

Abstract
Brain and spinal cord injury may lead to permanent disability and death because it is still not possible to regenerate neurons over long distances and accurately reconnect them with an appropriate target. Here a procedure is described to rapidly initiate, elongate, and precisely connect new functional neuronal circuits over long distances. The extension rates achieved reach over 1.2 mm/h, 30-60 times faster than the in vivo rates of the fastest growing axons from the peripheral nervous system (0.02 to 0.04 mm/h)28 and 10 times faster than previously reported for the same neuronal type at an earlier stage of development4. First, isolated populations of rat hippocampal neurons are grown for 2-3 weeks in microfluidic devices to precisely position the cells, enabling easy micromanipulation and experimental reproducibility. Next, beads coated with poly-D-lysine (PDL) are placed on neurites to form adhesive contacts and pipette micromanipulation is used to move the resulting bead-neurite complex. As the bead is moved, it pulls out a new neurite that can be extended over hundreds of micrometers and functionally connected to a target cell in less than 1 h. This process enables experimental reproducibility and ease of manipulation while bypassing slower chemical strategies to induce neurite growth. Preliminary measurements presented here demonstrate a neuronal growth rate far exceeding physiological ones. Combining these innovations allows for the precise establishment of neuronal networks in culture with an unprecedented degree of control. It is a novel method that opens the door to a plethora of information and insights into signal transmission and communication within the neuronal network as well as being a playground in which to explore the limits of neuronal growth. The potential applications and experiments are widespread with direct implications for therapies that aim to reconnect neuronal circuits after trauma or in neurodegenerative diseases.
Keywords: Neuroscience, Issue 124, Axonal growth, axonal guidance, microfluidic chambers, neuronal cultures, neuronal networks, neuronal regeneration, pipette micromanipulation, synapse, rewiring neuronal circuits
Introduction
Injuries to the adult central nervous system (CNS) may lead to permanent disability due to multiple mechanisms that limit axonal regrowth1. Following injury, many CNS axons do not form a new growth cone and fail to mount an effective regenerative response2. Furthermore, damage and scar tissue surrounding CNS lesions significantly inhibit axonal growth1,2,3. Current therapies to promote CNS regeneration after injury have focused on enhancing the intrinsic growth potential of the injured neuron and on masking the inhibitors of axonal extension associated with myelin debris and the glial scar1,3. Despite this, the capacity to regenerate long axons to distant targets and to form appropriate functional synapses remains severely limited4,5,6,7.
In the present work, microbeads, pipette micromanipulation, and microfluidic devices are used to rapidly initiate, elongate, and precisely connect new functional neuronal circuits over long distances. Previous work has shown that poly-D-lysine-coated beads (PDL-beads) induce membrane adhesion followed by the clustering of synaptic vesicle complexes and the formation of functional presynaptic boutons8. It was also shown that when the PDL-bead is mechanically pulled away after presynaptic differentiation, the synaptic protein cluster follows the bead, initiating a new neurite9. The following procedure exploits this fact along with the ability to culture embryonic hippocampal neurons of rats into organized regions on a coverslip using polydimethylsiloxane (PDMS) microfluidic devices to precisely rewire a neuronal circuit.
These PDMS microfluidic devices are non-toxic, optically transparent and consist of two chambers connected by a system of microchannels. Once assembled on a coverslip, each device serves as a mold to guide neuronal growth and maintain healthy neuronal cultures on precise patterns for longer than 4 weeks in vitro.
Here, a framework is presented in which to investigate the limits of extension and functionality of the new neurite. New, functional neurites are created and positioned to controllably (re)wire neuronal networks. The extension rates achieved are faster than 20 µm/min over millimeter-scale distances and functional connections are established. These results show, unexpectedly, that the intrinsic capacity of these neurites for elongation is much faster than previously thought. This proposed mechanical approach bypasses slow chemical strategies and enables controlled connection to a specific target. This technique opens new avenues for the in vitro study of novel therapies to restore neuronal connectivity after injury. It also enables the manipulation and rewiring of neuronal networks to investigate fundamental aspects of neuronal signal processing and neuronal function in vitro.
Protocol
All procedures detailed below were approved by McGill University's Animal Care Committee and conformed to the guidelines of the Canadian Council of Animal Care.
1. Standardization of Neuronal Cultures Using Microfluidic Devices: Device Assembly
Select a suitable microfluidic device for the desired experiment. To connect neurons within the same population, use the Neuro Devices (Figure 1) and to connect neurons in different populations use the Co-Culture Devices (Figure 6).
Clean and prepare the desired number of sterile coverslips or glass bottom dishes. For best results on plastic surfaces use 35 mm dishes, on glass use 25 mm coverslips, or 35 mm glass bottom dishes. Select glass thickness based on the imaging system, for instance 0.15 mm.
Coat the dishes or coverslips with 0.5-1 mL of 100 µg/mL PDL for 2 h or overnight at room temperature. Note: Protocol can be paused here and resumed the following day if desired. Furthemore, dishes can be coated with borate buffer-diluted PDL, Poly-L-Lysine (PLL), laminin or any other cell adhesion molecule.
Wash the dishes twice with water (do not use phosphate-buffered saline (PBS) as salt crystals may block the channels), remove all the liquid and let it dry in a sterile environment such as a biosafety cabinet for 5-10 min or until the surface is completely dry. Note: Be careful to ensure that the coverslips are absolutely dry, as any remaining liquid will interfere with the adherence of the microfluidic systems.
