

Abstract
The rock material pentlandite with the composition Fe4.5Ni4.5S8 was synthesized via high temperature synthesis from the elements. The structure and composition of the material was characterized via powder X-ray diffraction (PXRD), Mössbauer spectroscopy (MB), scanning electron microscopy (SEM), differential scanning calorimetry (DSC) and energy dispersive X-ray spectroscopy (EDX). Two preparation methods of pentlandite bulk electrodes are presented. In the first approach a piece of synthetic pentlandite rock is directly contacted via a wire ferrule. The second approach utilizes pentlandite pellets, pressed from finely ground powder, which is immobilized in a Teflon casing. Both electrodes, whilst being prepared by an additive-free method, reveal high durability during electrocatalytic conversions in comparison to common drop-coating methods. We herein showcase the striking performance of such electrodes to accomplish the hydrogen evolution reaction (HER) and present a standardized method to evaluate the electrocatalytic performance by electrochemical and gas chromatographic methods. Furthermore, we report stability tests via potentiostatic methods at an overpotential of 0.6 V to explore the material limitations of the electrodes during electrolysis under industrial relevant conditions.
Keywords: Chemistry, Issue 124, inorganic chemistry, electrocatalysis, bulk electrode, iron nickel sulfide, hydrogen evolution reaction
Introduction
The storage of fluctuating renewable energy sources such as solar and wind energy is of significant social interest due to the gradual fade of fossil fuels and subsequent need of alternative energy sources. In this respect, hydrogen is a promising sustainable candidate for a molecular energy storage solution because of a clean combustion process.1 Additionally hydrogen could be used as fuel or as starting material for more complex fuels, e.g. methanol. The preferred way for a facile synthesis of hydrogen using carbon neutral resources is the electrochemical reduction of water using sustainable energies.
Currently, platinum and its alloys are known to be the most effective electrocatalysts for the hydrogen evolution reaction (HER) showing low over-potential, a fast reaction rate and operation at high current densities.2 However, due to its high price and low natural abundance, alternative non-noble metal catalysts are required. Among the vast amount of alternative non-precious transition metal catalysts,3 especially transition metal dichalcogenides (MX2; M = Metal; X = S, Se) have been shown to possess high HER activity.4,5,6,7 In this respect, we recently presented Fe4.5Ni4.5S8 as a highly durable and active 'rock' HER electrocatalyst. This naturally abundant material is stable under acidic conditions and shows a high intrinsic conductivity with a well-defined catalytic active surface.8
While numerous materials with high HER activities have been reported, the electrode preparation is often accompanied with multiple problems, e.g. reproducibility and satisfactory stabilities (>24 h). Additionally, since the intrinsic conductivity of transition metal based catalysts in bulk is usually high, electrode preparation requires nano-structured catalysts to allow for an efficient electron transfer. These catalysts are then converted into a catalyst ink containing binders such as Nafion and the catalyst. Afterwards, the ink is drop-coated on an inert electrode surface (e.g. glassy carbon). Whereas being reasonably stable at low current densities an increased contact resistance and mediocre adhesion of the catalyst on the electrode support is commonly observed at high current densities.9 Hence, the need for more sufficient preparation methods and electrode materials is evident.
This protocol presents a novel preparation procedure for highly durable and cost efficient electrodes using bulk materials. The prerequisite for such an electrode is a low intrinsic materials resistance. Fe4.5Ni4.5S8 fulfills this criterion and can be obtained from the elements via a simple high-temperature synthesis in sealed silica ampules. The obtained material is characterized with respect to its structure, morphology and composition using powder Xray diffractometry (PXRD), differential scanning calorimetry (DSC), scanning electron microscopy (SEM) and energy dispersive Xray spectroscopy (EDX). The synthesized material is processed to afford two types of bulk electrodes, namely 'rock' and 'pellet' electrodes. The performance of both electrode types is then investigated using standard electrochemical tests and H2 quantification performed via gas chromatography (GC). A comparison of the performance of both types of electrodes in comparison to commonly used drop-coating experiments is presented.
