

Abstract
Maternal immune activation (MIA) via infection during pregnancy is known to increase risk for autism spectrum disorder (ASD). However, it is unclear how MIA disrupts fetal brain gene expression in ways that may explain this increased risk. Here we examine how MIA dysregulates rat fetal brain gene expression (at a time point analogous to the end of the first trimester of human gestation) in ways relevant to ASD-associated pathophysiology. MIA downregulates expression of ASD-associated genes, with the largest enrichments in genes known to harbor rare highly penetrant mutations. MIA also downregulates expression of many genes also known to be persistently downregulated in the ASD cortex later in life and which are canonically known for roles in affecting prenatally late developmental processes at the synapse. Transcriptional and translational programs that are downstream targets of highly ASD-penetrant FMR1 and CHD8 genes are also heavily affected by MIA. MIA strongly upregulates expression of a large number of genes involved in translation initiation, cell cycle, DNA damage and proteolysis processes that affect multiple key neural developmental functions. Upregulation of translation initiation is common to and preserved in gene network structure with the ASD cortical transcriptome throughout life and has downstream impact on cell cycle processes. The cap-dependent translation initiation gene, EIF4E, is one of the most MIA-dysregulated of all ASD-associated genes and targeted network analyses demonstrate prominent MIA-induced transcriptional dysregulation of mTOR and EIF4E-dependent signaling. This dysregulation of translation initiation via alteration of the Tsc2–mTor–Eif4e axis was further validated across MIA rodent models. MIA may confer increased risk for ASD by dysregulating key aspects of fetal brain gene expression that are highly relevant to pathophysiology affecting ASD.
Introduction
Multiple etiological pathways contribute to increased risk for autism spectrum disorder (ASD). For example, many monogenic syndromes and other rare de novo variants have been identified that have high penetrance for ASD,1, 2, 3, 4, 5 with theoretically many others that have yet to be discovered.6 Interestingly, such rare high-confidence mutations tend to be significantly enriched in genes involved in synaptic functions, transcriptional regulation, and chromatin remodeling functions, and/or are downstream targets of the fragile X syndrome protein (FMRP) complex.1, 3 In contrast, common variants may also significantly contribute to a large proportion (up to 60%) of genetic liability for ASD,7, 8 suggesting that hundreds of genes, individually associated with a small risk, may underlie ASD etiology via a much larger collective effect that acts at the network level either alone or in combination with environmental factors. Supporting this model, evidence from twin studies suggest that while heritability is quite high,9 there is also a substantial environmental component for ASD susceptibility.10 Recent evidence11, 12, 13, 14, 15, 16, 17, 18, 19, 20 has also catalyzed the concept that genetic and non-genetic factors and their interaction, may act at very early periods of fetal brain development and potentially alter protein or gene expression regulation leading to shared pathways for complex ASD-related phenotypes. Thus, much can be learned about the biological processes and molecular mechanisms involved in ASD by modeling environmental risk factors and studying their effects on functional genomics during early developmental stages of fetal brain development.
One environmental fetal programming21, 22 factor known to alter early fetal brain development and increase the risk for ASD is maternal infection during pregnancy.17, 18, 19, 23, 24, 25, 26 The effects of prenatal maternal infection on fetal brain development can be studied with maternal immune activation (MIA) animal models.27, 28, 29, 30 MIA can be induced experimentally via immunogens, such as polyinosinic-polycytidylic acid (poly(I:C)) and lipopolysaccharide (LPS). Poly(I:C) attempts to mimic viral-like infections via toll-like receptor 3 (TLR3) signaling, which induces production of type I interferons (IFN-α and IFN-β). In contrast, LPS mimics bacterial-like infections via TLR4 signaling which stimulates downstream production and secretion of TNF-α from innate immune cells (e.g., macrophages).30 Both poly(I:C) and LPS affect maternal cytokine signaling (e.g., interleukin-6) that passes through the placenta to affect fetal brain development31 and blocking key pathways prevents MIA-induced neural and behavioral abnormalities in ASD model systems.32 The consequences of MIA include behavioral deficits of broad relevance to ASD33, 34, 35 as well as numerous ASD-relevant influences on the developing brain.36 These influences include upregulation of cell cycle gene expression31 and shortening of cell cycle as seen in ASD,37 over-production of neurons38 analogous to some cases of ASD,14 increased cortical thickness,38 increased brain size39 as seen in many ASD toddlers,40, 41 altered expression of genes involved in neuronal migration,31 cortical layering defects42 including focal patches of disorganized cortex32 analogous to reports in some ASD cases,11 decreased intrinsic excitability of neurons,43 microglia abnormalities and enhanced microglia priming39, 44 as seen in ASD,45, 46, 47 alteration of GABAergic signaling,48 cerebellar vermis defects49 and defects of prefrontal dendritic morphology.50
Despite the numerous links between MIA and ASD pathology, several key questions remain with regard to how MIA affects the developing fetal brain at genomic and epigenomic levels and how such influence maps onto known genetic risk mechanisms associated with ASD. For example, does MIA exert its influence via genes associated with ASD and if so, which classes of genetic variants are most highly affected? Can MIA induce transcriptomic pathology in the fetal brain that shares similarities with cortical transcriptome dysregulation that is present in children and adults with ASD?51,52,53 What functional genomic pathology is present in the MIA-induced fetal brain that is not present in older children and adults with ASD? Are there specific mechanistic pathways that MIA dysregulates that are highly relevant for ASD? A better understanding of these key mechanistic links can help to further understand how MIA may confer risk for later development of ASD. By better understanding these mechanistic links between MIA and ASD, this work may ultimately help lead towards development of potential therapeutic targets for specific environmental risk factors that may be more amenable to prevention and/or treatment later in life54, 55 than genetic etiologies. Furthermore, if MIA alters expression in pathways shared with those in Fragile X Syndrome for which advances in drug development are in progress, then drugs that successfully target those pathways in Fragile X Syndrome could potentially be re-purposed.