Place microfluidic devices with patterns facing up under UV light in a sterile environment (biosafety cabinet) for 10 min. Be sure to follow sterile procedures when working in the biosafety cabinet10.
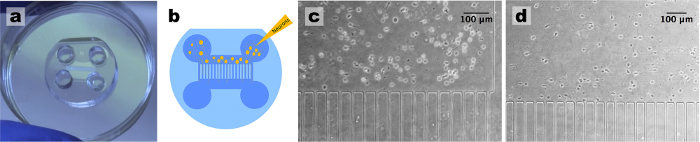
Using tweezers place a microfluidic device with pattern facing down in contact with the clean coverslip/dish. Use the tweezers to softly press the device so that it adheres to the glass. Note: The transparency of the adhered region will be visible when looking against the light. Make sure all corners are contacting the glass. Do this to all microfluidic devices. See Figure 1a .
- To fill the single population device with medium, point the pipette towards the channels and add 50 µL of complete cell medium supplemented with serum-free B-27 (volume ratio 1:50) and 500 µg/mL penicillin/streptomycin/glutamine (collectively called NBM) to the right upper well, and then add another 50 µL to the well lying diagonally to it. Do this for all devices, making sure that the medium flows between wells. Next, add 50 µL of medium to the remaining 2 wells. See Figure 2a .
- To fill the multiple population device with medium, point the pipette towards the channels and add 30 µL of complete NBM to the right wells, refer to Figure 6a. Do this for all devices, making sure that the medium flows between wells. Next, add 50 µL of medium to the remaining 4 wells.
Place the devices in a bigger plate with an open dish with autoclaved water (wet chamber) and place in the incubator (37 °C, 5% CO2 and 95% humidity) for 1-2 h while preparing the cell culture. See Figure 2b .

2. Plating Neurons in Microfluidic Systems
Following the protocol outlined in Ref.8, obtain dissociated hippocampal or cortical neurons from Sprague Dawley rat embryos (either gender).
Resuspend embryonic neurons in NBM at a concentration of 1-2 million neurons/mL. Verify cell concentrations in the microscope using a hemocytometer and following reference8. Adjust the cell concentration according to the desired cell density. To increase the chances of obtaining single hippocampal axons per channel, plate 10,000 neurons per device. To have multiple axons in the same channel, plate 60,000 neurons per device. Note: These numbers vary according to the neuronal type used.
Remove the medium from the microfluidic devices without emptying the wells. Leave approximately 5 µL in each.
- To plate cells in the single population device, add 50 µL of NBM to the lower right well. At this point the medium flows by itself to fill the other lower well. Add 20 µL of the concentrate cell solution into the top right well of the microfluidic device, as indicated in Figure 1b .
- To plate cells in the multiple population device add 20 µL of the concentrate cell solution into each of the right wells in Figure 6a.
Check in the microscope if cells are inside the chambers and place the devices in the incubator for 15-30 min to promote cell attachment to the substrate.
Check in the microscope if there are enough cells in the chambers. If more are needed, repeat steps 2.4 and 2.5.
Add 50 µL of NBM to the 2 top wells of the single population device and 20 µL of NBM into the same well as the cells were injected in the multiple population device. The media protrudes slightly to form a positive meniscus giving the wells a muffin top aspect. Again, see Figure 2a.
Maintain the cells at 37 °C, 5% CO2 and 95% humidity.
3. Maintaining the Neuronal Cultures
Remove NBM (roughly 30 µL with a pipette) from the cells and apply new pre-warmed NBM the day following their introduction to the devices (that is 1 d after step 2).
Check every 2 days if there is enough medium in each channel. If the muffin top is low just add more medium to the top wells.
Culture cells for at least 7 d before removal of the microfluidic devices. The cells can survive in these devices for several weeks. Remove the devices 1-2 days before experiments are performed on samples.
4. Removal of Microfluidic Devices
1 - 2 d before removal of microfluidic devices, add 2 mL of NBM prewarmed to 37 °C to each sample dish, flooding the chambers, and maintain the devices in the incubator.
Use sterile tweezers and one tip to remove the microfluidic devices from the coverslips leaving a patterned configuration of neurons. Use the tip to hold the coverslip in place and the tweezers to clasp the edge of the device at the bottom left corner of the well. Delicately apply torsion, raising the device up with the tweezers so that it peels off the coverslip. See Figure 2c-2d.
Every 2-3 days, replace half the NBM until the sample is used for experiments.
Before performing rewiring experiments on the sample, verify that neurites in the single population device channels and the neuronal populations in the multiple population device are isolated by examining the gaps between them in the microscope to ensure there are no filaments linking neuronal populations.
5. Preparing PDL-coated Beads
Add 2 x 50 µL drops of either 4, 10 or 20 µm polystyrene beads diluted in water (1:500) to 1 mL of PDL (100 µg/mL). Leave for at least 2 h at room temperature. Note: Protocol can be paused here and resumed the following day.
Centrifuge the solution at 8,820 x g for 1 min. Carefully remove supernatant without disturbing the beads accumulated at bottom of container.
Wash the beads twice with 1 mL of sterile 10 mM HEPES pH 8.4 solution.
