM | |
| Tris-HCl pH 8.1 | 16.7 mM | |
| Sodium chloride (NaCl) | 167 mM | |
| DMA solution | Dimethyl adipimidate (DMA) | 10 mM |
| Phosphate-buffered saline (PBS) | 1x | |
| Elution buffer | Sodium dodecyl sulfate (SDS) | 1% (w/v) |
| Ethylenediaminetetraacetic acid (EDTA) pH 8.0 | 10 mM | |
| Tris-HCl pH 8.0 | 50 mM | |
| High salt buffer | Sodium dodecyl sulfate (SDS) | 0.1% (w/v) |
| Triton X-100 | 1% (v/v) | |
| Ethylenediaminetetraacetic acid (EDTA) pH 8.0 | 2 mM | |
| Tris-HCl pH 8.1 | 20 mM | |
| Sodium chloride (NaCl) | 500 mM | |
| IMDM medium | Iscove's modified Dulbecco's medium (IMDM) | 1x |
| Fetal bovine serum (FBS) | 2% (v/v) | |
| Penicillin/Streptomycin | 1x | |
| Peptone primatone | 0.3 mg/mL | |
| Insulin solution human | 4.8 mg/mL | |
| Minimum essential medium non-essential amino acids (MEM NEAA) | 1x | |
| LiCl salt buffer | Lithium chloride (LiCl) | 0.25 M |
| IGEPAL-CA630 | 1% (v/v) | |
| Ethylenediaminetetraacetic acid (EDTA) pH 8.0 | 1 mM | |
| Tris-HCl pH 8.1 | 10 mM | |
| Low salt buffer | Sodium dodecyl sulfate (SDS) | 0.1% (w/v) |
| Triton X-100 | 1% (v/v) | |
| Ethylenediaminetetraacetic acid (EDTA) pH 8.0 | 2 mM | |
| Tris-HCl pH 8.1 | 20 mM | |
| Sodium chloride (NaCl) | 150 mM | |
| Protein A Sepharose beads washing buffer | Tris-HCl pH 8.0 | 20 mM |
| Sodium chloride (NaCl) | 500 mM | |
| Ethylenediaminetetraacetic acid (EDTA) pH 8.0 | 2 mM | |
| Sodium dodecyl sulfate (SDS) | 0.1% (w/v) | |
| IGEPAL-CA630 | 1% (v/v) | |
| SDS Lysis buffer | Sodium dodecyl sulfate (SDS) | 1% (w/v) |
| Ethylenediaminetetraacetic acid (EDTA) pH 8.0 | 10 mM | |
| Tris-HCl pH 8.1 | 50 mM | |
| TE buffer | Tris-HCl pH 8.0 | 10 mM |
| Ethylenediaminetetraacetic acid (EDTA) pH 8.0 | 1 mM |
Table 1: List of the buffers and the medium used in this protocol.
| ON | OFF | |
| Peak power | 150.0 | 2.5 |
| Duty factor | 15.0 | 15.0 |
| Cycles/burst | 500 | 500 |
| No. of cycles | 28 |
Table 2: Shearing settings used in this protocol. These conditions have been optimized for a mature mouse T-cell line.
| Antibody | Supplier | Amount of antibody/immunoprecipitation | Amount of cells/immunoprecipitation | Washing conditions |
| H3 | Abcam (ab1791) | 2.5 mg | 5 x 106 | Once in low salt buffer, twice in High salt buffer, twice in LiCl salt buffer and three times in TE buffer |
| H3K4me1 | Abcam (ab8895) | 2.5 mg | 5 x 106 | Once in low salt buffer, twice in High salt buffer, twice in LiCl salt buffer and three times in TE buffer |
| H3K4me3 | Diagenode (pAb-003-050) | 2.5 mg | 5 x 106 | Once in low salt buffer, twice in High salt buffer, twice in LiCl salt buffer and three times in TE buffer |
| H3K27ac | Diagenode (pAb-174-050) | 2.5 mg | 5 x 106 | Oce in low salt buffer, twice in High salt buffer, twice in LiCl salt buffer and three times in TE buffer |
| IgG | Diagenode (C15410206) | Variable* | Variable* | Variable* |
| * In the case of the IgG control, the amount of both antibody and cells, as well as the washing steps, have to mirror the conditions of the other immunoprecipitations. |
Table 3: Antibodies and washing conditions used in this study.
| Gene | forward primer | reverse primer | probe |
| Gapdh promoter (0 kb) | 5'-GGG TTC CTA TAA ATA CGG ACT GC-3' | 5'-CTG GCA CTG CAC AAG AAG A-3' | 68 |
| Dtx1 enhancer (+26 kb) | 5'-CTC TGG GTT GTA GGG GAC AG-3' | 5'-GCA TGG GAA CTG TGT TAC AGA A-3' | 27 |
| Gene desert | 5'-CAA TGC ATG GGT CCA GAT TT-3' | 5'-ATT GGC ACG GAA GTA GTG CT-3' | 94 |
Table 4: Primers and probes used for qPCR in this study.