In this work, we leverage bioinformatic and statistical approaches on available MIA gene expression data to investigate several key hypotheses about how MIA may dysregulate the fetal brain transcriptome in ways relevant to ASD. We first test the two hypotheses that MIA-induced effects may directly downregulate the expression of genes known to be associated with ASD and may indirectly alter protein targets downstream from two master regulatory genes of high penetrance for ASD (i.e. FMR1 and CHD8). We then test the hypothesis that MIA dysregulates the fetal brain transcriptome in ways that are similar to cortical transcriptome dysregulation observed in children and adults with ASD. We also heavily focus on how similarities in atypical biological systems (i.e. gene co-expression networks) in ASD and MIA can manifest in key pathways that are critically important for ASD and also reveal which early MIA-induced functional genomic pathologies are not detectable in the mature ASD brain. Finally, we independently induce MIA in mice to validate gene expression alterations in one prominent molecular pathway critical for protein translation processes during early fetal brain development and relevant to ASD pathophysiology.
Materials and methods
ASD and MIA Cortical transcriptome datasets
The primary MIA dataset was a rat model microarray dataset downloaded from Gene Expression Omnibus (GEO; Accession ID: GSE34058) and was previously published on by Oskvig and colleagues.31 This dataset applied a LPS manipulation for the MIA-inducing event on gestational day 15, which in humans corresponds to near the end of the first trimester of pregnancy. Gene expression was measured at 4 hours (4h) post-LPS injection on Affymetrix Rat GeneChip 1.0 ST chips. Raw data were also acquired and analyzed from the 24 hours (24h) post-LPS injection data reported by Oskvig et al., (Miles Herkenham, personal communication). Data were preprocessed from the raw CEL files with background adjustment, quantile normalization and summarization of probe intensities on log2 scale, using functions from the MATLAB Bioinformatics toolbox (i.e. rmabackadj.m, quantilenorm.m, rmasummary.m). We also analyzed two ASD cortical transcriptome datasets. The first was a microarray dataset from Voineagu et al.52 (GEO Accession ID: GSE28521) comprising frontal (BA9) and superior temporal cortex (BA41/42) tissue. The second dataset was an RNAseq dataset from Gupta et al.51 comprising frontal (BA44; BA10) and occipital cortex (BA19) tissue (http://www.arkinglab.org/resources/) (http://arkinglab.org/upload/RNASeq_Gupta/Samples104BGenes-EDASeqFull). For each ASD dataset we utilized the already preprocessed and quality controlled datasets publicly available in order to be as congruent as possible with prior published work.
Differential expression analyses
Differential expression (DE) analyses were performed in R. For the MIA rat dataset, we used sva56, 57 and limma packages58 for the DE analyses. Specifically, we utilized sva to determine a number of surrogate variables for inclusion as covariates in linear models via limma. For the ASD data-sets, we utilized linear mixed-effect models (i.e. lme function within the nlme R package) to model fixed-effect variables of diagnosis, RIN, age, sex, PMI, brain region and median 5-prime to 3-prime bias (specific to the Gupta dataset) and the random-effect of subject. False discovery rate (FDR) correction for multiple comparisons was achieved using Storey’s method for FDR control59, 60 implemented by the qvalue function in R.