Resuspend the PDL-coated beads in 200 mL of 10 mM HEPES pH 8.4 solution.
6. Preparing Micropipettes
Prepare pipettes from glass capillary tubes (1 mm inner diameter, 1.5 mm outer diameter) using a horizontal electrode puller. Adjust settings so the outer tip of the pulled micropipette is ~ 2-5 µm. Before pulling, ensure the glass tubes are clean.
Fix pipettes to glass slides for storage, and ensure that the tip does not contact the surface of the slide as the tip is fragile. Store at room temperature in a covered container to protect from dust. Use pipettes the same day they are pulled.
7. PDL-bead Adhesion to Neurons
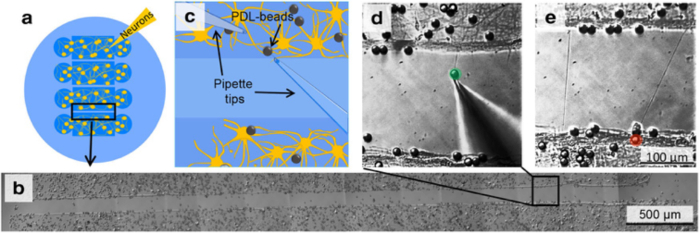
Add 40-60 µL of PDL-coated beads prepared in step 5 to a cell culture prepared in step 4. Center the pipette tip over the neurons, which are faintly visible on the coverslip, and add the beads (See Figure 3).
Return the sample to the incubator for 1 h to promote the formation of synaptic contacts8,9.
After the incubation, remove any un-adhered beads by gently washing the culture with pre-warmed NBM.

8. Preparing Physiological Saline Solution (for Room Temperature Experiments)
Prepare physiological saline solution by combining the ingredients listed in references7,8. This is to regulate the cell environment outside the incubator.
Verify osmolarity and pH levels as indicated in references7,8.
Continuously infuse solution with O2 to minimize pH fluctuations while conducting experiments.
Heat to room temperature.
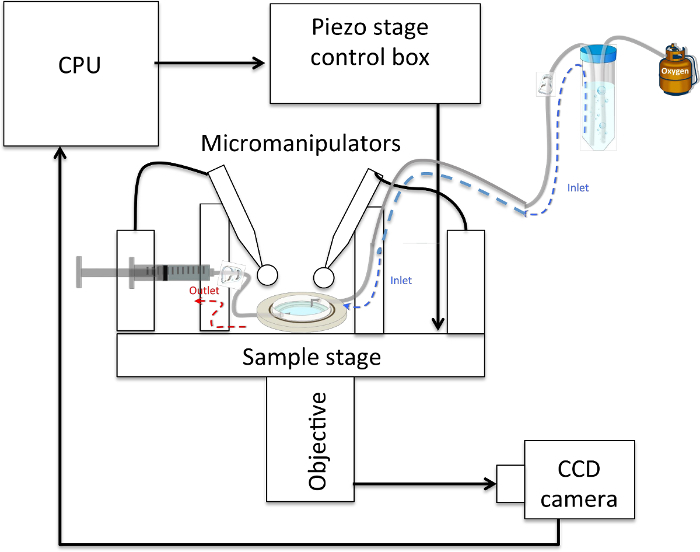
- Set up the perfusion system by inserting one end of a plastic tube (optional dimensions) in O2-infused physiological solution and fixing the other end to a needle inserted in the sample holder. Place the tubing and solution higher than the sample (See Figure 4).
- Disconnect the tube from the needle and connect it to a syringe. Use the syringe to exert pressure and draw liquid, filling the tube. Seal with a roller clamp and reconnect the needle.
9. Bead Micromanipulation
Install sample in an experimental set-up such that cells can be accessed from above by two micropipettes mounted in micromanipulators and accessed optically below, for instance with the 40X-phase objective (numerical aperture of 0.6) of an inverted optical microscope. In this configuration, mount a CCD camera for image capture on the side port of the microscope. Connect each pipette to 1 mL syringes via plastic tubing. At this step, replace NBM with physiological saline solution (1-2 mL) (See Figure 4).
During experiments, continuously perfuse cells with the physiological saline solution prepared in step 8 at a rate of 0.5-1 mL/min.
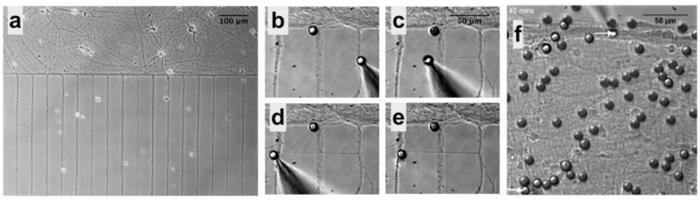
Select a PDL-bead NOT attached to a neuron in the field of view. Align the bead with a micropipette tip by focusing onto the bead then up to the micropipette. Bring the tip down as close as possible to the bead by monitoring it through the microscope.
Apply negative pressure with the 1 mL syringe connected to the pipette to pick up the bead. Maintain negative pressure throughout the experiment.
10. Pulling Neurites
Select a PDL-bead attached to a neuron in the field of view and attach it to the second micropipette using suction as described in steps 9.3-9.4.
Pull the PDL-bead-neuron complex by slowly (~0.5 µm/min) moving either the micromanipulator or the sample stage by 1 µm and pause for 5 min to allow neurite initiation.