Protocol
1. High-temperature Synthesis of Fe 4.5 Ni 4.5 S 8
NOTE: The herein described procedure for the synthesis of Fe4.5Ni4.5S8 is adopted from the literature.8,10 The strict application of the reported heating ramps is of high importance to prevent formation of phase impurities and defects of the silica ampule.
Mix iron (1.66 g, 29.8 mmol), nickel (1.75 g, 29.8 mmol) and sulfur (1.70 g, 53.1 mmol) thoroughly in a mortar and transfer the mixture to a silica ampule (10 mm diameter).
Evacuate the ampule overnight at 10-2 mbar.
Seal the ampule and place it in a tubular furnace.
Increase the temperature from room temperature (RT) to 700 °C at 5 °C/min followed by an isothermal step for 3 h.
Increase the temperature to 1100 °C within 30 min and keep it isotherm for 10 h.
Slowly cool the sample to RT by switching off the furnace. Crack the ampule to collect the solid product. Make sure to separate the Fe4.5Ni4.5S8 completely from silica glass fragments.
2. Physical Characterization
Mount a 10 mm x 5 mm x 3 mm piece of Fe4.5Ni4.5S8 rock on the sample holder and place in the vacuum chamber of the SEM instrument. Record the SEM images at 650X and 6,500X magnification at 20 kV. Simultaneously, use the same sample for EDX analysis at 4.4 kV.
For the collection of PXRD data, apply finely ground powder of Fe4.5Ni4.5S8 and mount it on an amorphous silicon wafer using silicon grease. Mount the wafer on the sample holder and collect the data in a continuous scan mode from 10-50° at a scan rate of 0.03° per 5 s using Cu-Kα radiation (λ = 1.5418 Å).
For Mössbauer analysis finely ground powder is used and placed in a polyoxymethylene (POM) cup. Record zero-field Mössbauer spectra at 25 °C using a 57Co radiation source in a Rh-matrix.
For DSC analysis, finely ground powder is placed in a tared α-Al2O3 crucible. Perform DSC measurements in the range from RT to 1,000 °C recording the heating and cooling curve at a rate of 10 °C/min. Perform the experiment under a flow of high purity nitrogen.
3. Preparation of 'Rock' Electrodes
Solder a copper wire to a wire ferrule.
Cut the Fe4.5Ni4.5S8 bulk material into smaller pieces (approx. 5 mm x 5 mm x 5 mm).
Place the small piece of Fe4.5Ni4.5S8 in the ferrule in a way that approx. 2 mm of material sticks out of the ferrule.
Mantle the ferrule and copper wire with 100 mm of Teflon tubing.
Seal the tip of the electrode with two-component epoxide glue and dry the electrode overnight under ambient conditions.
Grind off the tip until the shiny surface (metallic finish) of the Fe4.5Ni4.5S8 is exposed. Further polish with fine grade sand paper (20, 14, 3 and 1 µm grit) to obtain a smooth surface.
Clean the surface with deionized water and let it dry on air.
4. Preparation of 'Pellet' Electrodes
NOTE: Custom-built Teflon casings with a brass rod were used as contact for 'pellet' electrodes (3 mm diameter).
Grind 50 mg of material to obtain a fine powder of the Fe4.5Ni4.5S8 material.
Fill the finely ground powder into a compressing tool (3 mm in diameter) and press the material with a maximum weight force of 800 kg/cm2.
Remove the pellet from the mold using a distance holder.
Apply a two-component silver-epoxide glue on the brass rod in the cavity of the Teflon casing. Avoid any pollution of the tip of the Teflon-casing.
Place the pellet in the Teflon casing. The flat side of the pellet must stick out ~1 mm.
Remove any pollution on the Teflon casing with a paper tissue.
Verify the contact between the brass wire and the Fe4.5Ni4.5S8 pellet with a voltmeter to assure proper conductivity.
After 12 h of curing the two-component glue at 60 °C, cool down the electrode to ambient temperature.
Polish the electrode with sand paper (20, 14, 3 and 1 µm grit) to obtain a shiny flush flat surface within the Teflon case.
Clean the surface with deionized water and let it dry under ambient conditions.
