

Abstract
Neurodegenerative diseases often have a devastating impact on those affected. Retinal ganglion cell (RGC) loss is implicated in an array of diseases, including diabetic retinopathy and glaucoma, in addition to normal aging. Despite their importance, RGCs have been extremely difficult to study until now due in part to the fact that they comprise only a small percentage of the wide variety of cells in the retina. In addition, current isolation methods use intracellular markers to identify RGCs, which produce non-viable cells. These techniques also involve lengthy isolation protocols, so there is a lack of practical, standardized, and dependable methods to obtain and isolate RGCs. This work describes an efficient, comprehensive, and reliable method to isolate primary RGCs from mice retinae using a protocol based on both positive and negative selection criteria. The presented methods allow for the future study of RGCs, with the goal of better understanding the major decline in visual acuity that results from the loss of functional RGCs in neurodegenerative diseases.
Keywords: Bioengineering, Issue 125, retinal ganglion cells, RGC, flow cytometry, neurodegeneration, glaucoma, aging
Introduction
RGCs are terminally differentiated neurons, and therefore, primary cells are required for experimentation. The development of a protocol for the isolation and enrichment of primary murine retinal ganglion cells (RGCs) is fundamental to revealing the mechanisms of RGC health and degeneration in vitro. This is especially important for studies that seek to generate potential therapies to promote RGC function and to minimize their death. The degeneration of RGCs is associated with retinal degenerative diseases, such as glaucoma, diabetic retinopathy, and normal aging. Although the specific cellular mechanisms underlying RGC loss are unclear, a series of risk factors have been identified. Lack of oxygenation at the optic nerve head1,2,3 causes RGC death4 and acts as the disturbance of the homeostasis between the activation of excitatory and inhibitory receptors within individual RGCs5,6. A series of challenges impede progress towards the use of these cells for in-depth studies. First, the number of RGCs present in a murine retina is small. RGCs account for less than 1% of total retinal cells7,8,9. Second, most RGC-specific markers are intracellular proteins10,11,12. Selection based upon these markers leaves the cells non-viable, which precludes downstream functional analyses. Finally, currently available protocols are lengthy and lack standardization13,14. Early RGC isolation protocols were based on immunopanning methods. Barres et al.15adapted the classic immunopanning technique and added a second step, which excluded monocytes and endothelial cells from the bulk of retinal cells prior to positive selection based upon immunopositivity to anti-thymocyte antigen (aka Thy1), a cell-surface marker. Years later, Hong et al. combined magnetic bead isolation techniques with cell sorting strategies to isolate RGCs with higher purity16. The use of magnetic beads is still used in many scientific applications. Together, magnetic beads and flow cytometry protocols improved the purity of isolated cells. However, these purification systems have not yet been standardized for the isolation of murine RGCs from dissociated retinae.
Flow cytometry is a powerful analytical method that measures the optical and fluorescence characteristics of cell suspensions. Cells are analyzed both quantitatively and qualitatively with a high level of sensitivity, providing a multi-dimensional analysis of the cell population. Cellular discrimination is based upon two main physical properties: cell size or surface area and granularity or internal complexity17. A multi-dimensional analysis can be performed by combining antibodies tagged with fluorochromes that have similar excitation wavelengths and different emissions. Flow cytometry is fast, reproducible, and sensitive. Multitpe lasers permit even greater multi-dimensional analyses of single cells by flow cytometry. Thus, it is an attractive methodology for the study of cytological specimens. Fluorescence activated cell sorting (FACS) uses the multi-dimensional phenotypic differences identified by flow cytometry to sort individual cells into distinct subpopulations.
In the last decade, multiple surface and intracellular proteins have been identified as potential biomarkers for the selection of cells, including neurons. Initial studies that sought to isolate RGCs from rats used Thy1 as a ganglion cell marker. Unfortunately, Thy1, aka CD90, has multiple isoforms in other rodent species18,19,20 and is expressed by multiple retinal cell types19,20, making it a non-specific marker for RGCs. Another surface marker, CD48, is found on monocytic populations in the retina, including macrophages and microglia. Using these two surface markers, a modified RGC signature-Thy1+ and CD48neg cells-was developed15,16,21,22. Unfortunately, these two selection criteria are not sufficient to select for a highly enriched RGC population. To address this unmet need, a flow cytometry protocol was developed23 based on multi-layered positive and negative selection criteria using known cell surface markers to enrich and purify primary murine RGCs.
Protocol
All procedures detailed in the following protocol were approved by the Institutional Animal Care and Use Committee (IACUC) review board at the University of Tennessee Health Science Center (UTHSC) and followed the Association for Research in Vision and Ophthalmology (ARVO) Statements for the Use of Animals in Ophthalmic and Vision Research, in addition to the guidelines for laboratory animal experiments (Institute of Laboratory Animal Resources, Public Health Service Policy on Humane Care and Use of Laboratory Animals).
1. Preparation of Instruments, Solutions, and Media
Note: All information about materials, reagents, tools, and instruments reported in the protocol are specified in the Table of Materials.
Autoclave all dissection instruments and store them in a sterile area. Use the following instruments: 4 standard forceps (2 long and 2 short) and 2 scissors, as well as 2 forceps (1 long and 1 short) and 1 scissor for the dissection; keep an extra set as a backup.
- Prepare 100 mL of sterile PBS/1% FBS solution to use during washes, immunolabeling procedures, and cell sorting steps. Keep the solution chilled at 4 °C. Note: Do not add sodium azide (NaN3) to the solution, as it can be toxic to live cells.
- Prepare 100 mL of PBS/1% FBS with 99 mL of PBS and 1 mL of FBS.
- Prepare 100 mL of sterile neural cell medium supplemented with 3% FBS (see the Table of Materials) for use as collection and culture medium. Keep cell culture medium sterile at 4 °C. Only warm it to room temperature (RT) prior to use.
- Prepare 100 mL of neural cell medium supplemented with 3% FBS using 97 mL of neural cell medium and 3 mL of FBS.
Pre-chill collection tubes (15-mL tubes) pre-coated with 5 mL of collection medium by placing them in an ice bucket. Only use polypropylene tubes to prevent the cells from adhering to the tube surface.
Place 40-mm dishes, 70-µm nylon strainers, syringes, pestles for the cell strainer, and sterile polypropylene tubes in the biosafety cabinet. Sterilize all items prior to the procedures and maintain them in a sterile manner throughout the procedures. Note: All steps after the collection of the retinae will be performed in the biosafety cabinet.