Repeat step 10.2 twice. Note: The first 3 µm have to be pulled very slowly to guarantee experimental success, which occurs over 95% of the time.
Pull the PDL-bead-neuron complex by slowly (~0.5 µm/min) moving either the micromanipulator or the sample stage by 2 µm and pause for 5 min to allow neurite elongation.
After successful initiation and neurite extension for the first 5 µm, pull the neurite at 20 µm/min over millimeter-scale distances. Note: Pulling can be performed continuously or in steps and at varying rates. See Figure 5b-5c .
11. Connecting Neurons
Select a region rich in neurites and lower the PDL-bead-neurite complex so that it physically contacts it. Use other beads to gauge tip height above coverslip surface. See Figure 5d.
Leave the PDL-bead-neurite complex in contact with the target neurite while manipulating the second micropipette. Lower the second pipette with the second PDL-bead on top of the newly formed neurite about 20 µm form the first bead. Use the second PDL-bead to push the new neurite filament towards the target cell.
Hold both beads in place for at least 1 h. Verify the absence of focal swelling, a thickening of the neurites contacting the bead, with the microscope16.
During this time, use perfusion to slowly change the medium of the sample from physiological saline to pre-warmed, CO2-equilibrated NBM.
Release the bead from the second pipette by releasing suction. If the new neurite remains attached, release the first bead as well. See Figure 5e .
Gently remove saline solution and replace with NBM (~2 mL).
Carefully place the sample back in the incubator to strengthen the neuronal connection for future experiments. This connection is stable for >24 h7.
12. Verifying the Functionality of the New Connection via Whole-cell Paired Patch Clamp Recordings
Representative Results
Embryonic rat hippocampal neurons are cultured in microfluidic devices to enable precise positioning of cells, PDL-beads and micromanipulators. The first step is to properly assemble the microfluidic device on a glass coverslip or dish. It is essential that the microfluidic device be well attached to the substrate to avoid cells exiting the chambers and moving under the parts of the device that should be sealed (Figure 1a). To maintain healthy cultures for several weeks, it is important to prevent medium evaporation by checking the cell medium every 2-3 days and preserving a positive meniscus of medium (Figure 2a). Medium evaporation is also avoided by keeping both the cells and an open dish of water inside a larger plate (Figure 2b). The microfluidic devices can be removed at any time. For optimal results, NBM should be added to the cell-device system at least 1 d before device removal. This minimizes cellular stress as the neurons are in contact with medium at ideal temperature and pH when the devices are removed. When the microfluidic devices are slowly peeled off the dish (Figure 2cand 2d) cells will remain in the patterned position (Figure 3 and Figure 5a).
Two types of microfluidic devices are used: Neuro Device and Co-Culture Device. The first enables easy identification of axons, dendrites and cell bodies. The soma remain in the top soma chamber while axons and dendrites grow along the microfluidic channels (Figure 5a) towards the axonal chamber.
It is recommended that PDL-beads be added to cells at a ratio of 10:1 and that most beads that have not adhered to the culture be removed by washing the cells once with NBM following the 1 h incubation (Figure 3). Following PDL-bead adhesion to neurites, the PDL-bead-neurite complex is pulled and can be extended over large distances. The newly formed neurite can be precisely connected to neurites or soma millimeters away (Figure 5b-5e). The success rate for pulling a new neurite for the first 3 µm at speeds lower than 1 µm /min is >95% (n = 206). The success rate for connecting the new neurite to another cell is 70% (n = 30). The connection is very fragile in the first 18 h, mainly because the new neurite is a filament of less than 1 µm in diameter anchored by a 10 µm PDL-bead. If the dish is rapidly moved during the first minutes of contact, the resulting turbulence of the medium may cause the bead to roll and consequently the adhesion to be lost. However, if the sample is not shaken, in 30 min the bead attaches to the neurites on the dish and the new connection remains for at least 48 h. See Figure 4 for a schematic of the set-up.
The PDL-bead position on the culture can also be used to easily identify the location of the connected neurite (Figures 6d-6e). After initiation, extension and connection of a new neurite, a second, non-adherent PDL-bead, picked up via the micromanipulator, is used to create a second adhesion site between the initiated neurite and the second neuronal population. The second PDL-bead is placed on top of the new neurite, compressing it such that the new neurite just contacts the second neuronal population11. This will promote adhesion with the second neuronal population (Figure 5f).
The multiple population device enables the growth of 4 isolated neuronal populations on the same dish. Each neuronal population is confined to a 4 x 7 mm rectangle separated from other neuronal populations by 100 or 200 µm gaps (Figure 6a). Healthy neurons can grow inside the devices for several weeks. Usually, the neuronal populations remain isolated up to 48 h after removal of the device. After this 48 h period, neurons tend to grow towards neighboring neuronal populations and form natural connections. Before connecting two isolated populations, it should be verified that the populations are indeed truly isolated by examining the entire gap with the microscope to establish that there is no link between the two neuronal populations (Figure 6b).