Weighted gene co-expression network analysis
Analyses of gene networks organized by co-expression patterns was implemented with the WGCNA package in R.61 For datasets with multiple probes per gene, we collapsed probes by selecting the probe with the highest mean expression value across the full dataset as implemented with the collapseRows function in R.62 For the MIA dataset, we ran a signed WGCNA analysis where the soft power threshold was set to maximize R2 scale-free topology model fit as it plateaued above 0.8, and thus was set to 22. Soft power thresholded adjacency matrices were then converted into a topological overlap matrix and a topological overlap dissimilarity matrix (i.e. 1-topological overlap matrix). The topological overlap dissimilarity matrix was then input into agglomerative hierarchical clustering using the average linkage method. Gene modules were defined from the resulting clustering tree and branches were cut using a hybrid dynamic tree cutting algorithm (deepSplit=2).63 Modules were merged at a cut height of 0.2 and the minimum module size was set to 30. For each gene module a summary measure called the module eigengene (ME) was computed as the first principal component of the scaled (standardized) module expression profiles. Genes that cannot be clustered into any specific module are left within the M0 module, and this module is not considered in any further analyses. For the ASD datasets, we ran a signed consensus WGCNA analysis in order to detect consensus modules for cross-dataset comparisons (implemented with the blockwiseConsensusModules function).64 All of the parameters were set identically to the MIA analysis except for the soft power thresholds, which were set to 14 for both data-sets, based on similar criteria of maximizing R2 scale-free topology model fit. To test for DE at the level of ME variation we used linear mixed-effect models identical to those implemented in the DE analyses (i.e. same fixed and random effects). To identify MEs with replicable DE across both ASD datasets, we utilized t-statistics from the linear mixed models to compute replication Bayes Factor (repBF) statistics65 that quantify evidence for or against replication (see here for R code: http://bit.ly/1GHiPRe). Replication Bayes Factors greater than 10 are generally considered as strong evidence for replication. To identify replicable modules, we first considered modules that possessed a significant effect passing FDR59 q<0.05 within the Voineagu dataset, and then also required these modules possess significant effects in the Gupta dataset (FDR q<0.05), and that this evidence quantitatively produces evidence for replication with a replication Bayes Factor statistic >10. To test ASD gene modules for preservation with the MIA dataset, we ran a module preservation analysis using the function modulePreservation and set the number of permutations to 200.
MetaCore GeneGO enrichment analyses
In order to understand what molecular processes our gene lists were enriched in, we used MetaCore GeneGO software (https://portal.genego.com/) to perform all enrichment tests. These analyses were done at the level of ‘Process Networks’ within MetaCore.
Gene set enrichment analyses
All gene set overlap analyses were implemented using the sum(dhyper()) function in R. The background set size for all enrichment analyses was set to the total number of probes within the MIA dataset (i.e. 22071). For example, if there were 4959 MIA-downregulated genes and 35 ASD-associated genes, and 20 of the 35 ASD-associated genes were in the list of MIA-downregulated genes, the analysis examining enrichment of ASD-associated genes in the MIA-downregulated genes would be implemented with this line of code in R: sum(dhyper(20:35, 4959, (22071-4959), 35)).
Network analysis of PI3K–TSC1/2–mTOR–EIF4E axis
The analysis of the predicted targets of module M25 was performed by querying the Metacore GeneGO database. All genes from the M25 module (61 genes) were used as bait to search the database for canonical interacting partners using one interaction distance (the 'no filtering' option was used). This search yielded networks with 3257 genes in total. We saved this gene list and ran enrichment analysis to learn about the biological processes possibly affected by M25 dysregulation.
Similarly, to quantify the predicted effects of MIA and M25 dysregulation on the PI3K–TSC1/2–mTOR–EIF4E signaling pathway, we queried the Metacore GeneGO database to identify the shortest canonical network connecting these five key regulatory genes. These genes were thus used as bait and the shortest canonical network with a maximum number of two steps in the path was selected. Eighty-four nodes and their interactions were exported from Metacore, and the canonical network was reproduced in Cytoscape (http://www.cytoscape.org). Exporting the network in Cytoscape facilitated the color coding of the genes to display the overlap with the M25 targets and the differentially expressed genes from the MIA and ASD cortices as well as the ASD-associated genes.
Mouse MIA model experiment
All animal studies were performed in accordance with NIH guidelines for the use of animals and all procedures were reviewed and approved by the Stanford Institutional Animal Care and Use Committee. Timed pregnancies of Jackson Laboratory C57BL/6 J mice were obtained by housing a female and a male overnight. The individual mouse was separated the next morning and defined the mid-day of that day as embryonic day 0.5 (E0.5). The pregnant females were identified by body weight gain during the time course of pregnancy. To induce MIA responses, at E12.5, the pregnant dams were injected intraperitoneally with LPS from Escherichia coli 055:B5 (L4524, Sigma-Aldrich, St Louis, MO, USA) at doses 60 μg kg−1 dam’s body weight. Control dams were injected with saline (SAL, vehicle) only. This resulted in seven LPS-treated offspring (4 female, 3 male) and six SAL offspring (3 female, 3 male).