| Problem | Causes | Solution |
| Chromatin is under sheared and fragments are too big. | Too many cells have been used. | Reduce the number of cells. |
| The shearing is too low. | Increase the number of shearing cycles. | |
| Increase the shearing peak power, the duty factor and/or the cycles/burst. | ||
| Chromatin is over sheared and fragments are too small. | Too few cells have been used. | Increase the number of cells. |
| The shearing is too high. | Reduce the number of shearing cycles. | |
| Reduce the shearing peak power, the duty factor and/or the cycles/burst. | ||
| Too low recovery over IgG control and/or negative control (high background). | Not enough chromatin used for the experiment. | Increase the amount of chromatin. |
| Amount of antibody is too low. | Increase the amount of antibody. | |
| Amount of antibody is too high. | Reduce the amount of antibody. | |
| The antibody has low specificity. | Change the antibody. | |
| Washing conditions are too stringent. | Reduce the number of washing. | |
| Reduce the stringency of the washing buffers. | ||
| Washing conditions are not stringent enough. | Increase the number of washing. | |
| Increase the stringency of the washing buffers. | ||
| Too much DNA was pipetted in the qPCR. | Reduce the amount of DNA pipetted in the qPCR. | |
| qPCR conditions are not optimal. | Optimize the qPCR conditions. | |
| Reduce the number of cycles. | ||
| No product or product is too low in the qPCR reaction of the input sample. | Not enough DNA was pipetted in the qPCR. | Increase the amount of DNA pipetted in the qPCR. |
| qPCR conditions are not optimal. | Optimize the qPCR conditions. | |
| Increase the number of cycles. | ||
| Not enough chromatin used for the experiment. | Increase the amount of chromatin. | |
| No signal in the positive control and/or panH3 ChIP. | Not enough chromatin used for the experiment. | Increase the amount of chromatin. |
| Amount of antibody is too low. | Increase the amount of antibody. | |
| The antibody has low specificity. | Change the antibody. | |
| Elution is sub-optimal. | Be sure to frequently vortex the samples to maintain the beads in solution. |
Table 5: Troubleshooting.
Discussion
ChIP is a valid technique to investigate whether proteins or their PTMs are enriched at specific genomic regions. ChIP assay results are often challenging to interpret due to biological or technical reasons. The biological reasons are manyfold and include weak or indirect binding of proteins to DNA. There are also technical limitations, such as limited antibody specificity and inefficient cell lysis or chromatin shearing. A troubleshooting guide (Table 5) can help the reader to solve the problems that may be encountered with ChIP assays.
This protocol, making use of a cooling focused ultrasonicator device, allows one to efficiently shear the chromatin of a mature mouse T-cell line. The main critical step of the protocol is represented by the shearing of the chromatin, which must always be optimized for each cell type. It is suggested to vary the number of cells and the number of sonication cycles. Furthermore, the shearing efficiency is negatively influenced by the presence of SDS precipitates in the samples, which may form during the lysis of the cells on ice with SDS lysis buffer. In this case, it is best to incubate the sample after lysis at room temperature for a few minutes to reduce the presence of SDS precipitates. It is recommended to optimize the protocol to obtain sheared fragments between 200 and 500 bp and to always evaluate the shearing quality of the samples before proceeding with the immunoprecipitation. Additionally, the fixation time, amount of antibody and/or cells, and the washing conditions must be optimized for each antibody.
One more critical step in making a ChIP procedure successful is the choice of the antibody, as low-specificity antibodies significantly reduce the efficiency of the immunoprecipitation. Previously, a unified quality test procedure allowed for the evaluation of the specificity of several antibodies available on the market19. The screening pipeline was based on dot blot, Western blot, and ChIP, providing a list of high-quality reagents suitable for ChIP19. More recently, the specificity of several commercially available antibody was evaluated using high-density histone peptide microarray platforms, allowing for the establishment of a database on antibody specificity20. The reader can use this useful tool to identify the most specific antibodies that can be used for ChIP experiments.
This protocol has not been tested for primary T-cells and, in this case, the reader should refer to other published protocols that successfully perform ChIP on primary T-cells3,4,21. Notably, this protocol has been successfully used to perform ChIP on a mouse progenitor T-cell line, as well as on a mouse endothelial cell line.
5 x 106 cells should be used for each immunoprecipitation for the analysis of histone marks. Because this protocol can also be suitable, with some optimization, to perform ChIP on TFs or cofactors, it is best to increase the number of cells used for the immunoprecipitation in these cases. In order to reach the required number of cells for each experiment, more aliquots of sheared lysate can be pooled together before diluting the chromatin in the dilution buffer.