After connection, samples were incubated for 24 h and electrical whole cell paired patch clamp recordings were conducted to investigate whether the newly formed neurite used to connect two isolated neuronal populations was functional and able to transmit electrical signals. A neuron in population one, located less than 100 μm radius from the site where the induced neurite was initiated, was selected to record presynaptic action potentials (PAPs). This neuron was considered the presynaptic cell. Postsynaptic excitatory or inhibitory activity was recorded from a neuron in population two, on the other side of the gap, located in less than 100 μm radius of the micromanipulated connection (Figure 7a-7c). Recordings derived from the mechanically-induced connections were analyzed and compared to those from naturally connected neuronal populations and non-connected populations (Figure 7). The electrical responses after PAP recorded from neurons connected naturally and by micromanipulation are significantly higher and temporally correlate with the presynaptic activity (Figure 7).
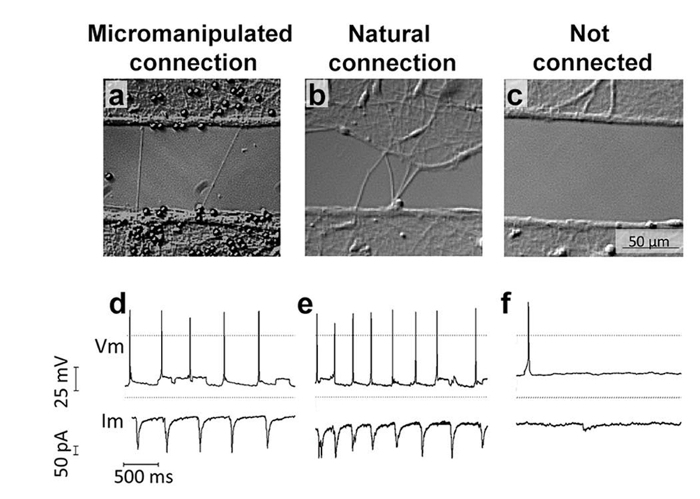
Figure 1: Standardization of Neuronal Cultures using Microfluidic Devices7. (a) Device assembly: when the microfluidic devices are properly assembled on a dry surface all chambers are visible. (b) Cell plating: plate the cells on the top right well and cells should move towards the left well. (c) Cell density: just after plating, check in the microscope if the concentration of cells is adequate. (d) After 1 d in culture, hippocampal neurons are well adhered close to the microchannels and start forming neurites. Please click here to view a larger version of this figure.
Figure 2: Maintenance of healthy neuronal cultures for several weeks7. (a) Add medium every 2-3 d and keep a positive meniscus in the upper wells of the microfluidic chambers so cells will have a constant supply of nutrients. (b) Keep cells inside a larger plate with a dish containing water to reduce medium evaporation. (c) Use sterile tip and tweezers to (d) easily peel off the microdevices. Please click here to view a larger version of this figure.
Figure 3: Location of Pipette Depositing Beads into Cultures. Once the microfluidic device has been removed, neurons are visible on the coverslip. When depositing beads, position the pipette tip so that it is in the center of the cells. Please click here to view a larger version of this figure.
Figure 4: Schematic of a Typical Neurite-pulling Set-up. The sample rests on a (piezo-actuated) stage and can be accessed from above by 2 micropipettes held in micromanipulators and connected to 1 mL syringes via plastic tubing. The sample is accessed optically from below by an objective connected to a CCD camera that sends images to a CPU. An inlet tube feeds oxygenated physiological saline solution to the sample, which rests below it, and an outlet tube connected to a syringe allows the withdrawal of solution in the event of overflow. Please click here to view a larger version of this figure.
Figure 5: PDL-bead Adhesion to Neurons and Pipette Micromanipulation7. (a) Neurons remain organized in patterns after removal of microfluidic chambers allowing easy identification of soma and neurites. (b) Micromanipulation of PDL-beads adhered to neurites enables neurite initiation, extension (c) and connection (d), followed by release of the PDL-bead from the pipette (e). (f) After contacting two isolated neuronal populations (bottom arrow), a second PDL-bead and pipette micromanipulator (top arrow) is used to establish a second adhesion point, a few hundreds of microns apart form the first contact point, and guarantee functional connections. Please click here to view a larger version of this figure.
Figure 6. Initiation, Elongation and Connection of New Neurites to Connect two Isolated Populations using Multiple Population Device and Micromanipulation7. (a) The device isolates 4 neuronal populations between 3 gaps of 100 or 200 µm each. (b) Following removal of the device, select one gap and certify that no neurites connect the 2 individual populations. (c) Schematic of the experimental set-up as should be visible in the optical microscope, indicates the position of two micropipettes and the presence of PDL-coated beads. (d) By applying negative pressure to a pipette, a PDL-bead adhered to one neuronal population is pulled with the pipette tip, thereby initiating a new neurite. By maintaining the negative pressure in the pipette, the PDL-bead-neurite complex (green) can be pulled, elongating the neurite. (e) Pipette micromanipulation guides extension of the new neurite over the gap and the formation of a connection with a new neuronal population. To ensure the adhesion of the new neurite to the second population, a PDL-bead (red) is positioned with a second pipette on top of both theextended neurite and neuronal population. Please click here to view a larger version of this figure.
Figure 7: The Newly Induced, Elongated and Connected Neurite can Transfer Information Between two Isolated Neuronal Populations7. Isolated neuronal populations were cultured separated by a 100 µm gap in PDMS microdevices. Paired patch clamp recordings were performed in whole cell configuration from a neuron in population one and a neuron in population two (on the other side of the gap) when the two populations were connected through mechanical manipulation (a), allowed to naturally interconnect across the gap (b) or remain non-connected by maintaining the gap (c). Representative traces of paired recordings are shown for each condition (d-f). Please click here to view a larger version of this figure.