2. Enucleation
Note: A total of 10 young (5-7 weeks old) C57BL/6J mice were used in this experiment to isolate 1.0 x 106 RGCs with the phenotype CD90.2+CD48negCD15negCD57neg.
Euthanize the mice using CO2 inhalation followed by cervical dislocation. Always use a secondary euthanasia method to ensure that the euthanized animal is dead. Note: Isofluorane can be used as an alternative to CO2. Ketamine is not recommended, as it is associated with an increase in the intraocular pressure (IOP) when used as an inducer of anesthesia24,25.
Insert forceps under the globe of the eye, grip the optic nerve, and pull up; the globe will be enucleated, with the optic nerve intact. Place collected eyes in a vial containing PBS and keep the vial on ice until the next step. Note: Any personnel can carry out this step after receiving the appropriate training from the Laboratory Animal Care Unit (LACU).
3. Preparation of the Retinal Cell Suspension
Place the collected eye on the base plate of a dissection microscope to begin the corneal dissection. Dissect each eye individually.
Carefully hold the globe at the optic nerve base using forceps. For this step, use one long, and one short standard forceps.
Use a 30-gauge (30G) sharp needle to puncture the cornea to allow the aqueous humor to evacuate from the eye, making it easier to hold the eye with the forceps.
Hold the cornea with the forceps and use scissors to make a small incision in the cornea. Gently peel the cornea and sclera using the forceps. When the globe is peeled halfway, roll the retina and lens out using the forceps. Discard the cornea, sclera, and lens. Note: This method ensures that the retina completely detaches from the remainder of the eye.
- Place the retina in a small 40-mm Petri dish containing PBS/1% FBS. Note: As an alternative to the Petri dish, a cell culture dish can be used. Make sure to always keep the retinae moist, as in physiological conditions.
- Wash each retina three times with fresh sterile PBS/1% FBS within the biosafety cabinet.
- Place up to 12 retinae on a sterile 70-µm nylon strainer moistened with PBS/1% FBS. Using the back-end of a 10-mL syringe, gently macerate the retinae using a circular movement to detach the cells.
- Alternatively, use a pestle for the cell strainer maceration or use enzymatic digestion with a combination of 15 IU/mL papain, 5 mM L-cysteine, and 200 U/mL DNase I for 15 min at 37 °C, followed by inactivation with PBS/10% FBS. Note: No changes in the percentage of cells recovered were observed when enzymatic digestion was compared to maceration with either the back-end of the syringe or the pestle.
To transfer the isolated cells, place the sterile 70-µm nylon strainer over the polypropylene collection tube. Pass the collected cells through the strainer using a P1000 pipette. Rinse the strainer (step 3.6) with PBS/1% FBS to release any remaining cells and transfer them to the collection tube.
- Add PBS/1% FBS to achieve a final volume of 1 mL per retina. Centrifuge the cell suspension for 7 min at 200 x g and RT. Note: Depending on the number of macerated retinae (< 6 retinae), the cell pellet may be too small to be visible.
- Discard the cell supernatant and resuspend the cell pellet in PBS/1% FBS using a ratio of 1 mL per 5 retinae.
- Count the cells using a hemocytometer.
- Clean a glass hemocytometer and coverslip with 70% alcohol. Gently swirl the tube containing the cells to ensure that the retina cell suspension is evenly distributed. Place 20 µL of 0.4% trypan blue in a microcentrifuge tube and mix with 20 µL of the cell suspension.
- Mix gently and apply 10 µL of the 0.4% trypan blue/cell suspension mix to the hemocytometer, filling both chambers; capillary action will draw the 0.4% trypan blue/cell suspension mix under the coverslip. Using a microscope, count all trypan blue-negative cells; this number represents the live cells.
- Determine cell viability using the following formula: live cell count / total cell count = % viability. Note: If the viability of the cells is less than 95%, the use of fluorescent dye for cell viability discrimination is required during FACS.
Use a fluorescent viability dye that is non-permeant to live cells. Wash the cells with PBS to remove any trace of serum. Add 1 µL of fluorescent dye for cell viability discrimination per 5.0 x 106 cells in a 100-µL final volume. Incubate at RT for 15 min, protected from light. Add 10x the volume of PBS/1% FBS. Centrifuge for 5 min at 200 x g and RT.
Incubate the cell suspension overnight at 4 °C, if necessary. If this is done, fill up the cell suspension tube with neural cell medium rather than PBS/1% FBS. Place the tube horizontally and keep it at 4 °C.
4. Immunolabeling the Retinal Cells
Use the following conversion to determine the volume of antibody per cell number: 2 µL of antibody per 5.0 x 106 cells in a 100-µL volume.
Wash the cells and maintain them in PBS/1% FBS. Take a small aliquot of cells (5.0 x 106 cells) to use as a negative control (unlabeled) at the time of the sort setup. Note: This negative control is critical for the proper calibration of the cell sorter.
To minimize the non-specific binding of antibodies to cells that express the Fcγ receptors II and III, add 1 µL of an anti-mouse CD16/32 antibody per 1.0 x 106 cells in a 50-µL final volume using PBS/1% FBS. Incubate for 10 min at RT.
- Add the antibody cocktail to the sample and mix gently by pipetting. Incubate for 30 min in an ice bucket. Ensure that the ice bucket is covered, as light could compromise the experiment due to the photobleaching of the tagged fluorophores.
- Prepare the antibody cocktail using the following fluorescently tagged anti-mouse antibodies: CD90.2 AF-700, CD48 PE-Cyanine7, CD15 PE, and un-tagged CD57. Note: For each antibody, use the following concentrations per 5.0 x 106 cells: CD90.2 AF700, 1 µg; CD48 PE-Cyanine7, 0.4 µg; CD15 PE, 0.02 µg; and un-tagged CD57, 0.4 µg.
- Use volumes as per the following calculations (based on the commercially available antibodies listed in the Table of Materials): total of 5.0 x 107 cells to be labeled; 2 µL per 5.0 x 106 cells = 20 µL of each antibody; 4 different antibodies = 80 µL of antibody cocktail; final volume = [(5.0 x 107 total cells)/5.0 x 106 cells] x 100 = 1,000 µL ; 80 µL Abs + 50 µL of anti-mouse CD16/32 + 870 µL PBS/1% FBS = 1,000 µL. Note: This combination provided the optimal combination of fluorochromes for the instrument configuration. The following parameters summarize the emission and excitation: AF-700, emission of 719 nm when excited with a 638-nm red diode laser; PE-Cyanine7, emission of 767 nm when excited with a 488-nm blue diode laser; and PE, emission of 575 nm when excited with a 488-nm blue diode laser.
