

Abstract
Here, we present chimera assembly by plasmid recovery and restriction enzyme site insertion (CAPRRESI). CAPRRESI benefits from many strengths of the original plasmid recovery method and introduces restriction enzyme digestion to ease DNA ligation reactions (required for chimera assembly). For this protocol, users clone wildtype genes into the same plasmid (pUC18 or pUC19). After the in silico selection of amino acid sequence regions where chimeras should be assembled, users obtain all the synonym DNA sequences that encode them. Ad hoc Perl scripts enable users to determine all synonym DNA sequences. After this step, another Perl script searches for restriction enzyme sites on all synonym DNA sequences. This in silico analysis is also performed using the ampicillin resistance gene (ampR) found on pUC18/19 plasmids. Users design oligonucleotides inside synonym regions to disrupt wildtype and ampR genes by PCR. After obtaining and purifying complementary DNA fragments, restriction enzyme digestion is accomplished. Chimera assembly is achieved by ligating appropriate complementary DNA fragments. pUC18/19 vectors are selected for CAPRRESI because they offer technical advantages, such as small size (2,686 base pairs), high copy number, advantageous sequencing reaction features, and commercial availability. The usage of restriction enzymes for chimera assembly eliminates the need for DNA polymerases yielding blunt-ended products. CAPRRESI is a fast and low-cost method for fusing protein-coding genes.
Keywords: Molecular Biology, Issue 124, Chimeric protein coding gene assembly synonym DNA sequence insertion, restriction enzyme site insertion, plasmid recovery, pUC18/19 vector, antibiotic resistance gene disruption, genetic engineering, fusion protein, primary sigma factor, RpoD, SigA
Introduction
Chimeric gene assembly has been widely used in molecular biology to elucidate protein function and/or for biotechnological purposes. Different methods exist for fusing genes, such as overlapping PCR product amplification1, plasmid recovery2, homologous recombination3, CRISPR-Cas9 systems4, site-directed recombination5, and Gibson assembly6. Each of these offers different technical advantages; for example, the flexibility of overlapping PCR design, the in vivo selection of constructions during plasmid recovery, or the high efficiency of CRISPR-Cas9 and Gibson systems. On the other hand, some difficulties can arise while performing some of these methods; for example, the first two approaches rely on blunt-ended DNA fragments, and ligation of these types of products could be technically challenging compared to sticky-ended ligation. Site-directed recombination can leave traces of extra DNA sequences (scars) on the original, like in the Cre-loxP system5. CRISPR-Cas9 can sometimes modify other genome regions in addition to the target site4.
Here, we introduce chimera assembly by plasmid recovery and restriction enzyme site insertion (CAPRRESI), a protocol for fusing protein-coding genes that combines the plasmid recovery method (PRM) with the insertion of restriction enzyme sites on synonym DNA sequences, enhancing ligation efficiency. To ensure amino acid sequence integrity, restriction enzyme sites are inserted on synonym DNA sequence stretches. Among the benefits of CAPPRESI are that it can be performed using ordinary laboratory reagents/tools (e.g., enzymes, competent cells, solutions, and thermocycler) and that it can give quick results (when the appropriate enzymes are used). Relying on restriction enzyme sites that emerge from synonym DNA sequences can limit the selection of the exact fusion points inside the proteins of interest. In such cases, target genes should be fused using overlapping oligonucleotides, and restriction enzyme sites should be inserted onto the resistance gene of the vector.
CAPRRESI consists of seven simple steps (Figure 1): 1) selection of the cloning vector, pUC18 or pUC196; 2) in silico analysis of the wildtype sequences to be fused; 3) selection of breaking regions for chimera assembly and plasmid disruption; 4) in silico generation of synonym DNA sequences containing restriction enzyme sites; 5) independent cloning of the wildtype genes into the selected plasmid; 6) plasmid disruption by PCR, followed by restriction enzyme digestion; and 7) plasmid recovery using DNA ligation and bacterial transformation. Chimeric genes produced by this technique should be verified with sequencing.
The pUC18/19 vectors offer technical advantages for cloning and chimera assembly, such as small size (i.e., 2,686 base pairs), high copy number, advantageous sequencing reaction features, and commercial availability7. Here, an Escherichia coli host was used to assemble and handle the chimeras because bacterial cultures are cheap and grow fast. Given this, subsequent cloning of the fusion fragments into the final target plasmids will be needed (e.g., expression vectors as pRK415 in bacteria or pCMV in mammalian cells).
CAPRRESI was tested for fusing two primary sigma factor genes: E. colirpoD and Rhizobium etlisigA. Primary sigma factors are RNA polymerase subunits responsible for transcription initiation, and they consist of four domains (i.e., σ1, σ2, σ3, and σ4)8. The amino acid sequence length of proteins encoded by rpoD and sigA are 613 and 685, respectively. RpoD and SigA share 48% identity (98% coverage). These primary sigma factors were split into two complementary fragments between regions σ2 and σ3. Two chimeric genes were assembled according to this design: chimera 01 (RpoDσ1-σ2 + SigAσ3-σ4) and chimera 02 (SigAσ1-σ2 + RpoDσ3-σ4). DNA fusion products were verified by sequencing.
Protocol
1. CAPRRESI Protocol
NOTE: Figure 1 represents the overall CAPRRESI protocol. This technique is based on an in silico design and the subsequent construction of the desired chimeras.
- Selection of the cloning plasmid, pUC18 or pUC19.
- Select the pUC vector that best fits technical demands. NOTE: Both vectors have the same sequence, except for the orientation of the multiple cloning site. This is important for the design of oligonucleotides and the PCR plasmid disruption.