5. Electrochemical Testing of Electrodes
NOTE: The experiments were accomplished with a standard three-electrode setup using the Fe4.5Ni4.5S8 electrode as working electrode, Ag/AgCl (sat. KCl or 3 M KCl solution) electrode as reference electrode and Pt wire or Pt-grid as counter electrode. A gas-tight cell equipped with a stirring bar was filled with the electrolyte consisting of 0.5 M H2SO4 for all electrochemical experiments. The electrolyte was not exchanged during the electrochemical testing of an electrode. All potentials are referenced to ERHE (RHE = reversible hydrogen electrode) according to ERHE = EAg/AgCl + X + 0.059 pH with X = 0.197 V (saturated KCl) or X = 0.210 V (3 M KCl), unless noted otherwise.
- Preliminary steps
- Connect all three electrodes with the wires of the potentiostat.
- Add 25 mL of electrolyte (0.5 M H2SO4) into the electrochemical cell and adjust the electrodes to ensure that the electrodes are fully immersed into solution. Subsequently, switch on the potentiostat.
- Switch on the magnetic stirring.
- Electrochemical cleaning of the electrode surface
- Perform a cyclic voltammetry (CV) experiment to obtain fast overview on the electrochemical processes that can be observed.
- Set the potential range from 0.2 to -0.2 V with a scan rate of 100 mV/s (non-catalytic potential area). Further, set the number of cycles to 20.
- Start the cycling process and wait until the last cycle is finished. If at least the last 3 to 4 obtained cycles coincide, the electrochemical electrode cleaning is completed. In case of divergence add more cycles until stable curves are obtained.
- Measurement of the catalytic performance – linear sweep voltammetry
- Before starting the experiment determine the iR compensation value for the electrochemical setup.
- Select the program for linear sweep voltammetry (LSV) experiments and set the potential range from 0.2 to -0.6 V and the scan rate to 5 mV/s, including the iR drop into the experiment. Start the experiment.
- Repeat the linear sweep experiments to ensure reproducibility. In case of non-reproducible results start over from step 5.2.
- Stability measurement & quantification
- Perform a controlled potential coulometry experiment (CPC).
- Set the potential to -0.6 V with an experiment time of at least 20 h (72,000 s).
- Simultaneously collect gas samples with a gas tight syringe from the headspace of the sealed cell through a septum for every hour for at least 4 h of the experiment. Inject the samples into a GC instrument for quantification and determine the amount of hydrogen produced using a calibration curve recorded on this instrument.
- Estimation of the electrochemical surface area (ESCA) NOTE: Do not stir the electrolyte solution during this experiment.
- Determine the iR compensation to measure the resistance of the solution.
- Select a potential range between 0.1 and 0 V in the cyclic voltammetry experiment and set the scan rate to 10 mV s-1. Use the iR drop correction. Set the number of cycles for the experiment to 5.
- Repeat steps 5.4.1) to 5.4.2) for scan rates of 20, 30, 40, 50 and 60 mV s-1.
- From the obtained CV curves pick the fifth cycle for further interpretation.
- Determine the charging current density differences (Δj = ja jc) and plot these values as a function of the scan rate. The linear slope is equivalent to twice of the double-layer capacitance Cdl, which is proportional to the electrochemical surface area (ECSA).
- Electrochemical impedance spectroscopy (EIS)
- Record electrochemical impedance spectra in the frequency range from 50 kHz to 1 Hz at the corresponding open-circuit potential and an overpotential of 0.3 V.
- Plot the Nyquist plot from the received data to determine the charge transfer resistance.