After the 30-min incubation, bring volume up to 5 mL using PBS/1% FBS. Wash the samples by centrifuging them for 7 min at 200 x g and RT. Repeat the procedure once to remove all unbound antibody.
- Add 2 µL of secondary antibody (0.1 µg), which will bind the CD57 of 5.0 x 106 cells. Incubate for 30 min in an ice bucket, as in step 4.4. Wash the cells twice, as in step 4.5.
- Label the cells with secondary antibody if an un-tagged primary antibody was used in step 4.4. Note: The secondary antibody of choice for this configuration is listed in the Table of Materials; it has an emission wavelength of 421 nm. Use it with a bandpass filter of 450/50 nm when excited with the violet diode laser at 405 nm.
- Use volumes as per the following calculations (based on the commercially available antibody listed in the Table of Materials): 2 µL per 5.0 x 106 cells = 20 µL of secondary antibody; final volume = [(5.0 x 107 total cells)/5.0 x 106 cells] x 50 = 500 µL; 20 µL of Abs + 480 µL PBS/1% FBS = 500 µL.
Keep the labeled cells in PBS/1% FBS. Count the cells using a hemocytometer; use a final retinal cell concentration of 3.0 - 4.0 x 107 cells/mL. Note: Do not use cell culture medium to dilute the cells because the phenol red can increase the autofluorescence, thereby reducing the resolution between negative and positive cells. The final cell concentration per volume highly depends upon the volume flow rate of the instrument configuration.
- Use single-color controls during setup to minimize fluorescence spillover. Note: This step, also known as compensation, is applied to correct the noise created by the combination of fluorophores. Polystyrene microspheres in PBS/0.1% BSA/2 mM NaN3 with the capacity to bind fluorescently tagged immunoglobulin (Ig) isotypes from multiple species provide positive controls to set the compensation.
- Place 3 drops of the polystyrene microspheres into sterile FACS tubes, allotting one per fluorophore. Add 1 µg of the respective fluorophore to each tube. Incubate for 15 min at RT, protected from light. Add 3 mL of PBS/1% FBS to the sample tubes. Centrifuge for 5 min at 200 x g and RT. Carefully remove the supernatant and resuspend in 250 µL of PBS/1% FBS.
- To set up the negative controls, use polystyrene microspheres that do not bind to Ig. Carry out this step the day before, if necessary.
5. Cell Sorting Strategy
Note: Specific instructions for instrument setup for FACS with 355 nm, UV; 405 nm, violet; 488 nm, blue; and 640 nm, red lasers with 2, 2, 5, and 3 fluorescence channel distribution, respectively. The operating software was DIVA version 8.0.1. Cell sorting was performed with a 70 μm nozzle, 70 psi sheath pressure, 87.5 frequency, 48.6 amplitude with first drop breakoff at 333, gap asetting of 6, sort precision set to four-way purity with default (32) purity mask, and drop delay adjusted to 42.98 using beads.
Use 15-mL polypropylene conical tubes as collection vessels. Add 5 mL of the collection medium (cell culture medium) to each tube and rotate the tube to coat the walls. Note: This step prevents the cells from sticking to the sides of the collection tube. As an alternative, tube can be coated with undiluted FBS.
Take into consideration the efficiency of the cell sorter to estimate the final yield of cells. Note: The efficiency of the sort varies based on the flow cytometer used. RGCs with the CD90.2+CD48negCD15negCD57neg phenotype comprise about 1% of all retinal cells.
Have the cell sorting performed by a trained operator (usually in a core facility) . Ensure that the instrument is cleaned and sterilized with 70% ethanol prior to sample acquisition. Carefully wipe the outside of collection tubes with 70% ethanol as well.
If the cells have settled or aggregated, pipette the cells up and down and filter the suspension through a sterile 40-µm nylon strainer prior to acquisition. Make sure to control the temperature and keep it at 4 °C while the cell sorting is being performed; failure to do so will reduce the cell yield.
Discuss the details of the sort with the cell sorter operator prior to performing the experiments. Note: At the time of experiment, the cell sorter operator will have clicked a series of buttons from the Workspace toolbar of the acquisition software for the cytometer. These are the Browser, Cytometer, Inspector, Worksheet, and Acquisition Dashboard. The strategy of cell sorting used here is known as a 2-way sort. Two populations are collected: the enriched RGCs and all other cell types. If additional populations are to be collected, up to two additional populations can be isolated as a 4-way sort. If additional populations will be collected, the cell sorter requires different collection tubes and setup.
- Perform compensation to correct for spectral overlap from the fluorophores. Note: This process can be done manually by adjusting each setting, or it can be done automatically as part of the software in use. Most software for cell sorting equipment have the option of an automated compensation. It is considered the gold standard and the most accurate type of compensation. The negative (no fluorophore) sample and the single controls prepared with polysterene microspheres (Compensation beads) specifically manufactured to bind all relevant monoclonal antibodies used in the experiment will be used in this step.
- Select Experiment > Compensation Setup > Create Compensation Controls. Add the fluorophore-specific controls from the list displayed in the screen. Click OK. Note: A compensation tube for each of the controls will be displayed.
- Use an unlabeled control (no fluorophore, step 4.2) to verify the forward-scattered light (FSC), the side-scattered light (SSC), and to gate the initial population (P1).
- Install the negative control tube onto the cytometer and click Load. Verify that the population of interest is displayed and select it. This is population 1 (P1). Right-click the P1 gate and select Apply to All Compensation Controls. Click Record Data. Once the recording is done, click Unload and remove the tube.
- Install the next tube onto the cytometer, as displayed on the screen. Repeat step 5.6.2 until all data from the controls have been recorded.
- Select Experiment > Compensation Setup > Calculate Compensation. Save the setup and name the experiment. Click Link & Save. Note: Compensation is a necessary step, as it removes the spectrum overlap between the detectors.
- If manual compensation is needed, do so by adjusting the means of the positive signals so that they equal the negatives for each of the fluorochromes used. Note: This step is done by the cell sorter operation and may take 30 min.
- Gating strategy ( Figure 3A).
- Set up P1 by plotting FSC versus SSC; the FSC is indicative of the size and the SSC indicates the internal complexity of the cells. See Figure 3A.
- Draw a pseudocolor plot of SSC-H (height) versus SSC-W (width) using the selected population in step 5.7.1 (P1); the pseudocolor plot allows for the visual representation of the density of cells relative to one another. Note: A lower cell density is represented by blue and green, whereas red and orange areas represent high cell density.
- Perform this step to collect single cells only; the single cell gate is called P2. See Figure 3A.