- In silico analysis of the wildtype genes and pUC18/19 sequences.
- Obtain the DNA sequences of the wildtype genes and save them in a FASTA format file (e.g., genes.fas). NOTE: The sequences of pUC18/19 vectors are contained in the pUC.fas file (Figure 1, step 1).
- Determine which restriction enzymes do not cut the wildtype genes and pUC18/19 sequences by running the REsearch.pl script. Alternatively, perform a restriction enzyme site search with an online tool (Figure 1, step 2).
- Type the following into a terminal: perl REsearch.pl genes.fas perl REsearch.pl pUC.fas NOTE: These commands will create the following output files: genes_lin.fas, genes_re.fas, pUC_lin.fas, and pUC_re.fas.
- Select the appropriate restriction enzymes for cloning the wildtype genes into the multiple cloning site of the chosen pUC18/19 vector by comparing the genes_re.fas and pUC.fas files. Choose the orientation of the insert relative to the ampicillin resistance gene (ampR) of the pUC18/19 vector.
- Design forward and reverse oligonucleotides to amplify the coding region of the wildtype genes (external oligonucleotides). Include the proper restriction enzyme site at each 5' end of the oligonucleotides; this will define the insert orientation.
- Include a functional ribosome binding site in the forward oligonucleotides.
- Insert both wildtype genes in the same orientation inside the vector.
- Selection of the breaking regions for chimera assembly and plasmid disruption.
- Obtain the amino acid sequence of the wildtype and the ampR genes by running the translate.pl script (Figure 1, step 3).
- Type the following into a terminal: perl translate.pl genes.fas perl translate.pl ampR.fas NOTE: This script will create the following output files: genes_aa.fas and ampR_aa.fas.
- Globally align the two wildtype amino acid sequences with an appropriate software (e.g., MUSCLE)9.
- Select the desired break regions (3-6 amino acids) for chimera assembly based on the sequence alignment. Save them into a file in FASTA format (e.g., regions.fas).
- Locate the DNA sequences that code for the selected amino acid stretches on both wildtype genes.
- In silico generation of synonym DNA sequences containing restriction enzyme sites.
- Obtain all the synonym DNA sequences that code for each amino acid sequence stretch selected as breaking regions (Figure 1, step 4); these sequences were stored in the regions.fas file.
- Type the following into a terminal: perl synonym.pl regions.fas NOTE: This script will create the regions_syn.fas output file.
- Search for restriction enzyme sites found on synonym DNA sequences.
- Type the following into a terminal: perl REsynonym.pl regions_syn.fas NOTE: This script will create the regions_syn-re.fas output file.
- Choose one restriction enzyme that is shared between synonym sequences of both wildtype genes; this site will be used to assemble chimeras via restriction enzyme digestion. Verify that the chosen restriction enzyme cut neither the wildtype genes nor the pUC18/19 vector sequences.
- Compare the restriction enzymes found on the genes_re.fas, pUC_re.fas, and regions_syn-re.fas files.
- In silico substitute the originals for their synonym DNA sequences at the corresponding loci on both wildtype genes. Append synonym DNA sequences into the wildtype sequence file (genes.fas).
- Get the amino acid sequence of genes contained on the genes.fas file (perl translate.pl genes.fas).
- Align the amino acid sequences translated from wildtype and synonym DNA sequences. Verify that both sequences are the same.
- Repeat all the steps of this section with the ampR gene present on the pUC18/19 vector. NOTE: The goal is to disrupt the ampR gene (file ampR.fas) into two complementary parts. The restriction enzyme selected to disrupt the ampR gene should be different from the one used for chimera assembly.
- Independent cloning of the two wildtype genes into the selected pUC18/19 vector (Figure 1, step 3).
- Purify the chosen pUC18/19 plasmid DNA using a plasmid DNA purification kit.
- For example, transform E. coli DH5α with pUC18 plasmid and grow the transformants overnight at 37 °C on solid LB-0.3 mg/mL ampicillin (Amp). Pick a transformant colony, inoculate a new LB-0.3 mg/mL Amp plate, and grow it overnight at 37 °C.
- Take part of the previous culture and grow it in liquid LB-0.3 mg/mL Amp for 6-8 h at 37 °C. Extract the plasmid DNA using a kit.
- PCR amplify wildtype genes from the total genomic DNA using the appropriate external oligonucleotides. Use a high-fidelity DNA polymerase if possible. Select the DNA polymerase that maximizes the yield. Perform PCR according to the manufacturer's guidelines and the melting temperature of the oligonucleotides.
- Run PCR cycles, depending upon the DNA polymerase, amplicon size, template, and oligonucleotides used. For example, to amplify rpoD DNA, prepare a 50 µL reaction: 5 µL of 10x buffer, 2 µL of 50 mM MgSO4, 1 µL of 10 mM dNTP mix, 2 µL of each 10-µM oligonucleotide (Table 2), 2 µL of template DNA (E. coli DH5α total DNA), 35.8 µL of ultra-pure water, and 0.2 µL of high-fidelity DNA polymerase. Run the following PCR cycles: 1 min at 94 °C for the initial denaturation followed by 30 cycles of amplification (30 s at 94 °C to denature, 30 s at 60 °C to anneal the oligonucleotides, and 2 min at 68 °C to extend).
- Dissolve 1.2 g of agarose in 100 mL of double-distilled water by heating them in the microwave. Assemble a gel tray and comb and put them into the horizontal gel caster. Fill the gel tray with the melted agarose solution and let it solidify. Remove the comb.