qRT-PCR analysis
Whole fetal brains were homogenized in TRIzol reagent (Life Technologies, Carlsbad, CA, USA) using RNase-free disposable pestles (Kimble Chase, Vineland, NJ, USA) to extract total RNA. Following chloroform, 100% ethanol was added to precipitate the aqueous phase containing RNA. Then the aqueous phase was transferred onto a QIAgen RNeasy mini spin column and RNA/DNA was isolated with the QIAgen RNeasy mini kit (Qiagen, Venlo, Netherlands) following the manufacturer’s protocol. DNA was digested using DNase-I enzyme (Qiagen) for 15 min at RT. Nanodrop spectrophotometer (ThermoFisher Scientific, Waltham, MA, USA) was used to assess the quality and concentration of isolated RNA. RNA was stored at −80 °C until cDNA synthesis. To synthesize cDNA template, reverse transcription PCR reaction was performed on extracted RNA with MultiScribe reverse transcriptase (ThermoFisher Scientific) and random primer sets in the following condition: 25 °C 10 min, 37 °C 120 min, 85 °C 5 min. The synthesized cDNA was kept at −20 °C until qRT-PCR reaction. VeriQuest Probe qPCR Master Mix (Affymetrix, Santa Clara, CA, USA) was used in the following qPCR reaction with Fast Real-Time PCR systems (Applied Biosystems, AP7900HT, Waltham, MA, USA); 50 °C 2 min, 95 °C 10 min, 40 cycles of (95 °C 15 s, 60 °C 1 min). The FAM-conjugated TaqMan qRT-PCR primer sets used in the present study and ROX was used as reference. Percentage of mRNA expression was calculated by converting relative mRNA copy number from differences between Ct (Cycle threshold) values of Gapdh (a housekeeping gene) and the gene-of-interest. The relative mRNA expression levels in LPS-treated fetal brains were normalized by SAL-treated control levels.
Taqman qRT-PCR primers
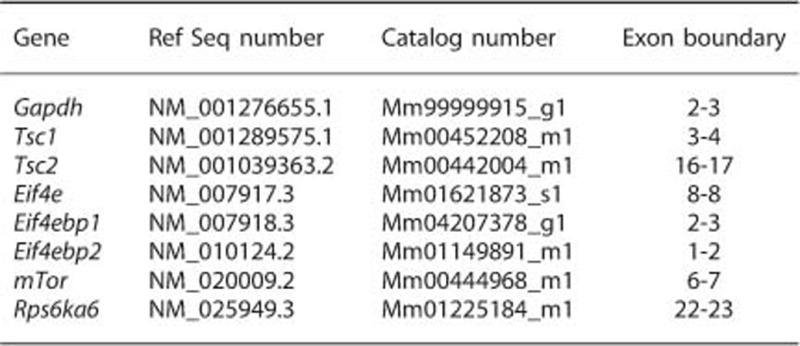
All statistical tests on qRT-PCR data employed one-tailed independent samples t-tests that do not assume equal variances (i.e. t.test function in R). The one-tailed predictions are justified by the directionality of DE observed in the rat microarray MIA dataset. Control for multiple comparisons was achieved by setting the FDR threshold to q<0.05.
Results
We evaluated MIA-induced DE in a rat dataset from Oskvig et al.,31 measured at 4 h post-LPS injection on gestational day 15. This manipulation in rat corresponds roughly to post-conception day 68 of human prenatal cortical development, which is analogous to the end of the first trimester of pregnancy66 (Supplementary Figure S1). Generally consistent with analyses in Oskvig et al., here we found evidence for massive MIA transcriptome dysregulation at 4 h post-LPS injection. At an FDR q<0.05 there were 6923 downregulated genes (7561 probes) and 4981 upregulated genes (5513 probes). Given this large degree of DE signal and to be more conservative in our approach, we utilized an FDR threshold of q<0.01, which yielded 4959 downregulated genes (5398 probes) and 4033 upregulated genes (4462 probes) (see Supplementary Table S1 for gene lists). In contrast to the 4 h dataset, a dataset measured 24 h post-LPS injection (24 h) yielded little DE signal, with only one downregulated gene (MAIN) and two upregulated genes (LCP2, RPL39) at FDR q<0.05, and no DE genes at FDR q<0.01. Given the majority of DE signal is present in the 4 h data-set, all further analyses will utilize this dataset.
To describe processes enriched in the 4 h MIA DE gene sets we used MetaCore GeneGO for pathway analysis. MIA-downregulated genes displayed enriched functions relevant to both early cortical development such as WNT/Hedgehog signaling and neurogenesis and later cortical development, such as axonal guidance and synaptogenesis (Figure 1a). In contrast, MIA-upregulated genes displayed predominant enrichment in processes that can play key roles in neurogenesis and early brain development, such as translation, cell cycle, DNA damage and proteolysis processes (Figure 2a) (see Supplementary Table S2 for full list of enrichments). This MIA-induced overexpression of early processes affecting protein synthesis, cell number, DNA integrity, and cell fate specification are in line with functions that would be expected to be normally active during late first trimester of human brain development, and are consistent with some hypotheses of the early neural abnormalities in ASD such as dysregulated neurogenesis.11, 14, 36, 37, 67
Figure 1.