- Repeat step 5.7.2 to plot FSC-H versus FSC-W using P2. Ensure that the selection of single cells as "doublets" or cell clumps contains both a positive and a negative marker, providing false positivity.
- Draw a pseudocolor plot of CD90.2 versus CD48. Select all CD90.2+CD48neg cells to eliminate monocytes from the RGC enrichment; call this gate CD90.2+CD48neg, or P3. See Figure 3A.
- Using the selected CD90.2+CD48neg population or P3 gate, draw a pseudocolor plot of CD57 versus CD15 to eliminate amacrine cell contaminants from the RGC enrichment. Gate the CD15negCD57neg population. See Figure 3A.
Define the populations to be collected for the sample in the "Sort Layout" window and proceed with the cell sorting; the sample to collect is CD90.2+CD48negCD15negCD57neg. Note: The population to be sorted is selected from the drop-down Add menu in the sort window. After adding the population, it will ask for target events or how many events are to be collected. At any time, the sort layout can be edited by clicking the Sort Location field containing the population. 0.9% ± 0.3 of RGCs with the phenotype CD90.2+CD48negCD15negCD57neg are obtained from young (5-7 weeks old) and 0.5% ± 0.3 and old (>12 months old) C57BL/6J mice23.
Using 25,000 cells, perform a purity check23. See Figure 3B-E. Note: The purity check is the process to re-analyze the sorted cells to verify the accuracy of the cell sort. A small aliquot of sorted cells will be loaded onto the cytometer to verify the effectiveness of the sort.
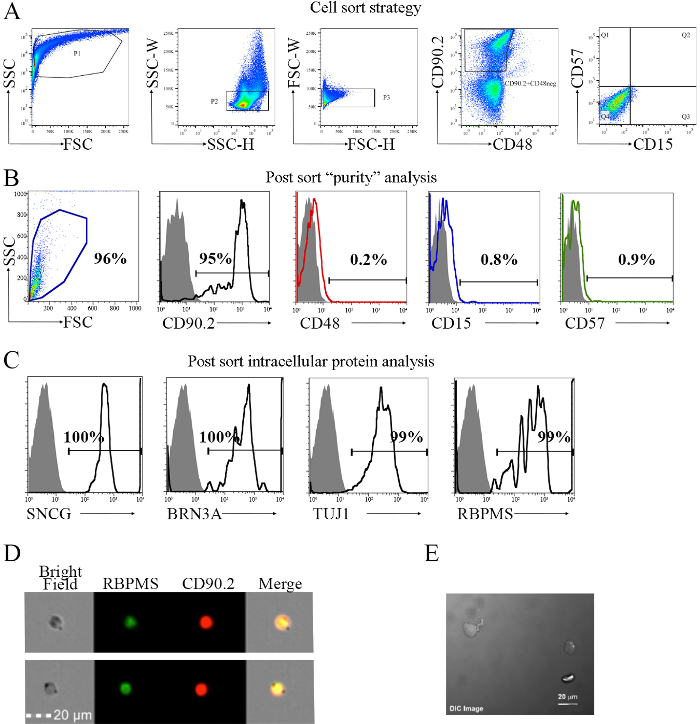
6. Confirmation on RGC Intracellular Markers
- Intracellular labeling.
- Fix sorted cells for 1 h and permeabilize at 4 °C to make the cells metabolically inactive and to allow the penetration of intracellular antibodies.
- Dilute the following antibodies in the permeabilization solution: anti-RNA binding protein with multiple splicing (RBPMS) at a 1:100 dilution in a 100 µg/mL stock; anti-synuclein gamma (SNCG), 1:100 in a 1 mg/mL stock; brain-specific homeobox/POU domain protein 3A (BRN3A), 1:100 in a 200 µg/mL stock; and anti-neuron-specific class III beta tubulin (TUJ1), 1:100 in a 1 mg/mL stock. Incubate the cells and antibody solutions for 1 h in a covered ice bucket.
- Wash the samples twice with PBS/1% FBS.
- Resuspend the cells in the appropriate AF488-tagged secondary antibody at a 1:200 dilution for 30 min in an ice bucket.
- Wash the samples as in step 6.1.3. Keep the cells in PBS/1% FBS (250 µL) until ready to analyze.
7. Validation of the Cell Sort by qPCR Analysis
Note: See Figure 4.
- RNA extraction23.
- Extract the RNA from 5.0 x 105 sorted cells by cell lysis and homogenization followed by the addition of chloroform.
- Transfer the upper, colorless phase to a clean microtube for alcohol precipitation followed by extract concentration in a spin column. Perform DNAse digestion on the column.
- Wash the column with RNase-free water for RNA elution.
- Assess the RNA concentration by spectrophotometry.
- cDNA synthesis and pre-amplification.
- Use 100 ng of RNA material and mix with a solution containing reverse transcriptase enzyme, recombinant ribonuclease inhibitor (to avoid RNA degradation), magnesium chloride (MgCl2), and deoxynucleotides.
- Mix the tube contents and incubate for 10 min at 25 °C followed by 60 min at 42 °C. Finish the reaction with a 5-min incubation at 85 °C. Store the resulting cDNA at -20 °C until ready to use. Note: cDNA material can be stored at -20 °C for up to a month if the pre-amplification step cannot be carried out immediately.
- Pre-amplification of cDNA.
- Pre-amplify cDNA material using a series of primers specific for multiple retinal cell types including: retinal ganglion cells, amacrine cells, astrocytes, Müller, bipolar, horizontal, photoreceptors, and retinal pigment epithelial cells, as detailed in Reference 23 and in the Table of Materials. Note: The pre-amplification reaction is prepared to increase the sensitivity of detection for quantification. The pre-amplification reaction contains a mixture of the primer mixes from the Table of Materials; 2.5 µL of cDNA, which started at 100 ng of RNA; reverse transcriptase enzyme; and nuclease-free water in a final volume of 10 µL.
- Carry out the enzyme activation step at 95 °C for 10 min, followed by 14 cycles at 95 °C for 15 s, followed by 60 °C for 4 min. Dilute the pre-amplified material 1:10 in Tris EDTA buffer and keep it at -20 °C until ready for use.
- 7.4 qPCR reaction.
- Prepare all qPCR reactions in a 10 µL final volume using the diluted, pre-amplified cDNA (2.5 µL), primers (listed in the Table of Materials), nuclease-free water, and a concentrate with DNA polymerase and deoxynucleotides.