- Place the gel into the electrophoresis chamber. Fill chamber with 1x Tris-acetate-EDTA buffer. Load 50 µL of PCR products into the gel wells. Electrophorese the samples (e.g., at 110 V for 1 h).
- After electrophoresis, stain the gel in 100 mL of double-distilled water containing 100 µL of 1 mg/mL ethidium bromide solution for 10 min with slow shaking. Wash the gel in double-distilled water for another 10 min in slow shaking; use a horizontal shaker. Caution: Ethidium bromide is a toxic compound; wear protective gloves and a cotton laboratory coat.
- Purify wildtype gene DNA from the corresponding bands of the gel using a kit. Visualize the bands by placing the stained gel in a UV chamber (recommended wavelength: 300 nm). Keep the exposure of the gel to a minimum. Use a DNA purification kit and follow the manufacturer's instructions. Caution: UV radiation is dangerous; wear a protective shield, glasses, and a cotton laboratory coat.
- Digest purified wildtype genes and pUC18/19 plasmid DNA with the restriction enzymes according to the manufacturer's instructions. Use fast digestion enzymes when possible. For example, digest 6 µL of pUC18 DNA (300 ng/µL) in 10 µL of ultrapure water, 2 µL of 10X buffer, 1 µL of KpnI restriction enzyme, and 1 µL of XbaI restriction enzyme. Leave the reaction at 37 °C for 30 min.
- Inactivate the restriction enzymes according to the manufacturer's guidelines. For example, inactivate the double-digestion reaction KpnI-XbaI at 80 °C for 5 min.
- Mix insert:vector DNA at a volumetric ratio of 3:1. Follow the manufacturer's instructions for a ligation reaction. Use fast ligation enzymes when possible (leave the reaction at 25 °C for 10 min). NOTE: Typically, the DNA concentration after purification using the kits is good enough for digestion and ligation reactions. For example, add 6 µL of rpoD DNA (KpnI-XbaI, 150 ng/µL), 3 µL of pUC18 DNA (KpnI-XbaI, 150 ng/µL), 10 µL of 2x buffer, 1 µL of ultrapure water, and 1 µL of T4 DNA ligase. Leave ligation reaction at 25 °C for 10 min.
- Transform the E. coli DH5α with 2-4 µL of the ligation reaction, as described in the Supplementary Materials. Plate the transformed cells into solid LB/Amp/X-gal/IPTG plates. Leave the plates overnight at 37 °C.
- Select white-colored transformant E. coli colonies and streak them on a new LB/Amp plate. Leave the plates overnight at 37 °C.
- Perform a colony PCR to choose candidates for sequencing the reaction, as described in the Supplementary Materials. Extract plasmid DNA from candidates. Alternatively, digest candidate plasmid DNA with the same restriction enzymes used for cloning the inserts.
- Plasmid disruption by PCR followed by restriction enzyme digestion.
- Design in silico forward and reverse oligonucleotides at break regions for wildtype and ampR genes, respectively. Substitute in silico the wildtype fragment for its corresponding synonym DNA sequence (obtained in step 1.4). NOTE: This synonym DNA sequence contains a restriction enzyme site. Forward and reverse oligonucleotides overlap at the restriction enzyme site. Oligonucleotides should range from 21-27 nucleotides, end with a cytosine or guanine, and contain a restriction enzyme site. A forward oligonucleotide has the same sequence as its target region on the template DNA. A reverse oligonucleotide is the reverse complementary sequence of the target region on the template DNA.
- Use the two wildtype gene constructions as the DNA template for PCR reactions (e.g., pUC18rpoD and pUC18sigA). Obtain two complementary parts of each construction (pUC18rpoDσ1-σ2, pUC18rpoDσ3-σ4, pUC18sigAσ1-σ2, and pUC18sigAσ3-σ4) (Figure 1, step 6).
- Load DNA samples into an agarose gel 1-1.2% [w/v]. Separate the PCR products from the DNA template by electrophoresis (e.g., 110 V for 1 h). Alternatively, digest the PCR reactions with DpnI to break the DNA template. NOTE: DpnI enzyme recognizes only methylated DNA sequences (5'-GATC-3').
- Purify the samples using a DNA purification kit. Follow the manufacturer's guidelines.
- Double-digest all the complementary DNA fragments with the appropriate restriction enzymes according to the manufacturer's directions. Use fast-digest restriction enzymes when possible. For example, double-digest the pUC18rpoDσ1-σ2 DNA fragment with AflII and SpeI using the manufacturer's protocol. Leave the digestion reaction at 37 °C for 15 min.
- Inactivate the restriction enzymes according to the manufacturer's guidelines. For example, inactivate AflII-SpeI at 80 °C for 20 min.
- Plasmid recovery using DNA ligation and bacterial transformation.
- Mix the proper DNA fragments in a volumetric 1:1 ratio to assemble the desired chimeric gene (Figure 1, step 7). NOTE: In this way, the integrity of the ampR gene is restored, producing a functional protein.
- For example, mix 4 µL of pUC18rpoDσ1-σ2 DNA (digested with AflII-SpeI, 150 ng/µL), 4 µL of pUC18sigAσ3-σ4 DNA (AflII-SpeI, 150 ng/µL), 10 µL of 2x buffer, 2 µL of ultrapure water, and 1 µL of T4 DNA ligase; the DNA concentration after kit purification is good enough for digestion and subsequent ligation. Leave the ligation reaction at 25 °C for 10 min.