Enrichment of maternal immune activation (MIA)-downregulated genes with classes of autism spectrum disorder (ASD)-associated genes and ASD cortical transcriptome downregulated genes. This figure describes MIA-downregulated genes and their enrichment within different classes of ASD-associated genes and genes that downregulated in the ASD cortical transcriptome. Panel a shows process level enrichments for all MIA-downregulated genes. Panel b shows enrichment odds ratios for different classes of ASD-associated genes (the * indicates enrichment passing false discovery rate (FDR) q<0.05, while the ~ indicates enrichment passing FDR q<0.1). Panels c and d show enrichment between downregulated genes in MIA and ASD cortical transcriptome datasets (panel c for the Voineagu dataset and panel d for the Gupta dataset). Panels e and f show process level enrichments for the common downregulated genes between MIA and ASD (panel e for the Voineagu dataset and panel f for the Gupta dataset).
Figure 2.

Maternal immune activation (MIA) effects on translation and transcriptional mechanisms. This figure shows results supporting the idea that MIA dysregulates processes involved in translation and transcription. Panel a shows process level enrichments for MIA-upregulated genes. Panel b shows enrichment odds ratios for MIA-downregulated or -upregulated gene sets with downstream FMRP and CHD8 targets (the * indicates enrichment test passing false discovery rate (FDR) q<0.05 threshold). Panels c and d show enrichment between upregulated genes in MIA and autism spectrum disorder (ASD) cortical transcriptome datasets (panel c for the Voineagu dataset and panel d for the Gupta dataset). Panels e and f show process level enrichments for the common upregulated genes between MIA and ASD (panel e for the Voineagu dataset and panel f for the Gupta dataset).
Classes of ASD-associated genes are enriched with MIA-downregulated genes
We next examined our first hypothesis that MIA-downregulated genes are substantially enriched in various classes of ASD-associated genes. Our first test of this hypothesis examined a class of genes identified by recent whole-exome sequencing studies as 'likely gene-disrupting' variants (LGD) (i.e. splice-site, nonsense or frameshift variants) (see Supplementary Table S1 for gene lists).1, 2, 3, 4 Remarkably, 57% (20/35) of LGD genes were present in the MIA-downregulated gene set, amounting to a substantial enrichment (OR=5.95, P=9.44e-6). When considering known ASD-associated missense variants,3 we also found substantial enrichment (33%, 48/145, OR=2.21, P=0.0021) (Figure 1b). We then further considered ASD-associated gene classes separated by the expert manually curated categories in the SFARI Gene database (http://gene.sfari.org/)68, 69 (see Supplementary Table S1 for gene lists). Here we also found that the MIA-downregulated genes are substantially enriched in several categories, with a gradient in enrichment that follows the strength of evidence implied by each category. The strongest enrichments by enrichment odds ratio were within the SFARI High Confidence category (75%, 12/16, OR=13.38, P=1.18e-5), followed by Syndromic (54%, 25/46, OR=5.32, P=2.57e-6), Strong (41%, 10/24, OR=3.18, P=0.0277) and Suggestive gene categories (40%, 27/67, OR=3.01, P=7.91e-4) (Figure 1b). These findings suggest that MIA may increase risk for ASD via downregulating at a very early stage of brain development the expression of many of the same genes that are known to be highly penetrant for ASD.
MIA dysregulates downstream targets of FMR1 and CHD8
The evidence of MIA downregulating expression of genes that are highly penetrant for ASD suggests that MIA might also exert important influence on downstream transcriptional programs of such genes. We tested this hypothesis with two such genes, FMR1 and CHD8,70, 71 because both are highly penetrant for ASD and are key master regulators of important neurodevelopmental processes, including mRNA translation, transport, or localization (FMR1)72, 73 and chromatin remodeling (CHD8) of hundreds of genes implicated in transcription, cell division, proteolysis, DNA integrity and signal transduction.67 Interestingly, both FMR1 and CHD8 themselves are not dysregulated by MIA. However, this allows for an interesting test of the hypothesis that although these key genes are not directly dysregulated by MIA, their downstream targets may still be impacted by MIA, and show evidence of substantial enrichment.
