

Abstract
Kinase activity is crucial for a plethora of cellular functions, including cell proliferation, differentiation, migration, and apoptosis. During early embryonic development, kinase activity is highly dynamic and widespread across the embryo. Pharmacological and genetic approaches are commonly used to probe kinase activities. Unfortunately, it is challenging to achieve superior spatial and temporal resolution using these strategies. Furthermore, it is not feasible to control the kinase activity in a reversible fashion in live cells and multicellular organisms. Such a limitation remains a bottleneck for achieving a quantitative understanding of kinase activity during development and differentiation. This work presents an optogenetic strategy that takes advantage of a bicistronic system containing photoactivatable proteins Arabidopsis thaliana cryptochrome 2 (CRY2) and the N-terminal domain of cryptochrome-interacting basic-helix-loop-helix (CIBN). Reversible activation of the mitogen-activated protein kinase (MAPK) signaling pathway is achieved through light-mediated protein translocation in live cells. This approach can be applied to mammalian cell cultures and live vertebrate embryos. This bicistronic system can be generalized to control the activity of other kinases with similar activation mechanisms and can be applied to other model systems.
Keywords: Developmental Biology, Issue 124, Optogenetics, protein-protein interactions, PC12 cell differentiation, Xenopus, embryonic development, cryptochrome, CRY2-CIBN, bicistronic, Raf/MEK/ERK kinase signaling cascade
Introduction
Growth factors are involved in a wide spectrum of cell functions, including proliferation, differentiation, migration, and apoptosis, and play pivotal roles in many biological events, including embryonic development, aging, and regulation of mental status1,2,3,4,5. Many growth factors signal through complex intracellular signaling cascades. These signaling events are often operated by reversible protein phosphorylation in a precisely regulated fashion6,7. Thus, an understanding of the signaling outcomes of protein kinases, which are responsible for protein phosphorylation, is fundamentally important.
Different growth factors act through a rather common intracellular signaling network, even though they stimulate distinct cellular responses8,9. Common intracellular mediators of receptor tyrosine kinases include Ras, Raf, extracellular signal-regulated kinase (ERK), mitogen-activated protein kinase (MAPK)/ERK kinase (MEK), phosphoinositide 3-kinase (PI3K), Akt, and phospholipase C gamma (PLCγ)10,11. Accumulating evidence suggests that signaling diversity and specificity depend upon the spatial and temporal regulation of signaling activity12. For instance, in rat pheochromocytoma cells (PC12), epidermal growth factor (EGF) stimulation, which results in cell proliferation, transiently activates the ERK pathway9. On the other hand, stimulation with nerve growth factor (NGF), which leads to cell differentiation, activates the ERK pathway in a sustained manner9,13. In cultured rat hippocampal neurons, transient signaling by brain-derived neurotrophic factor (BDNF) promotes primary neurite outgrowth, while sustained signaling leads to increased neurite branching14. During early embryonic development, phosphorylated ERK activity is temporally dynamic and is widespread across the embryo6. A recent genetic screen during early Xenopus embryogenesis showed that ERK and Akt signaling cascades, two downstream primary growth factor pathways, display stage-specific activation profiles7. Thus, an understanding of kinase signaling outcomes calls for tools that can probe the spatial and temporal features of kinase activity with sufficient resolution.
Conventional experimental approaches to probe the dynamic nature of signal transduction during development lack the desirable spatial and temporal resolution. For instance, pharmacological approaches utilize small chemical or biological molecules to stimulate or suppress signal transduction in cells and tissues. The diffusive nature of these small molecules makes it challenging to restrict their action to a specific region of interest15. Genetic approaches (e.g., transgenesis, the Cre-Lox system, or mutagenesis) often lead to the irreversible activation or repression of the target gene expression or protein activity16,17,18. The Tet-On/Tet-Off system19 offers improved temporal control of gene transcription but lacks strict spatial control because it relies on the diffusion of tetracycline. Recent developments in chemically induced protein dimerization20 or photo-uncaging21,22,23,24 have greatly enhanced the temporal control of signaling networks. The spatial control, however, remains challenging due to the diffusive nature of the caged chemicals.
Recent emerging optogenetic approaches, which harness the power of light to control protein-protein interactions, allow for the modulation of signaling pathways with high spatiotemporal precision as well as reversibility. Shortly after its initial success in controlling neuronal firing25,26,27, optogenetics has been extended to control other cellular processes, such as gene transcription, translation, cell migration, differentiation, and apoptosis28,29,30,31,32,33,34. A strategy using the photoactivatable protein pair Arabidopsis thaliana cryptochrome 2 (CRY2) protein and the N-terminal domain of cryptochrome-interacting basic-helix-loop-helix (CIBN) was recently developed to control Raf1 kinase activity in mammalian cells and Xenopus embryos35. CRY2 binds to CIBN upon blue-light stimulation, and the CRY2/CIBN protein complex dissociates spontaneously in the dark34. Blue light excites the CRY2 cofactor, flavin adenine dinucleotide (FAD), which leads to a conformational change in CRY2 and its subsequent binding to CIBN. Constitutively active (W374A) and flavin-deficient (D387A) mutants of CRY2 can be produced through mutations in the FAD-binding pocket: the CRY2W374A mutant binds to CIBN independent of light, whereas the CRY2D387A mutant does not bind to CIBN under blue-light stimulation36,37. The optogenetic system described in this protocol uses wild-type CRY2 and CIBN to induce protein translocation-mediated Raf1 activation in live cells. It is known that the membrane recruitment of Raf1 enhances its activity38. In this system, a tandem CIBN module is anchored to the plasma membrane and CRY2-mCherry is fused to the N-terminal of Raf135. In the absence of blue light, CRY2-mCherry-Raf1 stays in the cytoplasm, and Raf1 is inactive. Blue-light stimulation induces CRY2-CIBN binding and recruits Raf1 to the plasma membrane, where Raf1 is activated. Raf activation stimulates a Raf/MEK/ERK signaling cascade. Both CRY2- and CIBN- fusion proteins are encoded in a bicistronic genetic system. This strategy can be generalized to control other kinases, such as Akt, whose activation state can also be turned on by protein translocation in cells39. This work presents detailed protocols for implementing this optogenetic strategy in mammalian cell cultures and multicellular organisms.
Protocol
Animal research was conducted in accordance with guidelines set by the Illinois Institutional Animal Care and Use Committee (IACUC) and the University of Illinois Department of Animal Resources (DAR).
1. Optogenetic Induction of Protein Localization in BHK21 Mammalian Cell Culture
NOTE: Steps 1.1-1.3 provide a method to assemble a cell culture chamber for imaging with high-magnification objectives (e.g., 63X or 100X), which typically have short working distances. These objectives require a thin glass coverslip (e.g., #1.5, 170 µm thickness) as the imaging substrate. Alternatively, a glass-bottom cell culture dish/slide can be used. In such a case, steps 1.1-1.3 can be skipped.