- Transform E. coli DH5α with 3-5 µL of the chimera assembly ligation reaction (Supplementary Materials). Grow the transformant cells in LB/Amp/X-gal/IPTG plates overnight at 37 °C overnight.
- Select white-colored transformant E. coli colonies and streak them on a new LB/Amp plate. Leave the plates overnight at 37 °C.
- Perform a colony PCR to choose candidates for a sequencing reaction, as described in the Supplementary Materials. Visualize bands in a 1-1.2% (w/v) agarose gel by performing electrophoresis (described in step 2.3.2.1).
- Grow cultures from positive candidates. Extract plasmid DNA from them using a plasmid DNA purification kit.
2. Sequence Candidate Chimeric Constructions
Make sure the quality of the plasmid DNA is good enough for the sequencing reaction by following guidelines from the sequence service provider. Extract DNA using purification kits. For example, on the internet page of the sequence service provider, pay special attention to the recommended DNA concentration for the samples. Obtain the concentration of samples using a DNA quantification instrument.
Sequence candidate chimeric constructions with a sequencing service provider10.
Assemble sequencing reads with an in silico DNA assembler11.
Align assembled versus in silico-designed chimeric sequences using sequence alignment tools9,12. Verify that the chimeric gene was fused successfully.
3. Making Preparations
NOTE: All steps involving living cells should be performed in a clean laminar flow hood with the Bunsen burner on.
- Producing E. coli DH5α-competent cells.
- To produce E. coli-competent cells, follow published protocols13 or see the Supplementary Materials. Alternatively, use commercially available competent cells.
- Transforming E. coli DH5α-competent cells.
- For the chemical transformation of E. coli cultures, follow published protocols13 or see the Supplementary Materials. Alternatively, use electroporation for bacterial transformation. If commercially available competent cells are used, follow the manufacturer's guidelines.
- Purifying genomic and plasmid DNA from E. coli DH5α.
- Perform nucleic acid extraction, as stated in the Supplementary Materials, using commercially available kits or following published protocols13.
- Perform colony PCR.
- Use this technique to identify candidate transformant colonies for subsequent sequencing reaction. NOTE: This method does not represent a final verification of the integrity of the sequences. A detailed description of this technique is available in the Supplementary Materials.
- Preparing the solutions.
- See Table 1 for more information about solution preparation.
- Download the scripts and sequence files required for the CAPRRESI protocol.
- Download gene bank files containing the complete genome sequence of the desired species, extract the DNA sequence of the chosen gene using a genome browser, and save it in fasta file format. Download the Perl scripts required for the CAPRRESI protocol. Store the scripts and the sequence files in the same directory.
Representative Results
Figure 1 depicts CAPRRESI. Using this method, two chimeric genes were assembled by exchanging the domains of two bacterial primary sigma factors (i.e., E. coli RpoD and R. etli SigA). The DNA sequences of the rpoD and sigA genes were obtained using the Artemis Genome Browser14 from GenBank genome files NC_000913 and NC_007761, respectively. The DNA sequence of the pUC18 vector was obtained from the nucleotide database of the NCBI server. In silico analyses of restriction enzyme sites were done on DNA sequences by running the REsearch.pl Perl script (see step 1.2.2). These results were used for oligonucleotide design (step 1.2.3). External oligonucleotides included recognition sites for XbaI and KpnI restriction enzymes to clone wildtype genes into the pUC18 multiple cloning site. The forward oligonucleotide also included a ribosome binding site (Table 2). Genomic DNA was extracted from E. coli DH5α cells grown in LB/Nal broth (37 °C, 220-250 rpm) and R. etli CFN42 cells grown in PY/Nal/CaCl2 broth (30 °C, 220-250 rpm). These samples were used as DNA templates for wildtype gene amplification (step 1.5.2).
Wildtype genes were amplified using a Taq high-fidelity DNA polymerase, which was purified from agarose gels (DNA purification kit), double-digested with KpnI-XbaI restriction enzymes, and cloned into the pUC18 vector. Chemically competent E. coli DH5α cells were transformed with 5 µL of the ligation reaction corresponding to wildtype constructions pUC18rpoD and pUC18sigA (step 1.5.5). Transformant cells were selected on solid LB/Amp/X-gal/IPTG plates grown at 37 °C. Wildtype constructions were verified by Sanger sequencing10. Sanger sequence reads were in silico assembled using MIRA11 (step 1.5.8).
Amino acid sequences from wildtype genes were obtained by running the translate.pl script (step 1.3.1). After aligning wildtype genes with Clustal12, the amino acid regions TLV and NLR were selected on the ampicillin resistance gene (ampR) and wildtype primary sigma factors, respectively (step 1.3.3). These amino acid regions were used for calculating all the possible DNA synonym sequences that encode them by running the synonym.pl script (step 1.4.1). The REsynonym.pl script searched for restriction enzyme sites found on the previously generated synonym DNA sequences (step1.4.2). Those synonym regions that introduced the lowest number of changes as compared to the wildtype sequences were selected. For the primary sigma factor genes, wildtype sequences of RpoD (AACTTACGT) and SigA (AACCTTCGC) were exchanged for AACTTAAGG. The latter included an AflII site (bold). For the ampR gene, the wildtype sequence ACGCTGGTG was exchanged for its synonym, ACACTAGTG. The synonym DNA sequence inserted into the ampR gene contained a SpeI site (bold). The in silico substitution of the wildtype for the corresponding synonym sequences were done to verify the sequence integrity and the oligonucleotide design (steps 1.2-1.4).