Discussion
Using standard micromanipulation and innovative microfluidic devices, a new technique was developed to rapidly initiate, elongate and precisely connect new functional neuronal circuits over large distances. Pipette micromanipulation is a common tool in most neuroscience labs4,13. The real challenge to achieving reproducible and reliable results was standardization of healthy, precisely positioned neuronal cultures for the duration of the experiment (which can be on the order of weeks) through the development of microfluidic devices to organize cell cultures with micrometer precision. High quality cell cultures are the cornerstone of data validation. This contributes to easier and faster microscopy imaging and standardization of cultures and results. The microfluidic devices were designed to culture cells in vitro with similar organization as in vivo. The single population device enables the growth of long neurites and easy identification of axons, neurites and soma. The number of cells plated in the microfluidic chambers determines the neuronal density. Therefore, by plating fewer cells per device it is easy to identify single neurons close to the channels. The axon and multiple dendrites of one single neuron grow inside one channel. Dendrites grow at least 5 times slower than axons6,17 and after 2-3 weeks in vitro their growth in the channels is usually limited to 200 µm while fast-growing axons can reach beyond 2 mm17. Therefore, after adding PDL-beads to the samples, it is relatively easy to estimate whether the beads are adhering mostly to axons or to axons and dendrites (Figure 5b-5f ). There are higher chances of pulling new axons and dendrites when pulling the PDL-beads attached to neurites shorter than 200 µm, while there are greater odds of pulling only new axons when pulling PDL-beads attached to neurites longer than 500 µm. The multiple population device is useful for the reproducible growth of healthy separated populations for several weeks. This system of channels is useful for studying up to 4 different cell types or comparing the same cells with different treatments.
Moreover, a controlled and reproducible cell culture environment provides an ideal framework for cell analysis and manipulation. Precise positioning of cells on a dish facilitates identification of regions of interest as well as orientation and navigation through these areas of interest ultimately allowing unprecedented control over the neurite initiation site. A controlled cell distribution makes it easier to visualize the newly formed connection and much faster to find the new connection with the microscope in the days following incubation. In addition, reproducible configurations of cells enable easy and precise positioning of chemical cues, such as the PDL-coated beads, on the soma, dendrites or axons8. Taken together, imaging and analysis of cells grown in microfluidic devices are faster due to standard cellular organization reproduced in all dishes. Furthermore, the devices are made of biocompatible, transparent and removable material, enabling imaging at all visible wavelengths and cell survival inside the devices for several weeks. Miniaturization of cellular assays also helps gather more data with fewer cells. The low-volume consumption of the microfluidic devices reduces the number of cells needed per experiment and increases experimental efficacy. For instance, instead of spending several hours searching with a microscope for perhaps 1 or 2 isolated axons in a dish, the single population device can be used to immediately access over 100 isolated axons per cell sample. The microfluidic devices offer a miniaturized reliable and controlled cell culture environment favoring the analysis of rare samples with time to perform multiple tests.
Potential variations of the technique involve the direct attachment of the bead to the micropipette. In the current protocol, a method is described for attaching the bead to the tip via suction, but the bead can also be glued to the tip. Glue is the better option if a highly stable connection between the tip and the bead is desirable, for example if force measurements are made using the pipette as a sensor or if investigation of adhesion between PDL and the neurite is the main experimental objective. In another vein, suction is advantageous as it allows the release of the bead after placement without severing the neurite-bead complex thus enabling several connections to be made in parallel. Suction provides means for high throughput rewiring experiments, an improvement over other manipulation techniques such as atomic force microscopy (AFM)7,16. Both modifications of this method allow the neurite initiation site to be selected by simply maintaining a bead in contact with a dendrite so synapses are formed. However, the strategy of incubating several beads as described in step 7 saves time at the manipulation stage and is recommended if precise control over the initiation site is unnecessary in the experiment.
Future research could seek to address various instrumental limitations, namely temperature control and sample accessibility. Mechanical properties of the cellular membrane can significantly vary at different temperatures27. Ideally all experiments should be performed at 37 °C in neuronal medium and adequate conditions (correct CO2 pressure and humidity controls). However it was not possible to use a closed cell incubator, because access to the samples from the top is required for the micromanipulators, from the bottom for the microscope and from the side to move the stage. Therefore perfusion at room temperature was used. In a similar vein, since the set-up only has enough space to accommodate 2 micromanipulators and it takes almost 1 h to establish a single connection, there is a limit to the number of experiments one can perform. This issue could be addressed with a manipulator that pulls more than one bead at a time. Another potential improvement to this protocol is the capability to register the height of the bead-pipette complex from the sample surface. The inability to do this can lead to imprecisions when bringing down the second bead and placing it on the induced neurite to fix it. The bead is lowered on top of the new neurite in a neurite-rich region until the new neurite and those on the dish lie in the same focal plane. The new neurite is never between the bead and the dish, but always between a cushion of cell matter and the bead, therefore the compression is reduced.In addition, the new neurite is observed for 10 min in the microscope to evaluate if neurite focal swellings are formed near bead. As described in reference16, the presence of swellings indicates neurite degeneration. Nevertheless, since applying varying amounts of pressure to axons can lead to physiological changes16, future research could focus on adapting the pipette-probes by fixing reflectors to them and monitoring displacement perpendicular to the sample surface with AFM methods. Finally, perhaps the greatest challenge to this protocol is to test the functionality of the manipulated connection. The most direct, reliable and well established technique is paired whole-cell patch clamp recordings. However, the success rate of regular paired patch clamp recording is very low (<25%)18. Whole-cell patch clamp has several disadvantages including long set up time, limited number of experiments/day, limited recording time (~30 min), low experimental yield for paired patch clamp recordings and cell death after measurements. Due to these technical challenges the experimental yield of whole-cell patch clamp paired recordings after micromanipulation is very low. Better platforms and techniques are needed to more precisely stimulate, record and compare neuronal activity in natural and micromanipulated connections.