- Use the following instrument conditions to run the qPCR: a hold step of 50 °C for 2 min, followed by 95 °C for 10 min. Run a total of 40 cycles at 95 °C for 15 s followed by 60 °C for 1 min. Perform all measurements in replicates of three.
- Perform relative quantification using the comparative threshold (CT) after determining the values of CT for the housekeeping gene and the target genes in each sample. Calculate the relative fold change (Rq) using the following equation: Rq = 2T –∆C, where ∆CT = CT target gene – CT reference gene23. Note: The CT is defined as the PCR cycle at which the fluorescent signal of the reporter dye crosses an arbitrarily placed threshold26. The CT is inversely related to the amount of amplicon (PCR product) in the reaction.
Representative Results
The in-depth study of RGCs is impeded by many factors, including their low frequency and the lack of a robust and standardized methodology for their isolation. Figure 1 shows the methodology used for retinae isolation. Variations in the enucleation procedure exist based on the type of analysis, such as if the enucleation is part of in vivo experimentation27. Enucleation in this protocol is performed on euthanized mice. As shown in Figure 1A-B, forceps are placed under the eye and pulled up to cause minimal bleeding and to remove an eye globe with an intact optic nerve.
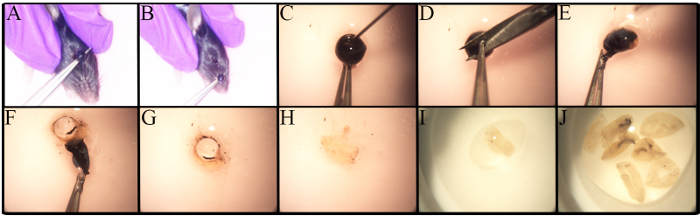
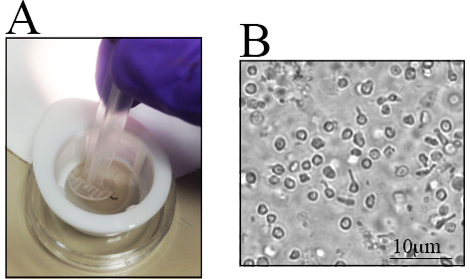
Differences exist in the number of RGCs in different mouse strains, especially in genetically altered mice23,28,29,30. Awareness of these differences is important when determining the number of mice to be used. Retinae from old C57BL/6J mice have fewer live retinal cells than their younger counterparts23. Therefore, retinal dissection must be carefully performed to maximize the cell yield. A step-by-step procedure for retinal dissection is presented in Figure 1C-J. Retinal cells are fragile. Thus, dissected retinae are placed in nylon strainers and are macerated with either the back end of a syringe, as shown in Figure 2A, or with a pestle for cell strainers. The maceration of cells directly in the cell strainer is fast and reduces cell clumps. A representative image of the cell suspension is illustrated in Figure 2B. Multiple inner retinal cells can be visualized. At this point, the RGCs have lost their signature morphology due to axotomy during cell isolation and the preparation of the cell suspension.
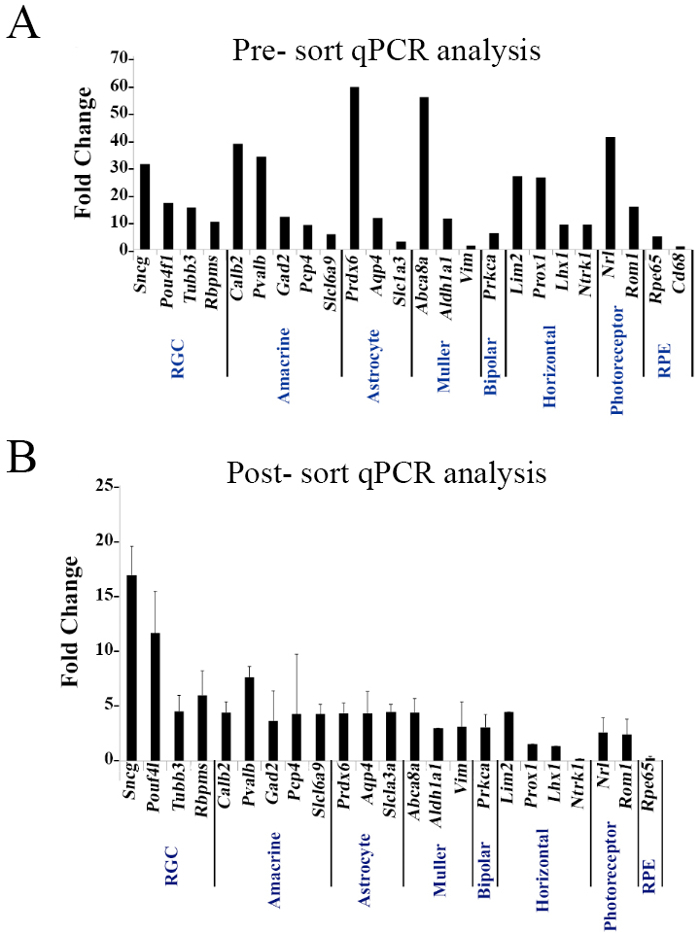
The most labor-intensive step of this methodology is the cell sorting setup. This phase is a critical step during multicolor FACS, as it maximizes signal-to-noise resolution. Figure 3A shows the gating strategy used for the isolation of RGCs. This strategy targeted the removal of contaminant cells from the cell suspension, which included monocyte, glial, amacrine, and photoreceptor cells. As part of the methodology, additional surface markers were confirmed by immunohistochemical analysis before they were used as part of the exclusion strategy. Previous data demonstrated that a small percentage of CD90.2+ cells are CD48+. Exclusion of these cells removed monocytes and possibly microglia from the retinae cell pool. It has been previously shown that the classic Thy1+CD48neg surface phenotype is not sufficient to identify and isolate murine RGCs23, as these cells express genes associated with amacrine, Müller, bipolar, horizontal photoreceptor, and retinal pigment epithelial cells (Figure 4A). This is further addressed by investigating additional markers for cell exclusion. CD15 has been described as a marker of amacrine and bipolar cells31, prompting its use as an additional marker for negative selection. Work from Uusitalo et al.32 described CD57 as an identification marker for glial cells and photoreceptors. Therefore, this antibody was added to the cell sorting strategy.
Next, these cells were characterized to validate the methodology for the isolation of murine RGCs. The phenotypes of the CD90.2+CD48negCD15negCD57neg sorted cells (Figure 3B) were evaluated for the expression of the following intracellular markers associated with RGCs10,11,12,33: SNCG, BRN3A, TUJ1, and RBPMS. As shown in Figure 3C, the sorted cells expressed all four RGC-associated intracellular proteins. Next, imaging flow cytometry was used in Figure 3D to show the intracellular localization of RBPMS and the cell surface expression of CD90.2. These results were tested in multiple cytometer systems, confirming the reproducibility and standardization. As shown in Figure 3E, some of the sorted cells began showing the morphology associated to RGCs after in vitro cell culture.