- Cleaning glass coverslips
- Place 30 glass coverslips in a coverslip holder.
- In a 600 mL plastic beaker, add 20 g of detergent and 400 mL of warm tap water. Place the beaker on a magnetic stirrer and stir until all detergent is dissolved. Remove the foam using tissue paper.
- Use the bottom of a 100 mm x 15 mm Petri dish as a spacer from the stir bar and as a surface for the coverslip holder. To maintain adequate flow during the cleaning process, make 3-4 draining holes in the Petri dish.
- To make the draining holes, heat up a soldering iron and burn through the plastic in a vented hood. CAUTION: Do not touch the heated soldering iron with bare hands.
- Place the coverslip holder on the Petri dish in the beaker. Stir for 2 h to overnight at room temperature.
- Wash three times with 500 mL of DI water in a 1,000 mL beaker. The detergent solution can be re-used.
- Pick up individual coverslips using forceps. Immerse the coverslips in 190-proof (95%) ethanol for 1 min.
- Dry the coverslips inside a sterile hood. Store the coverslips in a sterile plastic container until use.
- Making poly-L-lysine (PLL)-coated coverslips
- Make 0.1 M borate buffer by dissolving 1.24 g of boric acid and 1.9 g of disodium tetraborate in 400 mL of water. Adjust the pH to 8.5 with 10 M NaOH.
- Dissolve PLL in the borate buffer to a final concentration of 1 mg/mL. Aliquot the PLL solution in 50-mL sterile centrifuge tubes.
- Add 50 mL of PLL coating solution to a 10-cm tissue culture dish. Place the dish in a 37 °C incubator to warm up the PLL solution.
- Open the coverslip container (step 1.1.8) in a sterile hood.
- Immerse the cleaned coverslips one by one in the pre-warmed PLL solution in the tissue culture dish. Avoid bubble formation in order to immerse all surfaces.
- Place the dish with the coverslip in a 37 °C CO2 incubator overnight. Take out each coverslip and place it in a sterile coverslip holder. Rinse three times with autoclaved ultrapure water (18.2 MΩ·cm resistivity) in a 1,000 mL beaker in a sterile hood.
- Dry the coverslips in the coverslip holder in the sterile hood. Store the coverslips in a sterile plastic container until use.
- Assembling polydimethylsiloxane (PDMS) cell culture chambers
- In a plastic container, weigh 40 g of PDMS base and add 4 g of curing agent to achieve a 10:1 w/w ratio of PDMS and curing agent. Mix the PDMS/curing agent by stirring with a pipette tip for 2 min. Alternatively, use a centrifugal mixer.
- Use a beaker with a 4 inch diameter as a template to make a holder with a piece of aluminum foil. Place the aluminum foil holder in a 15 cm Petri dish. Place a 4 inch silicon wafer within this aluminum foil holder.
- Pour the mixed PDMS solution into the holder. Use a thin glass rod to touch the edge of the wafer gently. Remove any air bubbles trapped under the wafer.
- Insert the 15 cm Petri dish into a vacuum chamber to degas the PDMS solution. Continue degassing for about 20 min, until no bubbles are generated. In the meantime, preheat a convection oven to 65 °C.
- Carefully place the 15 cm Petri dish into the oven. Incubate for 2 h.
- Remove the plate from the oven. Use a glass rod to gently touch the edge of the PDMS and ensure complete crosslinking. If not, incubate longer, until the PDMS is solid and non-sticky.
- Remove the aluminum foil from the plate and peel it off the silicon wafer. Starting from the edge, carefully peel the cured PDMS off the silicon wafer. CAUTION: Avoid applying too much force, as it can break the wafer.
- Trim the PDMS into 20 mm x 30 mm rectangular pieces to fit the 24 mm x 40 mm coverslip with a razor blade.
- Use a chisel-shaped blade to cut a rectangular opening of 10 mm x 20 mm from the center of each PDMS piece.
- Clean the PDMS chambers using the detergent protocol described in step 1.1.
- Dry the PDMS chambers in a clean hood. Place the dry chambers in an autoclavable container covered with aluminum foil.
- Autoclave the container using gravity (121 °C, 15 psi) for 30 min and store the container at room temperature.
- BHK21 cell culturing and transfection
- Make 500 mL of medium for BHK21 cell culture by mixing 445 mL of DMEM with 50 mL of FBS and 5 mL of 100× Penn-Strep-Glutamine (10,000 U/mL penicillin, 10 mg/mL streptomycin, and 29.2 mg/mL L-glutamine). Ensure that the final concentration of FBS is 10%.
- Aliquot 10 mL of non-HEPES CO2-independent medium for live-cell imaging. NOTE: CO2-independent medium supports cell growth without a CO2 incubator and is ideal for imaging cells under atmospheric conditions. Refer to the Materials List for detailed information. In addition to the BHK21 cell line, other types of mammalian cells should also give similar results.
- Assemble a cell culture device by placing one autoclaved, sterile PDMS chamber (step 1.3) on a sterile PLL-coated coverslip (step 1.2) in a sterile hood.
- Place each device in a sterile 60 mm Petri dish.
- Use 0.5 mL of 0.25% trypsin to detach the cells from one well of a 12-well tissue culture plate. Count the cell density with a hemocytometer. Plate 20,000 BHK21 cells in the chamber (about 10,000 cells/cm2) with 200 µL of cell culture medium.
- Prepare DNA plasmid with a plasmid preparation kit (see the Materials List). NOTE: The design and functionality of the CRY2-mCherry-Raf1-2A-2CIBN-GFP-CaaX are shown in Figure 1A-1B.
- Twenty-four (24) h after cell plating, transfect the cells with 50-100 ng of CRY2-mCherry-Raf1-2A-2CIBN-GFP-CaaX plasmid, according to the manufacturer’s protocol.
- Three (3) hours after transfection, change the medium to 200 µL of fresh culture medium (step 1.4.1) and allow the cells to recover overnight by incubating the culture in a 37 °C, 5% CO2 incubator.
- Fluorescence live-cell imaging
- Turn on a computer, light source, microscope, and camera. Use a confocal fluorescence microscope (equipped with a 100X oil-immersion objective) for the rest of the protocol. NOTE: An inverted epifluorescence microscope can also be used.
- Replace the cell culture medium with 200 µL of CO2-independent medium.
- Set up the data-acquisition protocol before placing the cells on the microscope. Use 488-nm excitation FITC channel for optogenetic stimulation. Use 561 nm excitation TRITC channel to locate transfected cells and track the cellular localization of mCherry-labeled protein.