Across two FMRP target sets72, 73 (see Supplementary Table S1 for gene lists) we found that MIA-downregulated genes are highly enriched in FMRP targets (Darnell targets: OR=7.81, P=2.56e-127; Ascano targets: OR=2.07, P=1.75e-30). Of the MIA-upregulated genes, enrichment was apparent in one of the two FMRP target lists (Darnell targets: OR=0.22, P=1; Ascano targets: OR=1.86, P=1.15e-21) (Figure 2b). For CHD8 targets, we also examined two target sets derived from either midgestational human fetal brain tissue and human neural stem cells74 or from human neural progenitor cells75 (see Supplementary Table S1 for gene lists). The MIA-downregulated (OR=2.10, P=3.52e-25), but not MIA-upregulated (OR=1.19, P=0.84) genes were enriched in CHD8 targets identified in midgestation fetal brain tissue and human neural stem cells.74 In human neural progenitor cells both MIA-downregulated (OR=1.66, P=1.58e-7) and upregulated genes (OR=2.71, P=8.29e-87) were enriched in CHD8 targets75 (Figure 2b). Overall, this evidence supports our hypothesis that while MIA does not directly affect FMR1 or CHD8, two key genes with important transcriptional regulatory effects, it does potentially disrupt the same pathways by hitting their downstream targets.
MIA-dysregulated genes are also dysregulated in child and adult ASD cortical transcriptome
We next examined the hypothesis that MIA-dysregulated genes are also dysregulated in the child and adult ASD cortical transcriptome. To examine this, we re-analyzed two prior post-mortem ASD datasets from Voineagu et al.52 and Gupta et al.51 We found that ASD-downregulated genes in both datasets are substantially enriched in MIA-downregulated genes (Voineagu OR=1.26, P=0.024; Gupta OR=2.22, P=7.16e-21; see Figures 1c, d and Supplementary Table S1 for gene lists). These commonly downregulated genes are significantly enriched in biological processes such as Transport_Synaptic vesicle exocytosis, Development_Neurogenesis_Synaptogenesis, and Cell adhesion_Synaptic contact (Figures 1e and f). Similar to downregulated genes, ASD-upregulated genes in both datasets were significantly enriched in MIA-upregulated genes (Voineagu OR=1.57, P=9.56e-5; Gupta OR=1.69, P=3.84e-7; see Figures 2c, d and Supplementary Table S1 for gene lists). Genes commonly upregulated in MIA and ASD were enriched in translation initiation processes across both ASD datasets (Figures 2e and f). However, despite such statistically significant enrichment, it is very noteworthy that a large majority of down- and upregulated genes perturbing prenatal developmental processes in MIA, were not commonly dysregulated in the older child and adult ASD brain (Figures 1c, d, 2c and d). That is, 94–97% of MIA-downregulated genes and 96–97% of MIA-upregulated genes were not commonly dysregulated in older child and adult ASD cortical tissue. Thus, while a specific subset of genes are commonly dysregulated by MIA in early fetal development and in older children and adult ASD cortical tissue, many other MIA-dysregulated processes in fetal development are likely not captured in common by looking at older ASD cortical tissue far beyond critical prenatal stages of brain development (e.g., upregulated cell cycle processes with likely role in neurogenesis).
Translation and synaptic gene co-expression networks are highly preserved across MIA and ASD
We next tested whether systems-level transcriptome disruptions in MIA and ASD cortex are significantly similar or ‘preserved’. This approach goes beyond identifying overlap at the level of single genes and provides information about larger systems-level organization of the transcriptome and whether such dysregulated organization is similar across MIA and ASD cortical transcriptomic datasets. To do this, we implemented WGCNA to identify preservation of systems-level structure of gene networks in MIA and ASD cortical transcriptome datasets.61, 76 We specifically examined ASD co-expression modules for on-average DE in ME variation (i.e. systematic up- or downregulation along the main principal axis of variation for a given gene module), and determined whether such DE modules were preserved in network structure in MIA. Co-expression modules that are both dysregulated and highly preserved across both datasets are ideal candidates for pinpointing common systems-level biological disruption in both ASD and MIA.
We identified four consensus modules in ASD, M25, M3, M9 and M13, that show replicable on-average DE in both the Voineagu and Gupta datasets, and also showed moderate levels of preservation in the MIA dataset. M25 was replicably upregulated in post-mortem ASD cortical tissue, and was heavily enriched in translation initiation and translation elongation-termination (Figure 3a). The genes contributing to this enrichment are almost all exclusively ribosomal proteins (e.g., RPL, RPS genes) that make up the 40 and 60S ribosomal subunits (see Supplementary Figure S2). M25 was the top hit in terms of preservation median rank and was the most preserved of any of the replicable DE modules with Zsummary preservation statistics of 8.5 and 8.8 (indicating ‘moderate’ preservations), respectively, across Voineagu and Gupta ASD datasets (Figure 3c). Modules M3, M9 and M13 were replicably downregulated in Voineagu and Gupta ASD datasets, and were enriched in a variety of synaptic functions (Figure 3b). These modules also showed moderate levels of preservation primarily with the Zsummary statistics above 2 (Figure 3d). These results further strengthen the evidence that MIA dysregulates systems-level structure of transcriptome in a manner similar to the dysregulation present in the ASD cortical transcriptome, with emphasis on upregulation of translation initiation processes as the strongest preserved signal across MIA and ASD.