Lastly, a comparison of cells prior to enrichment and post-cell analysis was performed by qPCR analysis. Comparison of the Thy1+CD48neg phenotype to the CD90.2+CD48negCD15negCD57neg sorted cells revealed that the Thy1+CD48neg phenotype expresses genes associated with RGCs, but also with other retinal cells. However, the highly enriched sorted cell population (Figure 4B) showed a many-fold increase in the genes coding for the RGC-specific intracellular markers Sncg (SNCG), Pouf4l (BRN3A), Tubb3 (TUJ1), and Rbpms (RBPMS). Collectively, the mRNA and protein assessments validated the methodology.
Figure 1. Enucleation and Ocular Dissection for Retinal Isolation. Young C57BL/6J mice were euthanized prior to eye globe removal with CO2 and cervical dislocation. A) Place forceps under the eye and pull up the eye in one movement. B) The eye is removed, including the optic nerve. C-J)Step-by-step guide to remove the retina. C) A puncture is performed using a 30G needle prior to corneal removal to allow the aqueous humor to exit the eye. D) The cornea is held with forceps to make a small incision. E-F) The use of forceps allows for peeling off the cornea, retinal pigment epithelium, choroid, and sclera. The retina is detached from the sclera, rolled, and removed. G) The lens is removed and discarded. H-J) The collected retinae are placed in a small dish containing PBS/1% FBS to keep them moist at all times. Please click here to view a larger version of this figure.
Figure 2. Retinal Cell Suspension after the Maceration of Collected Retinae. The collected retinae are placed in a small dish to isolate the cells. A) Retinae are placed in a 70-µm nylon strainer and macerated using the back-end of a syringe. B) Representative image of the cell suspension, where distinct retinal cells are observed. The scale bar is 10 µm.
Figure 3. Sorting Strategy for the Isolation of Cells with the CD90.2+CD48negCD15negCD57neg Phenotype and Post-sorting Analysis. The sorting strategy is based on the inclusion of CD90.2 cells and the exclusion of CD48-, CD15-, and CD57-positive cells, which are contaminant cells. A) As a first step, plot size (FSC) and internal complexity (SSC) to obtain an overview of the cell population. Initial gated population (P1) is used to discriminate between single cells and clumped cells or aggregates using the SSC-height (H) versus width (W), P2. The selection of the single cells is used to choose the CD90.2+CD48neg cells. To confirm removal of all doublets, a plot of FSC-H versus FSC-W is performed, P3 (middle panel). Cells were labeled with AF700-conjugated anti-mouse CD90.2, PE-Cyanine7-conjugated anti-mouse CD48, PE-conjugated CD15, and anti-mouse CD57. As a secondary antibody to tag the anti-mouse CD57, anti-mouse BV421 was used. Population 3 (P3) was plotted in the fourth panel to select the CD90.2+CD48neg cells, removing the majority of contaminant cells. Next, a CD57 versus CD15 plot is generated using the selected CD90.2+CD48neg cells. Quadrant 4 (Q4) is selected, as it represents the CD90.2+CD48neg cells that are negative for both CD15 and CD57. The resulting phenotype of the gated population is CD90.2+CD48negCD15negCD57neg. B) Post-sort analysis of the surface markers used in A). Sorted cells are homogeneous in size, as shown in the first panel. The subsequent histograms show the percentage of each surface marker used in the sorting strategy detailed in A). A total of 95% of the cells are CD90.2+, as shown in the black line compared to the Ig control, represented by the solid histogram. These cells were gated to evaluate the percentages of CD48, CD15, and CD57, represented by the red, blue, and green lines, respectively. Results show minimal expression of these cell surface markers. C) Confirmation of the RGC phenotype by using the RGC-specific intracellular markers SNCG, BRN3A, TUJ1, and RBPMS. Black lines represent the percentage of cells expressing each intracellular marker. D) Representative images taken in an imaging cell sorter showing the intracellular localization of RBPMS, an RGC-specific intracellular marker, and the cell surface marker CD90.2. The scale bar is 20 µm. E) Representative image of sorted RGCs after 24 h in culture using a confocal microscope. The scale bar is 20 µm. Images B-E are adapted from previously published work with permission23. Images D-E were taken at 20X. Please click here to view a larger version of this figure.
Figure 4. Pre- and Post-sorting mRNA Analysis. Thy1+CD48neg and sorted cells with the phenotype CD90.2+CD48negCD15negCD57neg were assessed by qPCR analysis using a panel of 25 genes expressed by retinal cells. Target gene expression levels are presented as a Log2-fold change using Hprt as a housekeeping gene and water as a negative control. The calculation was done based of the ΔCT method. Mean ± SEM; n = 3 biological replicates were performed in triplicate. Figures obtained from previously published work with permission23.
Discussion
FACS is the technique of choice to purify cell populations. Other isolation methods include immunopanning, magnetic beads, and complement fixation depletion. The advantage of FACS over these other methodologies is based on the simultaneous identification of cell-surface markers with varying degrees of intensity. The fluorescent intensity of the molecule is proportional to the amount of protein expression. Until now, the isolation of RGCs was based solely on Thy1 (CD90) positivity and CD48 negativity15,16,22,34, regardless of the isolation method used. It has recently been shown that the Thy1+CD48neg phenotype is not sufficient to isolate a homogenous population of cells expressing RGC intracellular markers23. Identification of the RGC population is essential for their isolation, particularly because they comprise a small percentage of retinal cells7,8,9. The majority of RGCs are located in the innermost layer of the retina, while a small number are located in the inner plexiform layers (displaced RGCs35). Thus, tracing RGCs from the superior colliculi by stereotactic injections and tracing with hydroxystillbamidine (a retrograde tracer for outlining neurons) became attractive choices for many laboratories36,37,38. These systems require the injection of tracer, which, if not performed properly, may lead to some retinal regions left untraced. In addition, they are more technically demanding and, like other methodologies such as immunopanning, are lengthy. Immunopanning using the anti-Thy1 and -CD48 antibodies takes 48 h to complete and does not achieve more than 95% purity. This work describes a FACS-based methodology that offers a fast and reproducible protocol to isolate a homogeneous population of live RGCs with the CD90.2+CD48negCD15negCD57neg phenotype, without the use of any tracers, magnetic beads, or immunopanning techniques.