- Set the gain of FITC and TRITC channels as 120 and 200, respectively. Use a pixel-dwelling time of 2.2 µs and size of 512 x 512 pixels.
- Measure the power of the 488-nm light by placing a power meter close to the objective window. A total power of 2 µW (about 10,000 W/cm2 at the focus) is sufficient to induce CIBN-CRY2PHR association.
- Set up a timestamp acquisition with a 5 s interval and a total acquisition time of 2 min.
- Apply appropriate index-matching material (immersion oil) on the objective window. Use the phase-contrast mode to focus on the cells on the coverslip surface.
- Locate a transfected cell under 561 nm light by moving the microscope stage. CAUTION: Avoid using blue light during this step, as it will activate the association between photoactivatable proteins.
- Once a transfected cell is located, initiate data acquisition. NOTE: Successfully transfected cells should show fluorescence in both FITC (from 2CIBN-GFP-CaaX) and TRITC (from CRY2-mCherry-Raf1) channels (Figure 1C-D).
- Record a series of time-stamped images in both the FITC and TRITC channels (Figure 1D).
- To probe the spontaneous dissociation of the CRY2-CIBN protein complex, record another timestamp with the Txred channel alone for 20 min, with a 30 s interval.
- Save the time-stamped image for data analysis.
- Image analysis
- Open the images with an image analysis software that can extract the intensity from an image40.
- Select two representative snapshots: one prior to blue-light exposure and the other post-blue-light exposure.
- Draw a line across the cell spanning the background, the plasma membrane, and the cytoplasm. Project the intensity along this line. Save the values and plot them out to compare the difference before and after light exposure (Figure 1D).
- To analyze the kinetics of protein association, select one representative region of interest (ROI) in the plasma membrane, one ROI in the cytoplasm, and one ROI in the background.
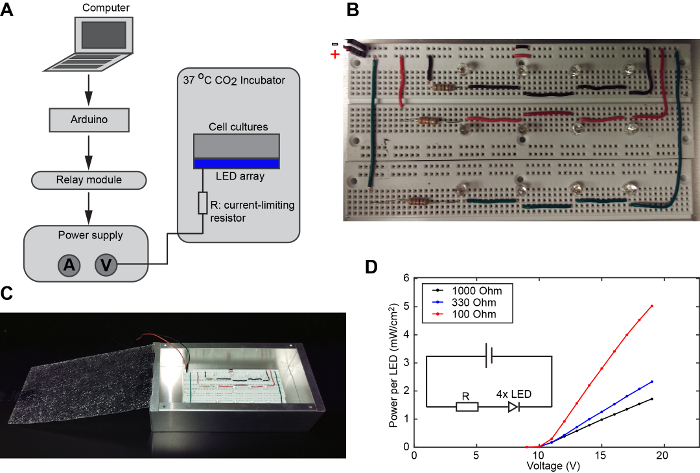
- Acquire the mean intensities of the plasma membrane (IPM), the cytoplasm (ICYT), and the background (IBKD) for the image stack.
- Calculate the ratio of membrane/cytosolic intensity for each image using the following equation:
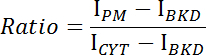
- Plot the ratio versus time and determine the binding or dissociation kinetics of CRY2-CIBN (Figure 1E-F).
2. Construction of an LED Array for Long-term Light Stimulation in a CO2 Incubator
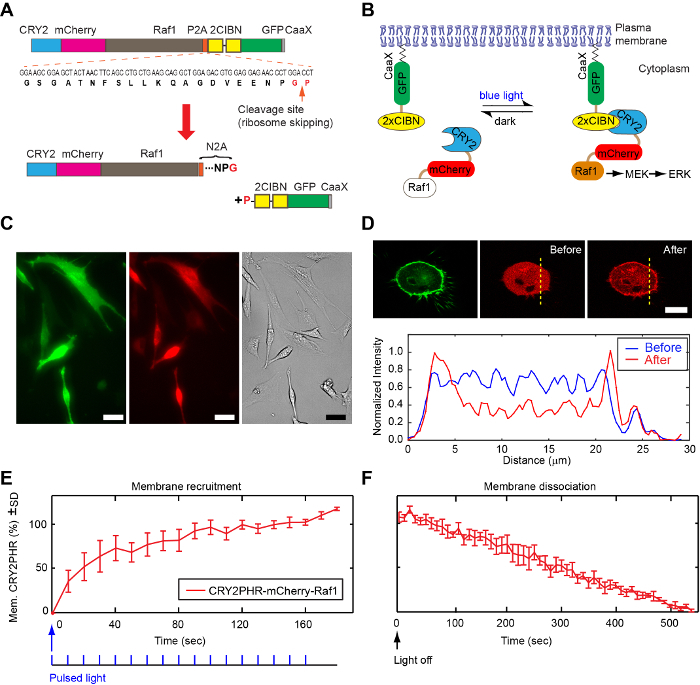
NOTE: The overall schematic of the experimental setup is shown in Figure 2A.
Make the LED array by inserting 12 blue LEDs into two breadboards and connecting current-limiting resistors. NOTE: With a 30 V power supply, four LEDs can be connected in series, and their brightness can be controlled by a current-limiting resistor.
Place the breadboards into an aluminum box. NOTE: The height of the aluminum box should be 2 in. This height is optimal for illuminating a 12-well tissue culture plate, because the size of the diverging light spot is the same as that of a single well.
Use two metal wires to connect to the power supply. Make sure that the length of the wire is sufficient when the light box is placed inside a CO2 incubator.
Use a transparent light diffuser as the cover of the light box (Figure 2C).
Calibrate the power output of each LED at a range of voltage inputs. Use a power of 0.2 mW/cm2 for the 24 h PC12 cell differentiation assay. Use a power of 5 mW/cm2 for live Xenopus embryos or explants assays.
3. Optogenetic Induction of PC12 Cell Differentiation
- Cell culturing and transfection
- Make 500 mL of medium for a rat pheochromocytoma (PC12) cell culture by mixing 407.5 mL of F12K with 75 mL of horse serum + 12.5 mL of FBS + 5 mL of 100× Penn-Strep-Glutamine (final concentrations of horse serum and FBS: 15% and 2.5%, respectively). Make low-serum medium by mixing 1 volume of full medium in 99 volumes of F12K medium.
- Plate PC12 cells in a 12-well plate at a density of 300,000 cells/well or 75,000 cells/cm2.
- Transfect the cells with CRY2-mCherry-Raf1-2A-2CIBN-GFP-CaaX 24 h after cell plating (similar to step 1.4.7). Use 1.2 µg of DNA for each well. Allow the cells to recover overnight in culture medium in a 37 °C incubator supplemented with 5% CO2. Check the transfection efficiency 16 h after transfection. NOTE: A 30-50% transfection efficiency should be achieved to ensure a sufficient cell count.