Representative Results
The successful synthesis of Fe4.5Ni4.5S8 possessing the Pentlandite structure is confirmed by powder X-ray diffraction experiments due to the prominent (111), (311), (222), (331) and (511) reflections being present (Figure 1a). A proper temperature control during the reaction, however, is the key to obtain phase pure materials. Notably, mono-sulfide solid solutions (mss), a common impurity of pentlandite materials11, have been observed, when the mixture was heated to 700 °C at a higher heating rate (e.g. 20 °C/min). An exemplary XRD pattern of such a sample is also depicted (Figure 1a, red). On the contrary, if an appropriate heating rate of 5 °C/min is applied, the appearance of mss can be significantly reduced if not even eliminated (Figure 1a, black). Mössbauer spectroscopy (Figure 1b) confirms the pentlandite structure of the Fe4.5Ni4.5S8 sample. The material reveals two distinct different iron sites with isomeric shifts of 0.13 (±0.02) and 0.50 (±0.02) mm/s as well as quadrupole couplings of 0.12 (±0.02) and 0.13 (±0.02), respectively (Figure 1b). This observation is in good agreement with literature reports and structural findings showing two different iron sites.12 Additionally, differential scanning calorimetry (DSC) (Figure 1c) revealed two main phase transitions at 612 °C and 861 °C, proving the absence of undesirable phases according to literature reports.13
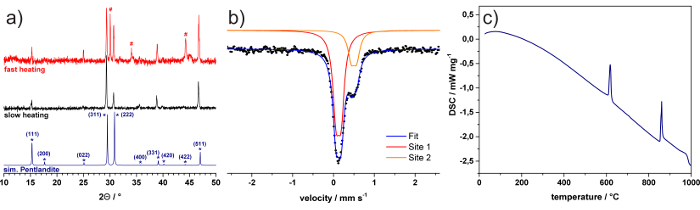
Figure 1: XRD and Mössbauer Spectroscopy. (a) XRD patterns of samples prepared with heating rates of 5 °C/min (black) and 20 °C/min (red). For reference, a pattern of Pentlandite simulated from single crystal diffraction data is shown (blue). Reflections corresponding to mss are marked with #. (b) Mössbauer spectrum and (c) DSC curve of Fe4.5Ni4.5S8. Parts of the figure were reproduced from a previous publication in Nat Comm.8 Please click here to view a larger version of this figure.
SEM images of polished Fe4.5Ni4.5S8 rock and pellet electrodes are depicted in Figure 2a. We employed EDX analysis to determine the elemental composition of the material on the surface. The EDX spectrum is shown in Figure 2b. The carbon peak in the spectrum yields from the use of carbon pads to mount the sample on the holder. From the EDX quantification – shown in Figure 2c – the ratio between iron and nickel was determined to be 1.06: 1.00. Assuming that the sum of iron and nickel is 9, the actual sum formula is Fe4.64Ni4.36S8.11. The deviation from the ideal formula Fe4.5Ni4.5S8 is within the margin of error. This result is consistent with the observation of a pure Pentlandite phase in the XRD pattern of the sample.

Figure 2: SEM and EDX analysis. SEM micrograph of (a) 'rock' and (b) 'pellet' electrodes. Scale bars = 1 µm. (c) EDX spectrum and (d) composition analysis of the 'rock' electrode. Parts of the figure were reproduced from a previous publication in Nat Comm.8 Please click here to view a larger version of this figure.
For electrochemical performance testing towards HER, first the electrodes have been prepared using a cut piece of rock and a pellet pressed from finely ground powder, respectively. The key steps of the 'rock' and 'pellet' electrode preparation are shown in Figures 3a and 3b, respectively. The standard testing procedure involves first, polishing and electrochemical washing of the electrodes. Then, linear sweep voltammograms (LSV) are recorded, followed by stability testing, coupled with product quantification. Finally, the electrochemical surface area (ESCA) is estimated. In Figure 3c, the LSV´s of both types' electrodes are shown. From the LSV the over potential for the HER is estimated to be -280 mV and -285 mV vs. RHE at a current density of 10 mA/cm2 for 'rock' and 'pellet' electrode, respectively. An exfoliated drop coated electrode did not show any improved electrocatalytic performance. Therefore, the influence of the preparation method on the performance is minor. Neglecting the drop-coating procedure, a similar behavior can be observed for the long-term stability of the electrodes (Figure 3d), whereas the 'pellet' shows a faster activation behavior as we have reported before for 'rock' Fe4.5Ni4.5S8 electrodes.8 Both electrodes show comparable current densities after the activation is finished. However, if a drop-coated electrode is exposed to comparable electrochemical conditions, the high amount of hydrogen produced, leads to detachment of the catalyst from the electrode support and thus to inactivation of the system. Figure 3d (inset) shows the amount of hydrogen produced depending on the time of electrolysis using a 'rock' electrode. Comparable hydrogen amounts can be observed for 'pellet' electrodes. From the slope of a linear fit of the GC quantification, a hydrogen production rate of 2.14 mmol H2 h-1 cm-2 is determined, which is – to our best knowledge – only beaten by platinum (11 mmol H2 h-1 cm-2) at comparable applied potentials.14 For determination of the ECSA, the charging current density difference was plotted as a function of the scan rate. The resulting graph is shown in Figure 3e. From the slope, only marginal differences in ECSA and hence the number of active sites, which are well in the margin of error of such measurements, can be observed. Therefore, both preparation types offer electrodes of similar performance. In Figure 3f an exemplary Nyquist plot from the EIS data of a 'rock' electrode is shown. Likewise, the 'pellet' electrodes reveal the same behavior. This plot reveals a very low charge-transfer resistance (Rct = 57.2 Ω) of the Fe4.5Ni4.5S8 bulk electrode, which is consistent with the high intrinsic conductivity of the material.