Synonym sequence changes were introduced by PCR using the appropriate oligonucleotides (Table 2) and wildtype constructions as DNA templates (step 1.5.2). Amplification reactions produced four complementary parts: pUC18rpoDσ1-σ2, pUC18rpoDσ3-σ4, pUC18sigAσ1-σ2, and pUC18sigAσ3-σ4. The complementary parts disrupted not only sigma factors, but also the ampR genes. Complementary parts were purified using a DNA purification kit (step 1.6.3).
These complementary DNA fragments were double-digested with AflII and SpeI restriction enzymes (step 1.6.4). According to CAPRRESI design, DNA fragments pUC18rpoDσ1-σ2 was ligated with pUC18sigAσ3-σ4 to obtain chimera 01, and pUC18sigAσ1-σ2 was ligated with pUC18rpoDσ3-σ4 to assemble chimera 02 (steps 1.7.1-1.7.3). Chemically competent E. coli DH5α cells were transformed with 5 µL of the ligation reaction of pUC18chim01 and pUC18chim02 (step 1.7.4). Transformant cells were selected on solid LB/Amp/X-gal/IPTG plates grown at 37 °C (step 1.7.5). Chimeric constructions were verified by Sanger sequencing10 (step 2). Sanger sequence reads were assembled using MIRA11, and its resulting files are listed in Table 3. Figure 2 shows the alignments (performed using MUSCLE)9 of assembled sequences of wildtype and chimeric genes at breaking regions.
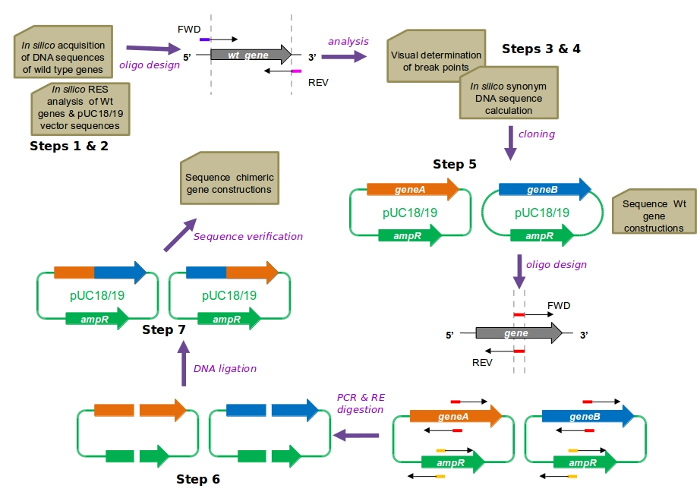 Figure 1: CAPRRESI overview. Representations: genes (thick arrows), functional plasmids (closed circles), and oligonucleotides (thin, black-tipped arrows). Restriction enzyme sites inserted into oligonucleotides appear in colors. Abbreviations: Wt (wildtype), FWD (forward oligonucleotide), REV (reverse oligonucleotide), ampR (ampicillin resistance gene), RES (restriction enzyme site), RE (restriction enzyme). Please click here to view a larger version of this figure.
Figure 1: CAPRRESI overview. Representations: genes (thick arrows), functional plasmids (closed circles), and oligonucleotides (thin, black-tipped arrows). Restriction enzyme sites inserted into oligonucleotides appear in colors. Abbreviations: Wt (wildtype), FWD (forward oligonucleotide), REV (reverse oligonucleotide), ampR (ampicillin resistance gene), RES (restriction enzyme site), RE (restriction enzyme). Please click here to view a larger version of this figure.
 Figure 2: Alignment of assembled wildtype and chimeric gene sequences. Assembled DNA sequences were aligned using MUSCLE9. Sequences were assembled with MIRA11. Synonym sequences appear in black. The AflII recognition site appears underlined. IUPAC nucleotide code: R (A or G) and K (G or T). Constructions: rpoD (red), sigA (blue), chimera 01 sequence (rpoDσ1-σ2-sigAσ3-σ4), and chimera 02 sequence (sigAσ1-σ2-rpoDσ3-σ4). Please click here to view a larger version of this figure.
Figure 2: Alignment of assembled wildtype and chimeric gene sequences. Assembled DNA sequences were aligned using MUSCLE9. Sequences were assembled with MIRA11. Synonym sequences appear in black. The AflII recognition site appears underlined. IUPAC nucleotide code: R (A or G) and K (G or T). Constructions: rpoD (red), sigA (blue), chimera 01 sequence (rpoDσ1-σ2-sigAσ3-σ4), and chimera 02 sequence (sigAσ1-σ2-rpoDσ3-σ4). Please click here to view a larger version of this figure.