- Change the medium to low-serum medium 24 h after transfection. Use 1 mL of medium per well of a 12-well plate.
- Light-induced PC12 cell differentiation
- Place the LED array in a CO2 incubator. Connect it to the power supply using a pair of wires. Set the power of the LED to 0.2 mW/cm2. Place the 12-well plate containing transfected PC12 cells (step 3.1.3) on the window of the LED array. Apply continuous light illumination for 24 h in a 37 °C incubator supplemented with 5% CO2.
- Fluorescence live cell imaging
- Follow steps 1.5.1-1.5.4 for microscope and sample preparation.
- Set up single-snapshot data acquisition. Use 200 ms for both the GFP and Txred channel.
- Capture images of the transfected cells in both the GFP and Txred channels. Record roughly 200 cells for each condition. Save the files for data analysis.
- Image analysis
- Count the percentage of differentiated cells over the total number of transfected cells.
- Use any cell-counting module in the image analysis software to count the cells.
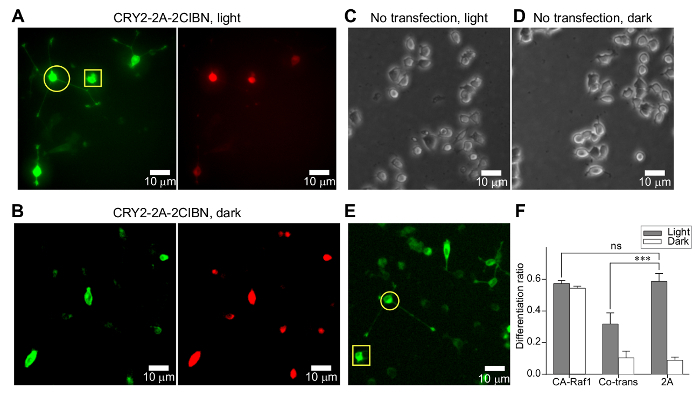
- Manually count the differentiated cells. NOTE: Differentiated cells are defined as those in which at least one neurite is discernibly longer than the cell body (Figure 3A-3B).
- Repeat step 3.4.3 with undifferentiated cells.
- Calculate the differentiation ratio using the following equation:
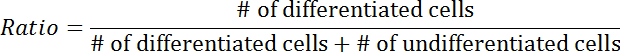
4. Optogenetic Control of Kinase Activity in Xenopus Embryos
- Preparation of buffers
- Prepare 1 L of 1x Marc's Modified Ringer (MMR): 100 mM NaCl, 2 mM KCl, 1 mM MgCl2, 2 mM CaCl2, and 5 mM HEPES; pH 7.5.
- Preparation of mRNA
- Digest the CRY2-mCherry-Raf1-2A-2CIBN-GFP-CaaX plasmid DNA (2 µg) with 1 µL of ApaI (50 unit) at 37 °C for 2 h.
- Precipitate linearized DNA in 60 µL of 100% ethanol. Spin at 12,000 × g for 5 min at room temperature to pellet the DNA. Carefully remove the supernatant and wash the pellet again with 60 µL of 70% ethanol. Spin at 12,000 × g for another 5 min at room temperature. Carefully remove the supernatant and resuspend the DNA pellet in 6 µL of RNase-free H2O.
- Perform in vitro transcription by incubating 1 µg of linearized DNA with 2 µL of supplied SP6 RNA polymerase, a ribonucleotide mixture (10 mM ATP, CTP, and UTP; 2 mM GTP; and 8 mM cap analog), and 20 µL of nuclease-free water at room temperature for 1 h.
- Remove the DNA template by DNase I digestion at 37 °C for at least 15 min and purify the synthesized RNA using a silica-membrane spin column.
- Stop the reaction and precipitate the RNA by adding 30 µL of nuclease-free water and 30 µL of LiCl precipitation solution. Mix thoroughly and chill for 30 min at -20 °C.
- Centrifuge at 4 °C for 15 min at 12,000 × g to pellet the RNA.
- Carefully remove the supernatant. Wash the RNA once with 1 mL of 70% ethanol and resuspend in 40 µL of nuclease-free water.
- Load 40 µL of RNA in a spin column. Centrifuge at 4 °C for 1 min at 12,000 × g to remove the unincorporated nucleotides and caps.
- Harvest Xenopus embryos
- Follow the protocol described by Sive et al.41 to obtain the Xenopus embryos.
- mRNA microinjection
- Fabricate the needles for microinjection by pulling glass capillaries with a capillary puller. Set the pulling program to: heat = 355, pull strength = 80, velocity = 50, and time = 100.
- Remove the jelly coat from the embryos by treating them with 3% cysteine (diluted in 0.2x MMR).
- Transfer the embryos to 3% polysucrose and 0.5x MMR solution for microinjection. Inject 500 pg to 1 ng of CRY2-mCherry-Raf1-2A-2CIBN-GFP-CaaX RNA into each embryo. NOTE: Injection of a dose higher than 1 ng results in blue light-independent activation of the MAPK signaling.
- Optogenetic stimulation of kinase activity
- Culture microinjected embryos in 3% polysucrose, 0.5x MMR solution at room temperature until they reach the mid-gastrula stage (stage 12). Then, culture the embryos in 0.2x MMR solution.
- Transfer the embryos or explants to a 12-well plate. Place the 12-well plate on the home-built LED array (step 2.5) for blue-light (475 nm) treatment.
- Place a mirror on the top of the 12-well plate to ensure full blue-light illumination of the embryos or explants.
- Tune the power of the blue light to 5 mW/cm2. NOTE: Blue-light treatment can be performed at any desired time, in either 3% polysucrose, 0.5x MMR solution, or 0.2x MMR solution.
- Harvest the embryos at any desired time. Use them for histological, Western-blot, or gene-expression analysis.
Representative Results
Ratiometric expression of photoactivatable protein pairs: Figure 1A shows the design of a bicistronic optogenetic construct, CRY2-mCherry-Raf1-P2A-CIBN-CIBN-GFP-CaaX (referred to as CRY2-2A-2CIBN), based on the porcine teschovirum-1 2A (P2A) peptide, which shows the highest ribosome-skipping efficiency among mammalian cell lines42. In previous work, it has been determined that the optimal ratio for CIBN-GFP-CaaX:CRY2-mCherry-Raf1 is 2:135. This configuration allows for sufficient membrane recruitment and activation of CRY2-mCherry-Raf1 (Figure 1B). Cells singly transfected with CRY2-2A-2CIBN show clear fluorescence in both the GFP and Txred fluorescent channels, either under epi-illumination (Figure 1C) or confocal microscopy (Figure 1D). Blue light-mediated membrane recruitment of CRY2-mCherry-Raf1 (Figure 1D) can be clearly detected in confocal microscopy. The association occurs within seconds after blue-light exposure (Figure 1E), and the CRY2-CIBN protein complex dissociates with a half-life of about 5.5 min (Figure 1F).