Figure 3.

Preservation of dysregulated gene co-expression network organization across maternal immune activation (MIA) and autism spectrum disorder (ASD). This figure shows ASD co-expression modules that are replicably dysregulated in ASD and preserved in network-level structure in the MIA dataset. Panels a and b depict gene co-expression modules that are replicably upregulated (a) or downregulated (b) in ASD cortical gene expression datasets. Scatter-boxplots show module eigengene (ME) expression levels with individual dots for each sample and boxplots that show the median and interquartile range (IQR; Q1=25th percentile, Q3=75th percentile), as well as the outer fences (Q1–(1.5IQR) and Q3+(1.5IQR)). Next to each scatter-boxplot are results from process level enrichment analysis on each module. Above these plots are replication Bayes Factor statistics indicating evidence in favor of replication (repBF>10 indicates strong evidence in favor of replication). Panels c and d show module preservation statistics (median rank and Zsummary) for preservation between ASD cortical gene modules (c, Voineagu dataset; d, Gupta dataset) and MIA gene modules. The horizontal lines on the preservation Zsummary plot indicate categories for evidence of preservation, with Zsummary statistics between 2 and 10 indicating ‘moderate’ evidence for preservation. Modules represented by black dots are not differentially expressed between ASD and Control brains. Modules represented by colored dots (not black) and without a specific number (e.g. M25) are differentially expressed but not significantly preserved between ASD and MIA. Colored modules M25, M13, M3 and M9 are differentially expressed and significantly preserved between ASD and MIA.
Activation of translation initiation processes dysregulates gene expression within members of the PI3K–TSC1/2–mTOR–EIF4E cascade in MIA and ASD
One common theme from the above DE and co-expression results of MIA and ASD transcriptomes is the presence of upregulated translation initiation processes. This common disruption suggests that either early environmental and/or genetic insults may lead to overlapping downstream effects via the dysregulation of translation pathways. Exaggerated cap-dependent translation is a well-known molecular mechanism regulating neurogenesis77 and contributing to synaptic and behavioral phenotypes associated with ASD and related neurodevelopmental disorders.78, 79 Key to this mechanism is the aberrant regulation of the PI3K–TSC1/2–mTOR signaling pathways, which in turn are responsible for the regulation of ribosomal protein S6 kinase (RPS6K) and EIF4E-binding partners (EIF4EBP1/2), acting to promote translation initiation.80 Loss-of-function of the TSC1/2 complex has been implicated in ASD etiology81, 82 and is an important upstream effector of mTOR-dependent signaling cascades.83 Dysregulation of mTOR-dependent signaling pathways has been found in ASD, Fragile X Syndrome, and Rett Syndrome.84 Enhanced mTOR signaling and cap-dependent translation initiation complex are found also in mouse models of human Fragile X Syndrome, characterized by the lack of FMRP.85, 86, 87 Of note, mTOR kinase plays pivotal roles as a master regulator to integrate environmental cues and to modulate translation, cell growth, survival and proliferation.84 Ultimately, these signaling pathways lead to the overexpression and activation of EIF4E-dependent mechanisms that have been directly linked to ASD, both in mouse models and humans.88 In support of this view, we have discovered evidence that downstream FMRP targets are dysregulated by MIA (Figure 2b) and that EIF4E displays the largest effect size of all ASD-associated genes (Cohen’s d=8.27).
To further explore the effects of our network-based findings in MIA and its relevance to ASD, we asked whether the upregulated translation initiation-enriched ASD-module M25, which is the strongest preserved DE module in MIA, would affect members of the PI3K–TSC1/2–mTOR signaling pathways. To examine this hypothesis, we first constructed a network of M25 targets using the MetaCore canonical database and identified 3257 M25 direct targets. Importantly, functional analysis of these targets displayed a top enrichment in the regulation of cell cycle phase transition (G1–S and G2–M) and several developmental processes comprising several key regulatory genes (e.g. AKT, JAK, NF-κB, PI3K, STATs, CDK, mTOR, NOTCH1, WNT and ERK/MAPKs; Supplementary Table S2) as well as cap-dependent translation regulatory genes (EIF4E and its binding partners; Supplementary Table S2). These findings suggest that the ASD-upregulated translation initiation-enriched M25 module, which is preserved in MIA, may influence expression and activity of both PI3K–TSC1/2–mTOR signaling and EIF4E-dependent genes with predicted early neurodevelopmental effects on the timing of cell cycle phases during neural progenitor cell divisions.