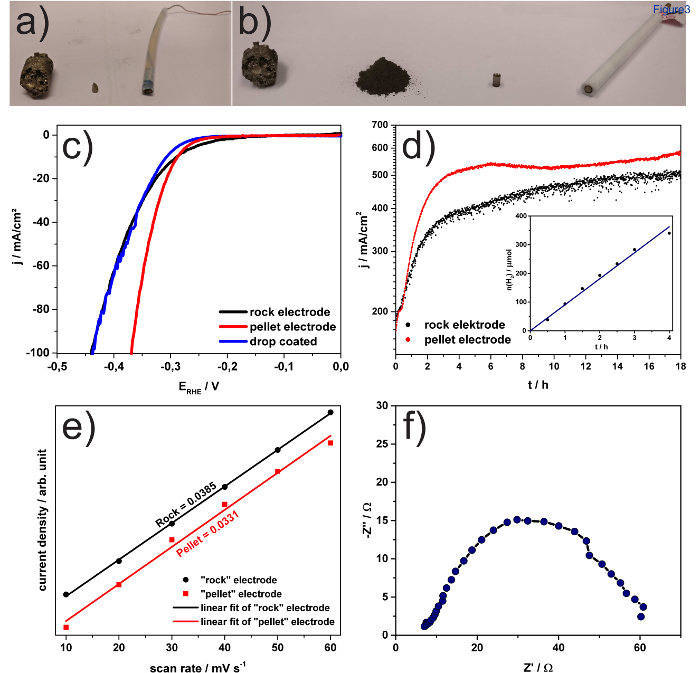
Figure 3: Electrode preparation and electrochemical analysis. Preparation steps of 'rock' (a) and 'pellet' (b) type Fe4.5Ni4.5S8 electrodes. Linear sweep voltammograms (c) at 5 mV s-1 of a 'rock' (black), a 'pellet' (red) and a drop coated (blue) electrode. (d) over 18 h at 0.6 V of 'rock' (black) and 'pellet' electrodes (red). Inset shows the hydrogen production over the first 4 h of electrolysis using a 'rock' electrode. The blue line indicates the linear fit of the data. (e) Charging current density differences as a function of the scan rate for 'rock' (black) and 'pellet' electrodes (red). The linear slope represents the electrochemical surface area (ECSA). f) Nyquist plot and the equivalent circuit at HER overpotential (η = 300 mV) of a 'rock' electrode. Parts of the figure were reproduced from a previous publication in Nat Comm.8 Please click here to view a larger version of this figure.