Light-controlled PC12 cell differentiation in the absence of nerve growth factor: An intriguing observation in growth factor signaling is that common downstream signaling pathways (e.g., ERK, PI3K-Akt, and PLCγ) elicit diversity and specificity in signaling responses. A better understanding of growth factor-mediated signaling can be achieved by enabling technology that allows for the precise spatiotemporal regulation of individual cascades. The optogenetic approach introduced in this protocol demonstrates one such technology that allows for the specific activation of the Raf/MEK/ERK signaling pathway with high temporal resolution. Because this approach bypasses the process of NGF binding to its membrane receptors, the signaling kinetics no longer depend on the endogenous receptors. Instead, precise kinetic control can be achieved by modulating the temporal profile of the stimulating light (Figure 2A). Thus, this approach opens up new possibilities to study the signaling kinetics of kinase activity. Additionally, this experimental setup provides an extremely simple and economic way to integrate the optogenetic approach into studies of intracellular signal transduction (Figure 2B-2D). For the PC12 cell differentiation assay, a 24-h continuous-light stimulation at 0.2 mW/cm2 is sufficient to induce significant neurite outgrowth (Figure 3A). Negative controls, including transfected cells without light (Figure 3B) and non-transfected cells with or without light (Figure 3C-3D), do not show significant neurite outgrowth. At this power, cells transfected with a constitutively active Raf1 (CA-Raf1) undergo normal differentiation (Figure 3E). Notably, cells transfected with the bicistronic optogenetic construct produce a significantly higher differentiation ratio than cells co-transfected with two constructs, reaching the value induced by CA-Raf1 (Figure 3F). The differentiation ratio is calculated by dividing the number of differentiated cells by the number of transfected cells guided by GFP fluorescence. Differentiated cells are defined as those with at least one neurite longer than the size of the cell body35. This enhancement in the differentiation ratio arises from: 1) a better delivery of photoactivatable proteins with the bicistronic system and 2) an optimized expression ratio between CIBN and CRY235.
The reversible optogenetic stimulation of the Raf/MEK/ERK signaling pathway in live Xenopus laevis embryos: X. laevis is a well-established model organism for studying gene transcription, signal transduction, and embryonic development. Previous work discovered that the activation of the Raf/MEK/ERK pathway leads to a cell fate change, resulting in the formation of an ectopic tail-like structure at the tailbud stage43. As a potent mesoderm inducer, overexpressed Raf1 can induce ectopic mesoderm during germ-layer specification, which occurs during the blastula and early gastrula stages, and result in the formation of ectopic tail-like structures later on. Alternatively, overexpressed Raf1 may trigger an epithelial-mesenchymal transition (EMT)-like event after germ-layer specification has completed and can directly transform ectoderm to neural and mesoderm lineages. This is followed by the proliferation and extension of these structures43. In these experiments, RNAs encoding MET receptor, Ras, and Raf1 were injected into embryos at the two-cell stage. Shortly after the RNA was injected into the embryo, the overexpressed MET/Ras/Raf1 began to constitutively activate the downstream signaling cascades. Therefore, it was technically challenging to bypass early developmental stages and activate Raf1 specifically after germ-layer specification. It was impossible to use such a strategy to determine if activation of the Raf/MEK/ERK signaling after germ-layer specification would induce a cell fate change, leading to the formation of the ectopic tail-like structure.
Optogenetics provides a mechanism to control the timing of Raf1 activity. When the RNA of CRY2-2A-2CIBN was injected into embryos at early developmental stages, Raf1 remained inactive until blue light was supplied. The animal cap assay was conveniently used to characterize the signaling outcome, because the basal ERK activity is low in animal caps. The light-mediated activation of the Raf/MEK/ERK signaling cascade induced a significant upregulation of xbra, a mesodermal marker, as determined by RT-PCR (Figure 4A) or whole-mount in situ hybridization (Figure 4B). Dynamic changes in the phosphorylation of ERK in response to blue light were also detected by Western-blot analysis (Figure 4C). In whole embryos, a mixture of CRY2-2A-2CIBN and n-β-gal RNAs was injected at the 8-cell stage into one of the dorsal animal blastomeres, which later give rise to dorsal anterior tissue. Compared with untreated embryos (Figure 4D-4E), those injected with CRY2-2A-2CIBN and treated with blue light formed ectopic tail-like structures (Figure 4F-4G). Injected embryos in the dark (Figure 4H) or uninjected under blue-light illumination (Figure 4I) did not form tail-like structure. Blue-light illumination after germ-layer specification induced significant tail-like structures (Figure 4J).
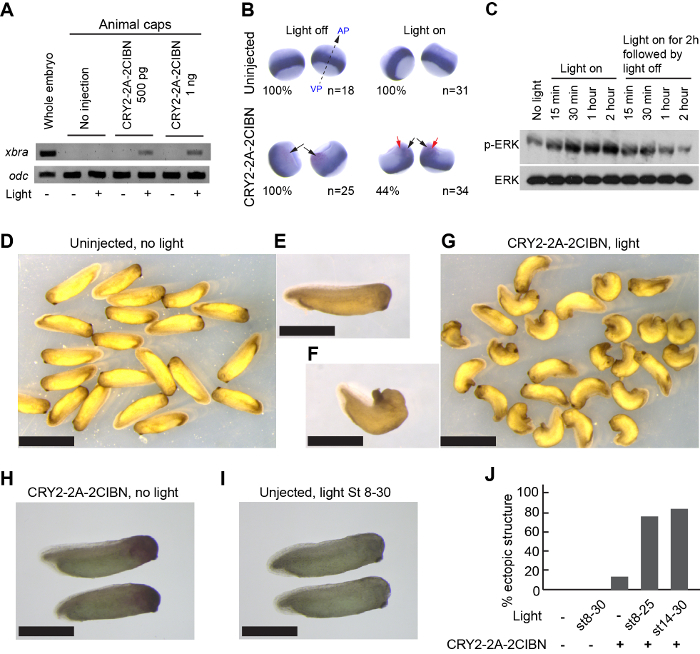
Figure 1: The mechanism of light-induced protein localization and activation of the kinase signaling pathway. (A) Design of a bicistronic construct, with an optimized CIBN-to-CRY2 ratio (2 CIBN:1 CRY2) that allows for the light-induced activation of the Raf/MEK/ERK kinase signaling pathway. Upon ribosomal skipping, one mRNA transcript generates two proteins: CRY2-mCherry-Raf1-N2A (ending with amino acids NPG) and proline-2CIBN-GFP-CaaX. (B) Light-induced binding between CIBN and CRY2 leads to membrane recruitment of CRY2-mCherry-Raf1, which activates the Raf/MEK/ERK signaling pathway. (C) Fluorescence images of BHK21 cells transfected with CRY2-2A-2CIBN under epi-illumination. Scale bar: 20 µm. (D) Confocal fluorescence imaging of cells transfected with CRY2-2A-2CIBN. The left panel shows cleaved 2CIBN-GFP-CaaX localized on the plasma membrane; the middle panel shows a snapshot of CRY2-mCherry-Raf1 before blue-light stimulation; the right panel shows a snapshot of CRY2-mCherry-Raf1 after 10 pulses of blue-light stimulation. The bottom panel shows normalized intensity profiles along a yellow dotted line across the cell, before and after light stimulation. Scale bar = 20 µm. (E-F) Kinetics for light-induced CRY2-CIBN association (E) and spontaneous dissociation (F) of the CRY2-CIBN protein complex in the dark. Figure 1A-B was adapted from reference35, reproduced with the permission of the Company of Biologists. Figure 1E-F was adapted from reference44 under the Creative Commons Attribution (CC BY) license. Please click here to view a larger version of this figure.