The following factors are required for the successful isolation of pure RGCs by FACS: 1) sort efficiency, which highly depends upon the equipment used; 2) optimal combination of antibody-tagged fluorochromes, to minimize noise; and 3) cell sorting setup. The sort efficiency is calculated by taking the number of target events selected for sorting divided by the number of target events detected, expressed as a percentage. The sort efficiency is a calculation provided by the equipment. This efficiency depends on the setup of the sorting system and the cell sorting mode. Choosing an optimal combination of fluorochromes is a complex process. Each fluorochrome has distinct properties and is characterized by its excitation and emission wavelengths. While the excitation is read with a laser, the emission is read by photomultiplier tubes, which are limited by the optical filters available in the FACS sorting equipment. Here, a combination of antibody-tagged fluorochromes PE, PE-Cyanine7, AF700, and BV421 is provided. This was determined after considering multiple fluorochrome combinations that provided the best resolution while reducing spectral overlap. Lastly, the sort setup is critical. In general, retinal cells are fragile. Thus, it is best to use a lower pressure to run the samples, to minimize the stress on the cells. It is critical to maintain the sample at 4 °C, because keeping murine RGCs for longer periods of time at room temperature can reduce the cell yield, especially when sorting large numbers of cells.
FACS-based sorting is an ideal methodology for the isolation of cells that make up a very small percentage of the cell suspension. The process, from retinal dissection to the completion of cell sorting, takes approximately 5 - 6 h, compared to immunopanning and tracer systems, which take days to complete. The multidimensional analysis of FACS and the ability of the equipment to collect several viable populations allows for further functional analyses of cells. The protocol described here is a powerful tool for the isolation of primary murine RGCs. Despite its multiple advantages, including its sensitivity, reproducibility, and the immediate identification of viable cells, there are some limitations. Firstly, it requires expensive instrumentation and a highly trained operator. Usually, the operator is an immunologist or a highly trained individual in the field, with whom is necessary to meet at the time of experiment setup. Nowadays, academic facilities have multiple core facilities, which may facilitate performing these types of experiments. Secondly, RGCs lose their typical morphology due to atoxomy, making them very small in size. At this time, it is not known if some of their genes may be modulated due to atoxomy.
The methodology presented here allows for the downstream analysis of RGC function in vitro and is a valuable tool to be used in the fields of visual and health sciences. Maintaining ganglion cell output to the brain is required for visual perception and is at risk in multiple diseases. These cells can be used for controlled in vitro experimentation, in both healthy and disease models. Electrophysiological, pharmacological, biochemical, and molecular studies can be performed on these cells, which is ideal for the development of future therapeutic targets.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
The authors would like to thank Mr. Tim Higgins, Senior Illustrator from the Department of Microbiology, Immunology and Biochemistry, for technical video assistance; Dr. Matthew W. Wilson for discussions and the members of the Jablonski and Morales-Tirado laboratories for their helpful comments. This work was supported by the Alcon Research Institute Young Investigator Award (VMM-T), the University of Tennessee Research Foundation (VMM-T), the National Eye Institute EY021200 (MMJ), the Gerwin Fellowship (VMM-T); the Gerwin Pre-doctoral Fellowship (ZKG), the Department of Defense Army Medical Research and Materiel Command (VMM-T), and the Unrestricted Grant from Research to Prevent Blindness.
References
- Flammer J, Orgul S. Optic nerve blood-flow abnormalities in glaucoma. Prog Retin Eye Res. 1998;17(2):267–289. doi: 10.1016/s1350-9462(97)00006-2. [DOI] [PubMed] [Google Scholar]
- Bathija R. Optic nerve blood flow in glaucoma. Clin Exp Optom. 2000;83(3):180–184. doi: 10.1111/j.1444-0938.2000.tb04912.x. [DOI] [PubMed] [Google Scholar]
- Osborne NN, Melena J, Chidlow G, Wood JP. A hypothesis to explain ganglion cell death caused by vascular insults at the optic nerve head: possible implication for the treatment of glaucoma. Br J Ophthalmol. 2001;85(10):1252–1259. doi: 10.1136/bjo.85.10.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JE. Circulation and axonal transport in the optic nerve. Eye (Lond) 2004;18(11):1089–1095. doi: 10.1038/sj.eye.6701574. [DOI] [PubMed] [Google Scholar]
- Tian N, Hwang TN, Copenhagen DR. Analysis of excitatory and inhibitory spontaneous synaptic activity in mouse retinal ganglion cells. J Neurophysiol. 1998;80(3):1327–1340. doi: 10.1152/jn.1998.80.3.1327. [DOI] [PubMed] [Google Scholar]
- Schmidt KG, Bergert H, Funk RH. Neurodegenerative diseases of the retina and potential for protection and recovery. Curr Neuropharmacol. 2008;6(2):164–178. doi: 10.2174/157015908784533851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreher B, Sefton AJ, Ni SY, Nisbett G. The morphology, number, distribution and central projections of Class I retinal ganglion cells in albino and hooded rats. Brain Behav Evol. 1985;26(1):10–48. doi: 10.1159/000118764. [DOI] [PubMed] [Google Scholar]
- Williams RW, Strom RC, Rice DS, Goldowitz D. Genetic and environmental control of variation in retinal ganglion cell number in mice. J Neurosci. 1996;16(22):7193–7205. doi: 10.1523/JNEUROSCI.16-22-07193.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon CJ, Strettoi E, Masland RH. The major cell populations of the mouse retina. J Neurosci. 1998;18(21):8936–8946. doi: 10.1523/JNEUROSCI.18-21-08936.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surgucheva I, Weisman AD, Goldberg JL, Shnyra A, Surguchov A. Gamma-synuclein as a marker of retinal ganglion cells. Mol Vis. 2008;14:1540–1548. [PMC free article] [PubMed] [Google Scholar]