Discussion
The synthesis of Fe4.5Ni4.5S8 was performed in a vacuum-sealed ampule to prevent oxidation of the material during synthesis. During the synthesis, temperature control is the key to obtain a pure product. The first, very slow heating step thereby prevents superheating of the sulfur, which might cause cracking of the ampule due to high sulfur pressure. Even more crucial is the prevention of phase impurities like mono-sulfide solid solutions (mss) by slow heating of the sample. The subsequent annealing step at 700 °C ensures equilibration of the system. Following, annealing at 1100 °C finally leads to the formation of a pure pentlandite phase, which is a high temperature phase of iron nickel sulfides.13
When preparing 'rock' electrodes, it is important that the piece of 'rock' sticks out of the ferrule approx. 2 mm to be able to cut enough material to obtain a smooth electrode surface (A = 0.135cm2) without exposing the ferrule. Epoxy glue is used to fixate the 'rock' in the ferrule as well as fixating the Teflon tubing, which prevents contact between the electrolyte and any parts of the electrode apart from the active material. Otherwise side reactions can take place and reproducibility is hampered. The application of the epoxy glue should be done in a way that no bubbles are enclosed in the glue. These bubbles might cause unwanted exposure of the ferrule or material. The epoxy glue should dry at least overnight to be fully processible. In case of 'pellet' electrodes, the Teflon casing serves a similar role as mentioned before for the 'rock' electrodes. Theused catalyst should be a well ductile material with good conductivity. Otherwise the material is not qualified to be pressed and instead conductive binder material has to be added to provide stability and conductivity. The gap dimensions between the case and the pellet must be negligibly small. It is thus better to prepare a pellet that is slightly bigger than the Teflon casing. The removal of excess silver epoxide glue is also important, to avoid the reaction of silver during electrochemical measurements. After curing the glued electrode at 60 °C, it is advisable to let the electrode cool down to room temperature before further preparation or the compressed material might crumble. To obtain a well-defined geometrical surface area (A = 0.071 cm2) without any cracks and indentations, the pellet has to stick out at least 1 mm. It has to be polished down until it is flush with the case. Since the pellet can be very brittle it might be helpful to use Teflon housings with a wall strength of at least 2 mm, so it provides sufficient support, while grinding and polishing. To provide a consistent quality and uniformity of the surface, it is reasonable to start with the coarsest and end up with the finest abrasive.
In general, for bulk electrodes as described herein it has to be noted that a conductive catalyst material is mandatory to ensure a fast electron transfer and prevent resistivity limitations. An approach to implement non-conductive catalyst materials into this electrode type could be accomplished by immobilizing the catalyst on a conductive substrate (e.g. carbon black, graphite) and preparing electrodes starting from this composite powder. This method, however, would exclude the possibility to prepare 'rock' electrodes. A further step to obtain high quality and especially more industrial relevant electrodes using a similar approach to the 'pellet' electrode is pressing the material directly on a current collector (e.g. Ni-mesh) and will be explored by us in future work.
Polishing the electrodes prior to testing the electrocatalytic properties is crucial to obtain reproducible results. The electrochemical cleaning of the electrode equilibrates the electrode and removes any adsorbed impurities from the electrode surface. Multiple LSV´s are recorded to ensure reproducibility of the data. In contrast to commonly reported stability tests, which are generally accomplished in a galvanostatic manner at relatively low current densities (mostly 10 mA/cm2), we opted to perform a potentiostatic stability test at high over potential, which leads to high current densities (>500 mA/cm2). The advantage of such a stress test is to push the material to its electrochemical and mechanical limits as well as providing a more realistic electrode test. For quantification the gas tightness of the electrochemical cell and syringe is crucial to obtain consistent results. Furthermore, a good calibration of the GC is mandatory. Stirring during this experiment provides a thorough mixing of the electrolyte and promotes desorption of hydrogen bubbles from the electrode surface. However, stirring must be turned off during ECSA experiments, since the correct build-up of the electrochemical double layer is required. From the ESCA, conclusions on the dependency of a materials activity and its active site count, which is proportional to the ESCA, can be obtained. Hence, ECSA analysis is a facile tool that enables a comparison of the performance of different catalysts.