| Solution | Components | Instructions | |
| Quantity | Compound | ||
| Luria-Bertani (LB) broth | 5 g | yeast extract | Dissolve all components in water. Autoclave. Store at room temperature until use. For solid LB broth, add 15 g of bacteriological agar to the recipe. |
| 10 g | casein peptone | ||
| 10 g | NaCl | ||
| up to 1 L | double distilled water | ||
| LB/Amp/X-gal/IPTG plates | 100 mL | melted solid LB broth | Add all components to temperate LB broth. Fill Petri dishes and wait until they solidify. Perform steps in a clean laminar flow hood with the Bunsen burner on. |
| 100 μL | ampicillin (Amp) [100 mg/mL] | ||
| 100 μL | X-gal [20 mg/ml] | ||
| 50 μL | 1 M IPTG solution | ||
| peptone yeast (PY) broth | 3 g | yeast extract | Dissolve all components in water. Autoclave. Store at room temperature until use. For solid PY broth, add 15 g of bacteriological agar to the recipe. For R. etli culture, add 700 μL of 1 M CaCl2 solution prior to use. |
| 5 g | casein peptone | ||
| up to 1 L | double distilled water | ||
| PY/CaCl2/Nal plates | 100 mL | melted solid PY broth | Add all components to temperate PY broth. Fill the Petri dishes and wait until they solidify. Perform steps in a clean laminar flow hood with the Bunsen burner on. |
| 700 μL | 1 M CaCl2 solution | ||
| 100 μL | nalidixic acid (Nal) [20 mg/mL] | ||
| 1 M calcium chloride (CaCl2) solution | 11.1 g | CaCl2 | Dissolve CaCl2 in water. Autoclave. Store at room temperature. |
| up to 100 mL | double distilled water | ||
| Tris-EDTA (TE) 10:1 pH 8.0 | 1 mL | 1 M Tris solution | Mix all components. Autoclave. Store at room temperature. |
| 400 μL | 0.25 M EDTA solution | ||
| up to 100 mL | double distilled water | ||
| TE 50:20 pH 8.0 | 5 mL | 1 M Tris solution | Mix all components. Autoclave. Store at room temperature. |
| 8 mL | 0.25 M EDTA solution | ||
| up to 100 mL | double distilled water | ||
| TE 10:1 10 mM NaCl solution | 58.4 mg | NaCl | Dissolve NaCl in TE 10:1. Autoclave. Store at room temperature. |
| up to 100 mL | TE 10:1 pH 8.0 solution | ||
| lysis solution I | 80 mg | lysozyme | Dissolve and mix all components in water. Filter sterilize using a 0.22 μm filter. Store at room temperature. |
| 2 mL | 0.5 M glucose solution | ||
| 400 μL | 0.5 M EDTA solution | ||
| 500 μL | 1 M Tris solution | ||
| up to 20 mL | sterilized double distilled water | ||
| lysis solution II | 1.2 mL | 5 M sodium hydroxide (NaOH) solution | Dissolve all components in water. Filter sterilize using a 0.22 μm filter. Store at room temperature. |
| 3 mL | 10% sodium dodecyl sulfate (SDS) solution | ||
| up to 30 mL | sterilized double distilled water | ||
| lysis solution III | 29.4 g | potassium acetate (CH3COOK) | Dissolve and mix all components in water. Filter sterilize using a 0.22 μm filter. Store at room temperature. |
| 11.5 mL | absolute acetic acid (CH3COOH) | ||
| up to 100 mL | sterilized double distilled water | ||
| 1 M magnesium chloride (MgCl2) solution | 9.5 g | MgCl2 | Dissolve MgCl2 in water. Autoclave. Store at room temperature. |
| up to 100 mL | double distilled water | ||
| 3 M sodium acetate (CH3COONa) solution | 24.6 g | CH3COONa | Dissolve CH3COONa in water. Autoclave. Store at room temperature |
| up to 100 mL | double distilled water | ||
| 10% SDS solution | 10 g | SDS | Dissolve SDS in water with a magnetic stirring bar at low velocity. Autoclave. Store at room temperature. |
| up to 100 mL | double distilled water | ||
| 3 M CH3COOK solution | 29.4 g | CH3COOK | Dissolve CH3COOK in water. Filter sterilize using a 0.22 μm filter. Store at room temperature. |
| up to 100 mL | sterilized double distilled water | ||
| proteinase K solution | 5 mg | proteinase K | Dissolve proteinase K in TE 50:20 solution. Heat at 37 ºC for 1 h. Store at -20 ºC. |
| up to 1 mL | TE 50:20 pH 8.0 solution | ||
| RNase solution | 10 mg | RNase | Dissolve RNase in water. Heat at 95 ºC for 10 min. Store at -20 ºC. |
| up to 1 mL | sterilized ultrapure water | ||
| 1 M isopropyl-β-D-thiogalactoside (IPTG) solution | 238.3 mg | IPTG | Dissolve IPTG in water. Filter sterilize using a 0.22 μm filter. Store at -20 ºC. |
| up to 1 mL | sterilized ultrapure water | ||
| 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal) solution | 20 mg | X-gal | Dissolve X-gal in water. Filter sterilize using a 0.22 μm filter. Store at -20 ºC. |
| up to 1 mL | sterilized ultrapure water | ||
| phenol:chloroform:isoamyl alcohol 24:24:1 solution | 24 mL | phenol | Mix all components. Store in an amber capped bottle at 4 ºC. CAUTION! Hazardous material. Wear protective glasses, gloves and cotton laboratory gown. Use an extraction hood. DO NOT turn on the Bunsen burner or any other source of fire during handling. Alternative: use commercial phenol:chloroform:isoamyl alcohol 25:24:1 solution |
| 24 mL | chloroform | ||
| 1 mL | isoamyl alcohol | ||
| 0.5 M glucose solution | 9 g | glucose | Dissolve glucose in water. Filter sterilize using a 0.22 μm filter. Store at room temperature. |
| up to 100 mL | sterilized ultrapure water | ||
| 0.5 M ethylenediaminetetraacetic acid (EDTA) pH 8.0 solution | 14.6 g | EDTA | Dissolve EDTA in water. Adjust pH with 5 M NaOH solution. Autoclave. Store at room temperature. |
| up to 100 mL | double distilled water | ||
| 0.25 M EDTA pH 8.0 solution | 7.3 g | EDTA | Dissolve EDTA in water. Adjust pH with 5 M NaOH solution. Autoclave. Store at room temperature. |
| up to 100 mL | double distilled water | ||
| 1 M Tris pH 8.0 solution | 12.1 g | Tris | Dissolve Tris in water. Adjust pH with absolute hydrochloric acid (HCl). Autoclave. Store at room temperature. |
| up to 100 mL | double distilled water | ||
| 5 M sodium hydroxide (NaOH) solution | 19.9 g | NaOH | Dissolve NaOH in water. Store at room temperature. CAUTION! Caustic compound. |
| up to 100 mL | sterilized double distilled water | ||
| 0.1 M NaOH solution | 399 mg | NaOH | Dissolve NaOH in water. Store at room temperature. |
| up to 100 mL | sterilized double distilled water | ||
| nalidixic acid (Nal) stock solution | 60 mg | nalidixic acid | Dissolve Nal in water. Filter sterilize using a 0.22 μm filter. Store at 4 ºC. |
| up to 3 mL | 0.1 M NaOH solution | ||
| ampicillin (Amp) stock solution | 300 mg | ampicillin | Dissolve Amp in water. Filter sterilize using a 0.22 μm filter. Store at 4 ºC. |
| up to 3 mL | sterilized double distilled water | ||
| 10x tris-acetate-EDTA buffer | 48.4 g | Tris | Dissolve all components in water. Autoclave. Store at room temperature. |
| 20 ml | 0.5 M EDTA solution | ||
| 11.44 ml | glacial acetic acid | ||
| up to 1 L | double distilled water |
Table 1: Solution preparation. Instructions for solution preparation.