- Nadal-Nicolas FM, et al. Brn3a as a marker of retinal ganglion cells: qualitative and quantitative time course studies in naive and optic nerve-injured retinas. Invest Ophthalmol Vis Sci. 2009;50(8):3860–3868. doi: 10.1167/iovs.08-3267. [DOI] [PubMed] [Google Scholar]
- Kwong JM, Caprioli J, Piri N. RNA binding protein with multiple splicing: a new marker for retinal ganglion cells. Invest Ophthalmol Vis Sci. 2010;51(2):1052–1058. doi: 10.1167/iovs.09-4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bergen NJ, et al. Recharacterization of the RGC-5 retinal ganglion cell line. Invest Ophthalmol Vis Sci. 2009;50(9):4267–4272. doi: 10.1167/iovs.09-3484. [DOI] [PubMed] [Google Scholar]
- Wood JP, Chidlow G, Tran T, Crowston JG, Casson RJ. A comparison of differentiation protocols for RGC-5 cells. Invest Ophthalmol Vis Sci. 2010;51(7):3774–3783. doi: 10.1167/iovs.09-4305. [DOI] [PubMed] [Google Scholar]
- Barres BA, Silverstein BE, Corey DP, Chun LL. Immunological, morphological, and electrophysiological variation among retinal ganglion cells purified by panning. Neuron. 1998;1(9):791–803. doi: 10.1016/0896-6273(88)90127-4. [DOI] [PubMed] [Google Scholar]
- Hong S, Iizuka Y, Kim CY, Seong GJ. Isolation of primary mouse retinal ganglion cells using immunopanning-magnetic separation. Mol Vis. 2012;18:2922–2930. [PMC free article] [PubMed] [Google Scholar]
- Julius RS. The sensitivity of exponentials and other curves to their parameters. Comput Biomed Res. 1972;5(5):473–478. doi: 10.1016/0010-4809(72)90053-5. [DOI] [PubMed] [Google Scholar]
- Reif AE, Allen JM. The Akr Thymic Antigen and Its Distribution in Leukemias and Nervous Tissues. J Exp Med. 1964;120:413–433. doi: 10.1084/jem.120.3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Noguchi T, Tsukada Y. Regional, cellular, and subcellular distribution of Thy-1 antigen in rat nervous tissues. Neurochem Res. 1981;6(5):507–519. doi: 10.1007/BF00964390. [DOI] [PubMed] [Google Scholar]
- Haeryfar SM, Hoskin DW. Thy-1: more than a mouse pan-T cell marker. J Immunol. 2004;173(6):3581–3588. doi: 10.4049/jimmunol.173.6.3581. [DOI] [PubMed] [Google Scholar]
- Sahagun G, Moore SA, Fabry Z, Schelper RL, Hart MN. Purification of murine endothelial cell cultures by flow cytometry using fluorescein-labeled griffonia simplicifolia agglutinin. Am J Pathol. 1989;134(6):1227–1232. [PMC free article] [PubMed] [Google Scholar]
- Shoge K, et al. Rat retinal ganglion cells culture enriched with the magnetic cell sorter. Neurosci Lett. 1999;259(2):111–114. doi: 10.1016/s0304-3940(98)00918-5. [DOI] [PubMed] [Google Scholar]
- Chintalapudi SR, et al. Isolation and Molecular Profiling of Primary Mouse Retinal Ganglion Cells: Comparison of Phenotypes from Healthy and Glaucomatous Retinas. Front Aging Neurosci. 2016;8:93. doi: 10.3389/fnagi.2016.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagdeve NG, Yaddanapudi S, Pandav SS. The effect of different doses of ketamine on intraocular pressure in anesthetized children. J Pediatr Ophthalmol Strabismus. 2006;43(4):219–223. doi: 10.3928/01913913-20060701-03. [DOI] [PubMed] [Google Scholar]
- Ding C, Wang P, Tian N. Effect of general anesthetics on IOP in elevated IOP mouse model. Exp Eye Res. 2011;92(6):512–520. doi: 10.1016/j.exer.2011.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Aerts J, Nys J, Arckens L. A highly reproducible and straightforward method to perform in vivo ocular enucleation in the mouse after eye opening. J Vis Exp. 2014. p. e51936. [DOI] [PMC free article] [PubMed]
- Li X, et al. Loss of AP-2delta reduces retinal ganglion cell numbers and axonal projections to the superior colliculus. Mol Brain. 2016;9(1):62. doi: 10.1186/s13041-016-0244-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moshiri A, et al. Near complete loss of retinal ganglion cells in the math5/brn3b double knockout elicits severe reductions of other cell types during retinal development. Dev Biol. 2008;316(2):214–227. doi: 10.1016/j.ydbio.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danias J, et al. Quantitative analysis of retinal ganglion cell (RGC) loss in aging DBA/2NNia glaucomatous mice: comparison with RGC loss in aging C57/BL6 mice. Invest Ophthalmol Vis Sci. 2003;44(12):5151–5162. doi: 10.1167/iovs.02-1101. [DOI] [PubMed] [Google Scholar]
- Jakobs TC, Ben Y, Masland RH. CD15 immunoreactive amacrine cells in the mouse retina. J Comp Neurol. 2003;465(3):361–371. doi: 10.1002/cne.10845. [DOI] [PubMed] [Google Scholar]
- Uusitalo M, Schlotzer-Schrehardt U, Kivela T. Ultrastructural localization of the HNK-1 carbohydrate epitope to glial and neuronal cells of the human retina. Invest Ophthalmol Vis Sci. 2003;44(3):961–964. doi: 10.1167/iovs.02-0489. [DOI] [PubMed] [Google Scholar]
- Jackson CJ, Garbett PK, Nissen B, Schrieber L. Binding of human endothelium to Ulex europaeus I-coated Dynabeads: application to the isolation of microvascular endothelium. J Cell Sci. 1990;96 ( Pt 2):257–262. doi: 10.1242/jcs.96.2.257. [DOI] [PubMed] [Google Scholar]
- Pennartz S, Perraut M, Pfrieger F. Purification of retinal ganglion cells from postnatal rats by magnetic cell sorting. MACSmore. 2010;12(2):16–18. [Google Scholar]
- Nadal-Nicolas FM, et al. Displaced retinal ganglion cells in albino and pigmented rats. Front Neuroanat. 2014;8:99. doi: 10.3389/fnana.2014.00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadal-Nicolas FM, Salinas-Navarro M, Vidal-Sanz M, Agudo-Barriuso M. Two methods to trace retinal ganglion cells with fluorogold: from the intact optic nerve or by stereotactic injection into the optic tract. Exp Eye Res. 2015;131:12–19. doi: 10.1016/j.exer.2014.12.005. [DOI] [PubMed] [Google Scholar]
- Barnstable CJ, Drager UC. Thy-1 antigen: a ganglion cell specific marker in rodent retina. Neuroscience. 1984;11(4):847–855. doi: 10.1016/0306-4522(84)90195-7. [DOI] [PubMed] [Google Scholar]
- Chiu K, Lau WM, Yeung SC, Chang RC, So KF. Retrograde labeling of retinal ganglion cells by application of fluoro-gold on the surface of superior colliculus. J Vis Exp. 2008. [DOI] [PMC free article] [PubMed]