This protocol could also be modified to perform ChIP on endogenous proteins that do not bind directly to the DNA. In this case, pre-fixation with protein-protein crosslinkers (e.g., dimethyl adipimidate, DMA) may be required (see Appendix B for more information). The successful performance of ChIP on cofactors was previously done by overexpressing proteins that are fused to a biotin-tag. In this case, the protein was purified with streptavidin-conjugated magnetic beads, and a pre-fixation with DMA was performed22.
Disclosures
The authors have no competing financial interests.
Acknowledgments
We are grateful to P. Käse and T. Schmidt-Wöll for excellent technical assistance. This work was supported by the collaborative research grant TRR81 and the Heisenberg program (BO 1639/5-1) of the DFG (German Research Foundation), the Max Planck Society and the EXC 294 in Freiburg, and the Excellence Cluster for Cardio Pulmonary System (ECCPS) in Giessen to T.B.
References
- Rothenberg EV. The chromatin landscape and transcription factors in T cell programming. Trends Immunol. 2014;35(5):195–204. doi: 10.1016/j.it.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JA, Mortazavi A, Williams BA, Wold BJ, Rothenberg EV. Dynamic transformations of genome-wide epigenetic marking and transcriptional control establish T cell identity. Cell. 2012;149(2):467–482. doi: 10.1016/j.cell.2012.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauchy P, et al. Dynamic recruitment of Ets1 to both nucleosome-occupied and -depleted enhancer regions mediates a transcriptional program switch during early T-cell differentiation. Nucl Acids Res. 2016;44(8):3567–3585. doi: 10.1093/nar/gkv1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch F, et al. Transcription initiation platforms and GTF recruitment at tissue-specific enhancers and promoters. Nat Struct Mol Biol. 2011;18(8):956–963. doi: 10.1038/nsmb.2085. [DOI] [PubMed] [Google Scholar]
- Pekowska A, et al. H3K4 tri-methylation provides an epigenetic signature of active enhancers. EMBO J. 2011;30(20):4198–4210. doi: 10.1038/emboj.2011.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhille L, et al. High-throughput and quantitative assessment of enhancer activity in mammals by CapStarr-seq. Nat Commun. 2015;6:6905. doi: 10.1038/ncomms7905. [DOI] [PubMed] [Google Scholar]
- Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Heintzman ND, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459(7243):108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473(7345):43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostuni R, et al. Latent enhancers activated by stimulation in differentiated cells. Cell. 2013;152(1-2):157–171. doi: 10.1016/j.cell.2012.12.018. [DOI] [PubMed] [Google Scholar]
- Giaimo BD, Oswald F, Borggrefe T. Dynamic chromatin regulation at Notch target genes. Transcription. 2017;8(1):61–66. doi: 10.1080/21541264.2016.1265702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liefke R, et al. Histone demethylase KDM5A is an integral part of the core Notch-RBP-J repressor complex. Genes Dev. 2010;24(6):590–601. doi: 10.1101/gad.563210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung C, Mittler G, Oswald F, Borggrefe T. RNA helicase Ddx5 and the noncoding RNA SRA act as coactivators in the Notch signaling pathway. Biochim Biophys Acta. 2013;1833(5):1180–1189. doi: 10.1016/j.bbamcr.2013.01.032. [DOI] [PubMed] [Google Scholar]
- Oswald F, et al. A phospho-dependent mechanism involving NCoR and KMT2D controls a permissive chromatin state at Notch target genes. Nucl Acids Res. 2016;44(10):4703–4720. doi: 10.1093/nar/gkw105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrigoni L, et al. Standardizing chromatin research: a simple and universal method for ChIP-seq. Nucl Acids Res. 2016;44(7):e67. doi: 10.1093/nar/gkv1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essen D, Dullforce P, Brocker T, Gray D. Cellular interactions involved in Th cell memory. J Immunol. 2000;165(7):3640–3646. doi: 10.4049/jimmunol.165.7.3640. [DOI] [PubMed] [Google Scholar]
- White J, et al. Two better cell lines for making hybridomas expressing specific T cell receptors. J Immunol. 1989;143(6):1822–1825. [PubMed] [Google Scholar]
- Ghisletti S, et al. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity. 2010;32(3):317–328. doi: 10.1016/j.immuni.2010.02.008. [DOI] [PubMed] [Google Scholar]
- Egelhofer TA, et al. An assessment of histone-modification antibody quality. Nat Struct Mol Biol. 2011;18(1):91–93. doi: 10.1038/nsmb.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbart SB, et al. An Interactive Database for the Assessment of Histone Antibody Specificity. Mol Cell. 2015;59(3):502–511. doi: 10.1016/j.molcel.2015.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenouil R, et al. CpG islands and GC content dictate nucleosome depletion in a transcription-independent manner at mammalian promoters. Genome Res. 2012;22(12):2399–2408. doi: 10.1101/gr.138776.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein K, et al. Site-specific methylation of Notch1 controls the amplitude and duration of the Notch1 response. Sci Signal. 2015;8(369):ra30. doi: 10.1126/scisignal.2005892. [DOI] [PubMed] [Google Scholar]