Figure 2: Design and calibration of the home-built LED array. (A) Schematic of the experimental setup for the light-controlled kinase activity in live cells. (B) Circuit board for the home-built LED array. (C) A light box that can be used for long-term light stimulation in a CO2 incubator. The height of the aluminum box is 2 in. (D) Typical LED output power versus voltage. Values light power per LED in a circuit in which a current-limiting resistor and four LEDs are connected in series. Figure 2A was adapted from reference35, reproduced with the permission of the Company of Biologists. Please click here to view a larger version of this figure.
Figure 3: Optogenetic induction of PC12 cell differentiation. (A) Multi-channel snapshots of PC12 cells transfected with CRY2-2A-2CIBN after 24 h of blue-light stimulation (0.2 mW/cm2). A circle marks a differentiated cell. A square marks an undifferentiated cell. (B) Same as (A), except no blue-light stimulation was used. (C-D) Non-transfected PC12 cells under 24 h of blue-light stimulation (0.2 mW/cm2) (C) or dark incubation (D). (E) Representative images of cells transfected with Raf1-GFP-CaaX (CA-Raf1). The circle and rectangle mark differentiated and undifferentiated cells, respectively. (F) Differentiation ratios of PC12 cells transfected with CA-Raf1, co-transfected with CRY2-mCherry-Raf1 and CIBN-GFP-CaaX, and singly transfected with CRY2-2A-2CIBN. 24 h after transfection, cells were either exposed to light or incubated in the dark for another 24 h. The values represent the mean ± SD from four independent data sets. Only cells exposed to blue light showed significant differentiation. Figure 3F was adapted from reference35, reproduced with the permission of the Company of Biologists. Please click here to view a larger version of this figure.
Figure 4: Optogenetic induction of kinase activity in live Xenopus embryos. (A) RT-PCR results showing that exposure to blue light induced the expression of xbra, a pan-mesodermal marker, in CRY2-2A-2CIBN-injected animal caps at the early gastrula stage. (B) Whole-mount in situ hybridization of xbra in embryos. Light-induced activation of MAPK activity modulates the spatial distribution of xbra (red arrows in the bottom-right panel). Black arrows mark nuclear β-galactosidase as the lineage tracer. AP: animal pole. VP: vegetal pole. (C) Western-blot analysis showing that, in an animal cap assay, CRY2-2A-2CIBN induced blue light-dependent, reversible phosphorylation of ERK. (D) Images showing the morphology of normal embryos, without mRNA injection and without light treatments. (E) A zoomed-in image of a single embryo. (F-G) Zoomed-in and whole-field images for embryos injected with CRY2-2A-2CIBN mRNA and subjected to blue-light stimulation. Activation of Raf1 by treating CRY2-2A-2CIBN-injected embryos with blue light induces ectopic tail-like structures in the head region. (H-I) Negative controls, including embryos injected with CRY2-2A-2CIBN but under dark incubation (H) and uninjected embryos under light stimulation (I). All light stimulation experiments were conducted with a power of 5 mW/cm2. (J) Statistical analysis of the percentage of embryos showing ectopic tail-like structure under each condition. Scale bar = (D) and (G): 1 mm; (E-F) and (H-I) 0.5 mm. Figure 4A, C, E-F, and H-J were adapted from reference35, reproduced with the permission of the Company of Biologists. Please click here to view a larger version of this figure.
Discussion
When building the light box, the power of individual LEDs should be measured. Based on previous experience, the power output can vary between individual LEDs due to manufacturing variance. Select a set of LEDs that have a power output within 10% of each other. The number of LEDs, the current-limiting resistor, and the power input can be modified for different types of cell culture containers (e.g., a 6-well or 24-well plate). A 24 h of light illumination at a power of 0.2 mW/cm2 does not induce detectable phototoxicity44. If a higher power is used, consider using intermittent light to reduce heat generation and phototoxicity. The optogenetic system introduced in this protocol induces neurite outgrowth in PC12 cells under intermittent light stimulation comparable to that of continuous light stimulation, given that the dark interval between the two adjacent light pulses is less than 45 min44. Due to intrinsic differences in the kinetics of protein association and dissociation, the duration between light pulses must be tuned for each unique protein pair. Overexpression of cytosolic Raf1 does not cause PC12 cell differentiation without blue-light illumination. By controlling the amount of mRNA injected into each embryo, no significant embryonic phenotypes were observed in the dark.
Although a low-power blue light is sufficient to stimulate the association of CRY2- and CIBN-fusion proteins, the poor penetration depth of blue light limits its use in deep tissue stimulation. Although such stimulation has been achieved by delivering light via other devices (e.g., fiber optics), the approaches are invasive45. This issue can be addressed in two ways: by using a protein pair that responds to longer wavelength (e.g., the phytochrome-PIF6 protein pair) or by using two-photon excitation. Both approaches can provide deeper penetration, but they could suffer from other limitations. For example, the PhyB-PIF6 pair requires a synthetic cofactor to function, and two-photon microscopy requires costly instrumentation. The lack of tunability in the CRY2-CIBN interaction is another limitation to consider. Wild-type CRY2 spontaneously dissociates from CIBN with a half-life of 5.5 min in the dark34. Depending on the kinetics of the target signaling pathway, a shortened or prolonged dissociation half-life may be preferred. Mutations in CRY2 have been made that can either shorten the dissociation half-life to 2.5 min (W349R) or prolong it to 24 min (L348F)46. Alternatively, engineered photoactivatable protein pairs based on fungal receptor Vivid (VVD) and p/nMagFast1 and 2 show a dissociation half-life of 4.2 min and 25 s, respectively47. In this protocol, the wild-type CRY2-CIBN protein pair turns on the MAPK pathway within 5 min of light stimulation, and the activity decays back to the basal level within 30 min in mammalian cells44 and 1-2 h in Xenopus embryos35. For spatial control, confocal microscopy can focus the blue laser to the size of a diffraction-limit spot of about 200 nm in the image plane. By adjusting the size of field diagram in an epi-illumination microscope equipped with a non-coherent light source, it is possible to limit the illumination size to about 10 µm in diameter. Both methods should be able to achieve the subcellular control of optogenetic stimulation in cell cultures.