The importance of tension in axonal growth has been known since the early days of neuroanatomy - referring to it as passive stretching20. During early embryonic development neurites migrate through small distances to reach their targets. As most cells divide and duplicate, axons are subjected to continuous forces to elongate and adjust their length to embryonic growth20,21. Several groups have tried to test the limits of neurite growth by applying chemical cues and/or mechanical tension to neurons (for review see reference22). In these reports23,24,25,26 as well as during physiological growth, the neurites are pulled while attached to a substrate. The major difference to our setup is that in the present technique the new neurites only have two adhesion contacts; at the base the neurite is attached to the neuron and at the tip the neurite is attached to the bead. During elongation the components of the new neurite have the freedom to disperse in the most efficient way to accommodate the pulling forces. More importantly, this protocol describes how to reproduce these experiments without causing neurite rupture nor degeneration, based on previous studies on the formation of synaptic contacts8 and on the resistance of axons to pressure16 showing how to use micro- and nanotools to continuously pull the neurites with the appropriate force. The neurite extension rates described here (1.2 mm/h) are 30-60 times faster than the in vivo rates of the fastest growing axons from the peripheral nervous system (0.02 to 0.04 mm/h)28 . When compared to the same neuronal type in vitro, the axonal extension rates described here are 7.5 times faster than the growth rates described by other authors at an earlier stage of development (0.1 mm/h)4. Different types of neurons have been found to extend axons at different intrinsic rates that vary by several fold29. In addition, central nervous system axons usually lose the high rate of axonal growth after target innervation in vivo29 and 3 d of culture in vitro6. Therefore the current axonal extension technique should be tested with different neuronal types to better understand the limits of neurite extension.
Pipette micromanipulation and microfluidic devices are techniques demonstrated to create new functional neurites and to controllably position or (re)wire neuronal networks. This platform is ideal for systematic, standardized measurements. It introduces reproducibility and in vivo control into experiments on complex networks of neurons. The extension rates achieved are faster than 20 µm/min over millimeter-scale distances and functional connections are established. These results show, unexpectedly, that the intrinsic capacity of axons for elongation, including that of their cytoskeletal components, is much faster than previously thought10,11,12. This proposed mechanical approach bypasses slow chemical strategies and thus represents a paradigm shift for therapeutic developments to restore neuronal connectivity after injury and for micro-neuroengineering of artificial neural networks for their controlled study in vitro. These results also have a major impact on regenerative medicine and neuro-engineering approaches with direct implication for therapies that aim to reconnect neuronal circuits after trauma or in neurodegenerative diseases. This platform opens the door to obtaining data on neuron communication, signal modulation as well as growth and regeneration. It is a new way of mechanically regenerating the CNS and similar techniques may allow restoration of function after injury. Furthermore, this technique can be used to create systematically engineered neuronal networks as novel bioassay platforms for drug discovery and target validation. It is a precursor to the direct wiring of robust brain-machine interfaces.
Disclosures
The author Margaret H Magdesian is the CEO of Ananda Devices that produces instruments used in this Article.
Acknowledgments
We would like to thank Yoichi Miyahara for many helpful discussions and insights. MA and PG acknowledge funding from NSERC.