In summary, we synthesized Fe4.5Ni4.5S8, a highly conductive transition metal sulfide, and prepared two types of bulk electrodes using this material. The electrodes were tested towards hydrogen evolution reaction using standard electrochemical methods such as LSV, CPC and ECSA determination. The hydrogen evolution rate was determined using a GC and is among the highest observed for non-noble metal catalysts at low overpotential. The performance of both types of electrodes is similar with the 'pellet' electrode being slightly ahead in performance and especially electrode activation. This, however, might stem from an overestimated surface area in 'rock' electrodes. Notably, the 'pellet' electrodes have the advantage that the surface area can be more easily and more exactly determined and reproduced. This feature renders 'pellet' electrodes more valuable for comparable studies, while 'rock' electrodes are easier and cheaper to prepare. Future work will focus on the design of an up-scale version of 'pellet' electrodes.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank B. Konkena und W. Schuhmann for valuable scientific discussions. Financial support by the Fonds of the Chemical Industry (Liebig grant to U.-P.A.) and the Deutsche Forschungsgemeinschaft (Emmy Noether grant to U.-P.A., AP242/2-1).
References
- May MM, Lewerenz H-J, Lackner D, Dimroth F, Hannappel T. Efficient direct solar-to-hydrogen conversion by in situ interface transformation of a tandem structure. Nat Comm. 2015;6:8286. doi: 10.1038/ncomms9286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng W, et al. Correlating hydrogen oxidation and evolution activity on platinum at different pH with measured hydrogen binding energy. Nat Comm. 2015;6:5848. doi: 10.1038/ncomms6848. [DOI] [PubMed] [Google Scholar]
- Li X, Hao X, Abudula A, Guan G. Nanostructured catalysts for electrochemical water splitting: Current state and prospects. J. Mater. Chem. A. 2016;4(31):11973–12000. [Google Scholar]
- Merki D, Hu X. Recent developments of molybdenum and tungsten sulfides as hydrogen evolution catalysts. Energy Environ. Sci. 2011;4(10):3878. [Google Scholar]
- Kibsgaard J, Chen Z, Reinecke BN, Jaramillo TF. Engineering the surface structure of MoS2 to preferentially expose active edge sites for electrocatalysis. Nat Mater. 2012;11(11):963–969. doi: 10.1038/nmat3439. [DOI] [PubMed] [Google Scholar]
- Kong D, Cha JJ, Wang H, Lee HR, Cui Y. First-row transition metal dichalcogenide catalysts for hydrogen evolution reaction. Energy Environ. Sci. 2013;6(12):3553. [Google Scholar]
- Voiry D, et al. Enhanced catalytic activity in strained chemically exfoliated WS(2) nanosheets for hydrogen evolution. Nat Mater. 2013;12(9):850–855. doi: 10.1038/nmat3700. [DOI] [PubMed] [Google Scholar]
- Konkena B, et al. Pentlandite rocks as sustainable and stable efficient electrocatalysts for hydrogen generation. Nat Comm. 2016;7:12269. doi: 10.1038/ncomms12269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon HS, et al. Simple Chemical Solution Deposition of Co₃O₄ Thin Film Electrocatalyst for Oxygen Evolution Reaction. ACS Appl Mater Interfaces. 2015;7(44):24550–24555. doi: 10.1021/acsami.5b06189. [DOI] [PubMed] [Google Scholar]
- Xia F, Pring A, Brugger J. Understanding the mechanism and kinetics of pentlandite oxidation in extractive pyrometallurgy of nickel. Mine Eng. 2012;27-28:11–19. [Google Scholar]
- Drebushchak VA, Kravchenko TA, Pavlyuchenko VS. Synthesis of pure pentlandite in bulk. J Crystal Growth. 1998;193(4):728–731. [Google Scholar]
- Knop O, Huang C-H, Reid K, Carlow JS, Woodhams F. Chalkogenides of the transition elements. X. X-ray, neutron, Mössbauer, and magnetic studies of pentlandite and the π phases π(Fe, Co, Ni, S), Co8MS8, and Fe4Ni4MS8 (M = Ru, Rh, Pd) J Solid State Chem. 1976;16(1-2):97–116. [Google Scholar]
- Kullerud G. Thermal stability of pentlandite. The Canadian Mineralogist. 1963;7(3):353–366. [Google Scholar]
- Siracusano S, et al. An electrochemical study of a PEM stack for water electrolysis. Int J Hydrogen Energy. 2012;37(2):1939–1946. [Google Scholar]