The ability of optogenetics to reversibly interrogate signal transduction with high spatial and temporal resolution offers an entirely new opportunity to study dynamic intracellular signaling in live cells. Results from this protocol have revealed that Raf1 activation is primarily responsible for the induction of neuronal differentiation in PC12 cells44. The same pathway also causes the formation of secondary tail-like structures in developing Xenopus embryos. By controlling the timing of Raf1 activation, the tail-like structure can be induced after the stage of germ-layer formation35. The recently described LOVTRAP method utilizes two engineered proteins, Zdark and LOV2, which selectively bind to each other only in the dark to modulate the light-induced dissociation of a protein of interest (POI)48. Unlike light-mediated protein-protein association, which is used in this approach, LOVTRAP uses light to induce protein dissociation. This new modality is more flexible at modulating protein localization and activity in live cells. By carefully selecting the mode of activation within a signaling cascade, it is possible to dissect signal transduction in unconventional ways. For instance, it is possible to activate receptor tyrosine kinases without ligand-receptor binding49 or to induce a subset of ligand-elicited signaling cascades44. The strategy reported here can be generalized to control other kinases, such as Akt39 and GTPases (e.g., Rho GTPase), whose activation states can also be turned on by protein translocation. Optogenetics will continue to be applied to live multicellular organisms, as demonstrated by recent works featuring the optogenetic control of protein localization and signaling in Drosophila50, zebrafish51,52,53,54, and Xenopus embryos35.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
This work was supported by the University of Illinois at Urbana-Champaign (UIUC) and the National Institutes of Health (NIGMS R01GM111816).
References
- Schlessinger J, Ullrich A. Growth factor signaling by receptor tyrosine kinases. Neuron. 1992;9(3):383–391. doi: 10.1016/0896-6273(92)90177-f. [DOI] [PubMed] [Google Scholar]
- Thisse B, Thisse C. Functions and regulations of fibroblast growth factor signaling during embryonic development. Dev Biol. 2005;287(2):390–402. doi: 10.1016/j.ydbio.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Perrimon N, Pitsouli C, Shilo BZ. Signaling mechanisms controlling cell fate and embryonic patterning. Cold Spring Harb Perspect Biol. 2012;4(8):a005975. doi: 10.1101/cshperspect.a005975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salles FH, et al. Mental disorders, functional impairment, and nerve growth factor. Psychol Res Behav Manag. 2017;10:9–15. doi: 10.2147/PRBM.S104814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basson MA. Signaling in cell differentiation and morphogenesis. Cold Spring Harb Perspect Biol. 2012;4(6) doi: 10.1101/cshperspect.a008151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schohl A, Fagotto F. beta-catenin, MAPK and Smad signaling during early Xenopus development. Development. 2002;129(1):37–52. doi: 10.1242/dev.129.1.37. [DOI] [PubMed] [Google Scholar]
- Zhang SW, Li JJ, Lea R, Amaya E, Dorey K. A Functional Genome-Wide In Vivo Screen Identifies New Regulators of Signalling Pathways during Early Xenopus Embryogenesis. PloS one. 2013;8(11):e79469. doi: 10.1371/journal.pone.0079469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney C, et al. Growth factor-specific signaling pathway stimulation and gene expression mediated by ErbB receptors. J Biol Chem. 2001;276(25):22685–22698. doi: 10.1074/jbc.M100602200. [DOI] [PubMed] [Google Scholar]
- Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80(2):179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- Vandergeer P, Hunter T, Lindberg RA. Receptor Protein-Tyrosine Kinases and Their Signal-Transduction Pathways. Annu Rev Cell Biol. 1994;10:251–337. doi: 10.1146/annurev.cb.10.110194.001343. [DOI] [PubMed] [Google Scholar]
- Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141(7):1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter T. Signaling--2000 and beyond. Cell. 2000;100(1):113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- Qiu MS, Green SH. PC12 cell neuronal differentiation is associated with prolonged p21ras activity and consequent prolonged ERK activity. Neuron. 1992;9(4):705–717. doi: 10.1016/0896-6273(92)90033-a. [DOI] [PubMed] [Google Scholar]
- Ji Y, et al. Acute and gradual increases in BDNF concentration elicit distinct signaling and functions in neurons. Nat Neurosci. 2010;13(3):302–309. doi: 10.1038/nn.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luby-Phelps K. Cytoarchitecture and physical properties of cytoplasm: volume, viscosity, diffusion, intracellular surface area. Int Rev Cytol. 2000;192:189–221. doi: 10.1016/s0074-7696(08)60527-6. [DOI] [PubMed] [Google Scholar]
- Sauer B. Inducible gene targeting in mice using the Cre/lox system. Methods-a Companion to Methods in Enzymology. 1998;14(4):381–392. doi: 10.1006/meth.1998.0593. [DOI] [PubMed] [Google Scholar]
- Ling MM, Robinson BH. Approaches to DNA mutagenesis: an overview. Anal Biochem. 1997;254(2):157–178. doi: 10.1006/abio.1997.2428. [DOI] [PubMed] [Google Scholar]
- Gama Sosa MA, De Gasperi R, Elder GA. Animal transgenesis: an overview. Brain Struct Funct. 2010;214(2-3):91–109. doi: 10.1007/s00429-009-0230-8. [DOI] [PubMed] [Google Scholar]
- Wanka F, et al. Tet-on, or Tet-off, that is the question: Advanced conditional gene expression in Aspergillus. Fungal Genet Biol. 2016;89:72–83. doi: 10.1016/j.fgb.2015.11.003. [DOI] [PubMed] [Google Scholar]
- Karginov AV, Ding F, Kota P, Dokholyan NV, Hahn KM. Engineered allosteric activation of kinases in living cells. Nat Biotechnol. 2010;28(7):743–747. doi: 10.1038/nbt.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu QY, Deiters A. Optochemical Control of Deoxyoligonucleotide Function via a Nucleobase-Caging Approach. Accounts of Chemical Research. 2014;47(1):45–55. doi: 10.1021/ar400036a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbely E, Torres-Kolbus J, Deiters A, Chin JW. Photocontrol of tyrosine phosphorylation in mammalian cells via genetic encoding of photocaged tyrosine. J Am Chem Soc. 2012;134(29):11912–11915. doi: 10.1021/ja3046958. [DOI] [PubMed] [Google Scholar]