| No. | Code | Sequence | Length (nt) | Features |
| 1 | RpoD-FWD | GCTCTAGAGAAGGAGATATCATATGGAGCAAAACCCGCAGTCA | 43 | XbaI site; wild type gene amplification and vector cloning |
| 2 | RoD-REV | GGGGTACCTTAATCGTCCAGGAAGCTACG | 29 | KpnI site; wild type gene amplification and vector cloning |
| 3 | SigA-FWD | GCTCTAGAGAAGGAGATATCATATGGCAACCAAGGTCAAAGAG | 43 | XbaI site; wild type gene amplification and vector cloning |
| 4 | SigA-REV | GGGGTACCTTAGCTGTCCAGAAAGCTTCT | 29 | KpnI site; wild type gene amplification and vector cloning |
| 3 | AmpR-FWD | ACACTAGTGAAAGTAAAAGAT | 21 | SpeI site inserted, synonym mutation. Disruption of ampR gene |
| 4 | AmpR-REV | TCACTAGTGTTTCTGGGTGAG | 21 | SpeI site inserted, synonym mutation. Disruption of ampR gene |
| 5 | σ2RpoD-FWD | AACTTAAGGCTGGTTATTTCTATCGCT | 27 | AflII site inserted, synonym mutation. Construction of chim01-02 |
| 6 | σ2RpoD-REV | GCCTTAAGTTCGCTTCAACCATCTCTT | 27 | AflII site inserted, synonym mutation. Construction of chim01-02 |
| 7 | σ2SigA-FWD | AACTTAAGGCTCGTCATTTCAATCGCC | 27 | AflII site inserted, synonym mutation. Construction of chim01-02 |
| 8 | σ2SigA-REV | GCCTTAAGTTCGCTTCGACCATTTCCT | 27 | AflII site inserted, synonym mutation. Construction of chim01-02 |
Table 2: Oligonucleotide sequences. Restriction enzyme sites appear underlined. Ribosome binding site: bold.
| Construction | Sequence file | Quality file |
| chimera01 | chim01_out.unpadded.fasta | chim01_out.unpadded.fasta.qual |
| chim01_A3/B3/E2/F2/G2/H2.pdf | ||
| chimera02 | chim02_out.unpadded.fasta | chim02_out.unpadded.fasta.qual |
| chim02_C3/D3/E3/F3/G3/H3.pdf | ||
| rpoD | rpod_out.unpadded.fasta | rpod_out.unpadded.fasta.qual |
| sigA | siga_out.unpadded.fasta | siga_out.unpadded.fasta.qual |
Table 3: Sequence files obtained with the MIRA assembler and DNA sequencer. Sanger sequencing reads from chimeric and wildtype constructions were assembled with MIRA11 software using the mapping option. Chromatograms of chimera sequencing appear as PDF files.
Discussion
CAPRRESI was designed as an alternative to the PRM2. The original PRM is a powerful technique; it allows for the fusion of DNA sequences along any part of the selected genes. For PRM, wildtype genes should be cloned into the same plasmid. After that, oligonucleotides are designed inside wildtype and antibiotics resistance genes found on the plasmid. Plasmid disruption is achieved by PCR using blunt-ended, high-fidelity DNA polymerases and previously designed oligonucleotides. The ligation of complementary PCR products assembles chimeric genes and restores the antibiotics resistance cassette, resulting in functional plasmid recovery. PRM enables the construction of chimeric gene libraries on the same plasmid background. Unfortunately, the ligation of blunt-ended DNA sequences could be technically arduous. Chimera assembly by overlapping PCR fragments1 also requires that DNA polymerases yield blunt-ended products and DNA purification for each reaction. The chimeric genes assembled by this technique are linear DNA products. The cloning of each construction into the target vector could delay further analysis.