References
- Chew DJ, Fawcett JW, Andrews MR. The challenges of long-distance axon regeneration in the injured CNS. Prog. Brain. Res. 2012;201:253–294. doi: 10.1016/B978-0-444-59544-7.00013-5. [DOI] [PubMed] [Google Scholar]
- Bradke F, Fawcett JW, Spira ME. Assembly of a new growth cone after axotomy: the precursor to axon regeneration. Nat. Rev. Neurosci. 2012;13(4):189–193. doi: 10.1038/nrn3176. [DOI] [PubMed] [Google Scholar]
- Aguayo AJ, et al. Synaptic connections made by axons regenerating in the central nervous system of adult mammals. J. Exp. Biol. 1990;153:199–224. doi: 10.1242/jeb.153.1.199. [DOI] [PubMed] [Google Scholar]
- Lamoureux P, Ruthel G, Buxbaum RE, Heidemann SR. Mechanical tension can specify axonal fate in hippocampal neurons. J Cell Biol. 2002;159(3):499–508. doi: 10.1083/jcb.200207174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goslin K, Banker G. Experimental observations on the development of polarity by hippocampal neurons in culture. J Cell Biol. 1989;108(4):1507–1516. doi: 10.1083/jcb.108.4.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotti CG, Sullivan CA, Banker GA. The Establishment of polarity by hippocampal neurons in culture. J. Neurosci. 1988;8(4):1454–1468. doi: 10.1523/JNEUROSCI.08-04-01454.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magdesian MH, et al. Rapid mechanically controlled rewiring of neuronal circuits. J. Neurosci. 2016;36(3):979–987. doi: 10.1523/JNEUROSCI.1667-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucido AL, et al. Rapid assembly of functional presynaptic boutons triggered by adhesive contacts. J. Neurosci. 2009;29(40):12449–12466. doi: 10.1523/JNEUROSCI.1381-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez F, Thostrup P, Colman D, Grutter P. Dynamics of presynaptic protein recruitment induced by local presentation of artificial adhesive contacts. Dev. Neurobiol. 2013;73:1123–1133. doi: 10.1002/dneu.22037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JoVE Science Education Database. Cambridge, MA: JoVE; 2016. General Laboratory Techniques. An Introduction to Working in the Hood. https://www.jove.com/science-education/5036/an-introduction-to-working-in-the-hood. [Google Scholar]
- Strober W. Monitoring cell growth. Curr Protoc Immunol. 2001;A-3A doi: 10.1002/0471142735.ima03as21. [DOI] [PubMed] [Google Scholar]
- Beaudoin GM, 3rd, et al. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat. Protoc. 2012;7(9):1741–1754. doi: 10.1038/nprot.2012.099. [DOI] [PubMed] [Google Scholar]
- Bray D. Axonal growth in response to experimentally applied mechanical tension. Dev. Biol. 1984;102:379–389. doi: 10.1016/0012-1606(84)90202-1. [DOI] [PubMed] [Google Scholar]
- Lamoureux P, Buxbaum RE, Heidemann SR. Axonal outgrowth of cultured neurons is not limited by growth cone competition. J. Cell Sci. 1998;111:3245–3252. doi: 10.1242/jcs.111.21.3245. [DOI] [PubMed] [Google Scholar]
- Pfister BJ, Bonislawski DP, Smith DH, Cohen AS. Sketch-grown axons retain the ability to transmit active electrical signals. FEBS Lett. 2006;580:3525–3531. doi: 10.1016/j.febslet.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magdesian MH, et al. Atomic force microscopy reveals important differences in axonal resistance to injury. Biophys. J. 2012;103(3):405–414. doi: 10.1016/j.bpj.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polleux F, Snider W. Initiating and growing an axon. CSH Persp. Biol. 2010;2(4):001925. doi: 10.1101/cshperspect.a001925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debanne D, et al. Paired-recordings from synaptically coupled corticol and hippocampal neurons in acute and cultured brain slices. Nat. Protoc. 2008;3:1559–1568. doi: 10.1038/nprot.2008.147. [DOI] [PubMed] [Google Scholar]
- Qi G, Radnikow G, Feldmeyer D. Electrophysiological and Morphological Characterization of Neuronal Microcircuits in Acute Brain Slices Using Paired Patch-Clamp Recordings. J. Vis. Exp. 2015. p. e52358. [DOI] [PMC free article] [PubMed]
- Harrison RG. On the origin and development of the nervous system studied by the methods of experimental embryology. Proc. R. Soc. Lond. B. 1935. pp. 155–196.
- Weiss P. Nerve patterns: the mechanics of nerve growth. Growth 5 (Suppl. Third Growth Symposium) 1941. pp. 153–203.
- Gray C, et al. Rapid neural growth: calcitonin gene-related peptide and substance P-containing nerves attain exceptional growth rates in regenerating deer antler. Neurosci. 1992;50(4):953–963. doi: 10.1016/0306-4522(92)90218-q. [DOI] [PubMed] [Google Scholar]
- Heidemann SR, Bray D. Tension-driven axon assembly: a possible mechanism. Front. Cell Neurosci. 2015;9 doi: 10.3389/fncel.2015.00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray D. Axonal growth in response to experimentally applied tension. Dev. Biol. 1984;102:379–389. doi: 10.1016/0012-1606(84)90202-1. [DOI] [PubMed] [Google Scholar]
- Heidemann SR, Buxbaum RE. Mechanical tension as a regulator of axonal development. Neurotoxicology. 1994;15:95–107. [PubMed] [Google Scholar]
- Heidemann SR, Lamoureux P, Buxbaum RE. Cytomechanics of axonal development. Cell Biochem. Biophys. 1995;27(3):135–155. doi: 10.1007/BF02738107. [DOI] [PubMed] [Google Scholar]
- Pfister BJ, Iwata A, Meaney DF, Smith DH. Extreme stretch growth of integrated axons. J. Neurosci. 2004;24(36):7978–7983. doi: 10.1523/JNEUROSCI.1974-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG, Kocsis JD. The axon: structure, function and pathophysiology. Oxford University Press, USA; 1995. [Google Scholar]
- Cho EY, So KF. Rate of regrowth of damaged retinal ganglion cell axons regenerating in a peripheral nerve graft in adult hamsters. Brain Res. 1987;419:369–374. doi: 10.1016/0006-8993(87)90610-x. [DOI] [PubMed] [Google Scholar]
- Davies AM. Intrinsic differences in the growth rate of early nerve fibres related to target distance. Nature. 1989;337:553–555. doi: 10.1038/337553a0. [DOI] [PubMed] [Google Scholar]