- Gautier A, Deiters A, Chin JW. Light-activated kinases enable temporal dissection of signaling networks in living cells. J Am Chem Soc. 2011;133(7):2124–2127. doi: 10.1021/ja1109979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DP, et al. Genetic Encoding of Photocaged Cysteine Allows Photoactivation of TEV Protease in Live Mammalian Cells. J Am Chem Soc. 2014;136(6):2240–2243. doi: 10.1021/ja412191m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisseroth K. Optogenetics. Nat Methods. 2011;8(1):26–29. doi: 10.1038/nmeth.f.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci. 2005;8(9):1263–1268. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- Banghart M, Borges K, Isacoff E, Trauner D, Kramer RH. Light-activated ion channels for remote control of neuronal firing. Nat Neurosci. 2004;7(12):1381–1386. doi: 10.1038/nn1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Cui B. Optogenetic control of intracellular signaling pathways. Trends in Biotechnology. 2015;33(2):92–100. doi: 10.1016/j.tibtech.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischer D, Weiner OD. Illuminating cell signalling with optogenetic tools. Nat Rev Mol Cell Bio. 2014;15(8):551–558. doi: 10.1038/nrm3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B, Lin MZ. Optobiology: optical control of biological processes via protein engineering. Biochemical Society Transactions. 2013;41(5):1183–1188. doi: 10.1042/BST20130150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker CL. Manipulating cellular processes using optical control of protein-protein interactions. Prog Brain Res. 2012;196:95–117. doi: 10.1016/B978-0-444-59426-6.00006-9. [DOI] [PubMed] [Google Scholar]
- Toettcher JE, Gong DQ, Lim WA, Weiner OD. Light Control of Plasma Membrane Recruitment Using the Phy-Pif System. Method Enzymol. 2011;497:409–423. doi: 10.1016/B978-0-12-385075-1.00017-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoltowski BD, Gardner KH. Tripping the light fantastic: blue-light photoreceptors as examples of environmentally modulated protein-protein interactions. Biochemistry. 2011;50(1):4–16. doi: 10.1021/bi101665s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MJ, et al. Rapid blue-light-mediated induction of protein interactions in living cells. Nat Methods. 2010;7(12):973–975. doi: 10.1038/nmeth.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy VV, et al. Reversible optogenetic control of kinase activity during differentiation and embryonic development. Development. 2016;143(21):4085–4094. doi: 10.1242/dev.140889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, et al. Photoexcited CRY2 Interacts with CIB1 to Regulate Transcription and Floral Initiation in Arabidopsis. Science. 2008;322(5907):1535–1539. doi: 10.1126/science.1163927. [DOI] [PubMed] [Google Scholar]
- Li X, et al. Arabidopsis cryptochrome 2 (CRY2) functions by the photoactivation mechanism distinct from the tryptophan (trp) triad-dependent photoreduction. Proc Natl Acad Sci U S A. 2011;108(51):20844–20849. doi: 10.1073/pnas.1114579108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leevers SJ, Paterson HF, Marshall CJ. Requirement for Ras in Raf Activation Is Overcome by Targeting Raf to the Plasma-Membrane. Nature. 1994;369(6479):411–414. doi: 10.1038/369411a0. [DOI] [PubMed] [Google Scholar]
- Kohn AD, Takeuchi F, Roth RA. Akt, a pleckstrin homology domain containing kinase, is activated primarily by phosphorylation. J Biol Chem. 1996;271(36):21920–21926. doi: 10.1074/jbc.271.36.21920. [DOI] [PubMed] [Google Scholar]
- Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 2012;9(7):676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sive HL, Grainger RM, Harland RM. Early Development of Xenopus laevis: A Laboratory Manual. Cold Spring Harbor Laboratory Press; 2000. [Google Scholar]
- Kim JH, et al. High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PloS one. 2011;6(4):e18556. doi: 10.1371/journal.pone.0018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimura A, et al. Oncogenic Met receptor induces ectopic structures in Xenopus embryos. Oncogene. 2006;25(31):4286–4299. doi: 10.1038/sj.onc.1209463. [DOI] [PubMed] [Google Scholar]
- Zhang K, et al. Light-Mediated Kinetic Control Reveals the Temporal Effect of the Raf/MEK/ERK Pathway in PC12 Cell Neurite Outgrowth. PloS one. 2014;9(3):e92917. doi: 10.1371/journal.pone.0092917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanty SK, Lakshminarayananan V. Optical Techniques in Optogenetics. J Mod Opt. 2015;62(12):949–970. doi: 10.1080/09500340.2015.1010620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taslimi A, et al. Optimized second-generation CRY2-CIB dimerizers and photoactivatable Cre recombinase. Nature chemical biology. 2016;12(6):425–430. doi: 10.1038/nchembio.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano F, Suzuki H, Furuya A, Sato M. Engineered pairs of distinct photoswitches for optogenetic control of cellular proteins. Nat Commun. 2015;6:6256. doi: 10.1038/ncomms7256. [DOI] [PubMed] [Google Scholar]
- Wang H, et al. LOVTRAP: an optogenetic system for photoinduced protein dissociation. Nat Methods. 2016;13(9):755–758. doi: 10.1038/nmeth.3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KY, et al. Light-inducible receptor tyrosine kinases that regulate neurotrophin signalling. Nat Commun. 2014;5:4057. doi: 10.1038/ncomms5057. [DOI] [PubMed] [Google Scholar]
- Boulina M, Samarajeewa H, Baker JD, Kim MD, Chiba A. Live imaging of multicolor-labeled cells in Drosophila. Development. 2013;140(7):1605–1613. doi: 10.1242/dev.088930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Gomez G, Lin S, Lin C. Optogenetic control of transcription in zebrafish. PLoS One. 2012;7(11):e50738. doi: 10.1371/journal.pone.0050738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley CE, et al. Reversible Optogenetic Control of Subcellular Protein Localization in a Live Vertebrate Embryo. Dev Cell. 2016;36(1):117–126. doi: 10.1016/j.devcel.2015.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motta-Mena LB, et al. An optogenetic gene expression system with rapid activation and deactivation kinetics. Nature chemical biology. 2014;10(3):196–202. doi: 10.1038/nchembio.1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer HM, et al. Red Light-Regulated Reversible Nuclear Localization of Proteins in Mammalian Cells and Zebrafish. ACS Synth Biol. 2015;4(9):951–958. doi: 10.1021/acssynbio.5b00004. [DOI] [PubMed] [Google Scholar]