CAPRRESI benefits from many of the strengths of PRM and also simplifies the ligation of complementary DNA fragments by using restriction enzyme sites on synonym DNA sequence regions. For these reasons, CAPRRESI does not require blunt-ended DNA polymerases. The use of synonym DNA sequences restrains fusion sites for chimera assembly but enhances ligation efficiency. Another important modification introduced in CAPRRESI is that chimeras are assembled and handled in pUC18/19 vectors. These vectors possess several advantageous features, as stated previously. Chimeric DNA could be obtained by propagating the construction vector in E. coli hosts. Subsequent cloning of chimeric genes into the final plasmid (e.g., an expression vector) is sometimes required.
CAPRRESI also relies on the in silico handling of DNA and amino acid sequences. Ad hoc Perl scripts enable the selection of the appropriate restriction enzymes to clone wildtype genes, the calculation of all synonym DNA sequences corresponding to break regions (for chimera assembly), and the identification of restriction enzymes for simplifying plasmid recovery. Given these conditions, CAPRRESI is an alternative for fusing protein-coding genes. Critical steps of the CAPRRESI protocol are: 1) the selection of breaking regions and 2) the purification of PCR products. Breaking regions are stretches where synonym sequences are calculated, producing restriction enzyme sites that are not found on wildtype sequences. The use of restriction enzyme sites for chimera assembly eliminates the need for DNA polymerases that yield blunt-ended products and eases the ligation reaction. Not all regions will have usable restriction enzyme sites; for this reason, it is recommended to move breaking regions by one amino acid at a time until the best sequence stretch is found. If the in silico design does not allow for the movement of the breaking regions or the sequence stretches lack synonym DNA sequences with suitable restriction enzyme sites, it is recommended to fuse target genes using overlapping oligonucleotides and to introduce synonym DNA sequences into the ampicillin resistance gene (found on the vector) only. In this way, the genes can be fused at any part and the plasmid recovery relies on the ligation of one unique site (digested with only one restriction enzyme). The flexibility of CAPPRESI allows for it to be performed in combination with other techniques.
The purification of PCR products is performed to eliminate template DNA, decreasing the chances of parental plasmid contamination after the ligation of complementary DNA fragments. Fulfilling these two critical steps enhances bacterial transformation efficiency (the number of correctly assembled constructions), allowing CAPRRESI to be performed with either chemically competent (CaCl2) or electrocompetent E. coli cells. The first type of competent cells represents the cheapest source of E. coli cultures, employed for bacterial transformation in most laboratories. Because of all the features shown previously, CAPRRESI represents a useful option for assembling chimeras.
Moreover, CAPRRESI could work in combination with overlapping PCR fragments or plasmid recovery techniques. For example, instead of inserting two synonym sequences per complementary DNA portion, the desired breaking region (on the target wildtype genes) could use overlapping oligonucleotides to fuse intervening sequences. The introduction of synonym sequences could be restricted to the ampR gene of pUC18/19 vectors, leading to the use of only one restriction enzyme to restore plasmid integrity. These future applications could strengthen CAPRRESI by allowing for the fusion of any type of DNA sequences (i.e., not only protein-coding genes), without compromising ligation efficiency.
In order to test CAPRRESI, two chimeric sigma factors were assembled by exchanging regions of two wildtype primary sigma factor genes: rpoD (E. coli) and sigA (R. etli). All known primary sigma factors hold four domains, σ1 to σ48. Chimera 01 consists of RpoDσ1-σ2 and SigAσ3-σ4, while chimera 02 has SigAσ1-σ2 and RpoDσ3-σ4. The integrity of the chimeric constructions was verified by Sanger sequencing10 (Figure 2).
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
This work was supported by Consejo Nacional de Ciencia y Tecnología, CONACYT, México (grant number 154833) and Universidad Nacional Autónoma de México. The authors wish to thank Víctor González, Rosa I. Santamaría, Patricia Bustos, and Soledad Juárez for their administrative and technical advice.
References
- Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77(1):61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- Vos MJ, Kampinga HH. A PCR amplification strategy for unrestricted generation of chimeric genes. Anal. Biochem. 2008;380(2):338–340. doi: 10.1016/j.ab.2008.05.031. [DOI] [PubMed] [Google Scholar]
- Hawkins NC, Garriga G, Beh CT. Creating Precise GFP Fusions in Plasmids Using Yeast Homologous Recombination. Biotechniques. 2003;34(1):1–5. doi: 10.2144/03341st03. [DOI] [PubMed] [Google Scholar]
- Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32(4):347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy A. Cre recombinase: The universal reagent for genome tailoring. Genesis. 2000;26(2):99–109. [PubMed] [Google Scholar]
- Gibson DG, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods. 2009;6(5):343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- Norrander J, Kempe T, Messing J. Construction of improved M13 vectors using oligodeoxynucleotide-directed mutagenesis. Gene. 1983;26(1):101–106. doi: 10.1016/0378-1119(83)90040-9. [DOI] [PubMed] [Google Scholar]
- Gruber TM, Gross CA. Multiple sigma subunits and the partitioning of bacterial transcription space. Annu Rev Microbiol. 2003;57:441–466. doi: 10.1146/annurev.micro.57.030502.090913. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macrogen Inc. Seoul, Rep. Of Korea: 2016. Available from: http://dna.macrogen.com/eng/index.jsp. [Google Scholar]
- Chevreux B. MIRA: an automated genome and EST assembler. 2005. pp. 1–161. Available from: http://www.chevreux.org/thesis/index.html.
- Thompson JD, Gibson TJ, Higgins DG. Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinformatics. 2002. pp. 1–22. Chapter 2 (Unit 2.3) [DOI] [PubMed]
- Sambrook J, Russell DW. Molecular Cloning: A laboratory manual. CSHLP. 2001.
- Rutherford K, et al. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16(10):944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]