To directly compare and quantify the effects of MIA and M25 dysregulation on this signaling pathway, we queried the MetaCore database to generate the shortest canonical network-path encompassing the PI3K–TSC1/2–mTOR–EIF4E axis. This network included 84 genes. We then asked the question of whether this axis is enriched in differentially expressed (DE) MIA genes, DE ASD genes, genes that are M25 targets, or ASD-associated genes (LGD, Missense or SFARI) (see Figure 4). We found significant enrichments for genes DE in MIA (OR=4.006, P=7.14e-5) and for M25 targets (OR=22.09, P=1.89e-36) (Figure 4). Although only 9 of the 84 genes were DE in ASD brains resulting in a nonsignificant enrichment (OR=1.21, P=0.46), 10 genes of the 84 genes were those that are known to be ASD-associated (OR=9.22, P=4.62e-9) and of those 10, 8 were DE in MIA and/or ASD brains. Of note, we found that all key members of the signaling pathway (PI3K, TSC1, TSC2, mTOR, EIF4E) were dysregulated in MIA together with mTOR and EIF4E-binding partners (Figure 4). Altogether these findings provide compelling evidence that both MIA and ASD cortical transcriptome dysregulation involve the canonical PI3K–TSC1/2–mTOR axis regulating EIF4E-mediated cap-dependent translation.
Figure 4.

Canonical network encompassing the PI3K–TSC1/2–mTOR–EIF4E axis. This figure depicts all 84 genes comprising the shortest canonical network path of the PI3K–TSC1/2–mTOR–EIF4E axis, as defined by the MetaCore GeneGO database. The plot is arranged by cellular compartment for each protein in the network. Nodes were depicted in larger size if they are members of the autism spectrum disorder (ASD)-associated gene list compiled by SFARI Gene. Nodes with green borders are direct targets of the ASD co-expression module M25, which is ASD-upregulated and enriched in translation initiation and is preserved within the MIA dataset. Each node is colored on the inside to indicate directionality of differential expression (blue=downregulated, red=upregulated, gray=not differentially expressed). Rectangular shapes characterize all genes within this network. However, within each node a diamond shape indicate that the gene was differentially expressed in ASD brains. MIA, maternal immune activation.
Cross-species validation of MIA dysregulation of the Tsc1/2–mTOR–Eif4ebp1/2 axis
The network-level analyses suggest that MIA dysregulates the TSC1/2–mTOR–EIF4E signaling pathway. To directly test this hypothesis and validate inferences across model species, we performed a MIA mouse model validation experiment. Based on our findings in the rat model and literature evidence, we were interested specifically on validating mRNA expression changes for the key components of the TSC1/2–mTOR–EIF4E signaling pathway (Tsc1/2, mTor and Eif4e). mTOR has two main direct substrates, RPS6K and eukaryotic translation initiation factor 4E-binding proteins (EIF4EBP1/2), that are responsible for activating further downstream signaling pathways to regulate translational machinery.89 Thus we included Rps6k, and Ei4ebp1/2 genes in the validation experiment as well. Similar to the experimental design of the original rat MIA model dataset,31 we induced MIA in pregnant dams using LPS at gestational day 12.5 in mice as previously described90, 91 (see Materials and Methods). Mouse fetal brains were collected 2 h post-LPS injection and mRNA transcript levels were quantified by qRT-PCR and expression levels were normalized by SAL controls. Consistent with the MIA rat gene expression findings, we replicated the effect of significant MIA downregulation of Tsc2 (t=−2.91, P=0.012), mTor (t=−2.83, P=0.012), Eif4ebp1 (t=−3.77, P=0.0024) and Eif4ebp2 (t=−5.05, P=0.00078). We also replicated the MIA-induced upregulation of Eif4e (t=2.13, P=0.029). Replication of Tsc1 MIA-induced downregulation was observed, albeit at trend-level significance (t=−1.70, P=0.06), whereas Rps6ka6 was not differentially expressed (t=−0.12, P=0.54) (Figure 5). Together with the original discoveries in the rat MIA dataset, this cross-species validation (performed in MIA mouse model) strongly supports that MIA-induced transcriptional dysregulation of genes involves translation processes supported by the Tsc1/2–mTor–Eif4ebp1/2 axis.
Figure 5.

Cross-species validation of dysregulated gene expression within the Tsc1/2–mTor–Eif4ebp1/2 axis in a mouse MIA model. This figure summarizes the results from qRT-PCR analysis of gene expression along the Tsc1/2–mTor–Eif4ebp1/2 axis hypothesized to show dysregulation from the previous rat model data. Each gene is displayed with a scatter-boxplot and gene expression for each individual sample is plotted on the y-axis as % mRNA expression normalized to the average value in the SAL condition. Boxplots show the median and interquartile range (IQR; Q1=25th percentile, Q3=75th percentile), as well as the outer fences (Q1 – (1.5IQR) and Q3+(1.5IQR)). MIA, maternal immune activation; SAL, saline.
Discussion
In this study we examined animal models of MIA corresponding to late first trimester of gestation in humans in order to better understand how MIA may lead to increased risk for ASD via impairments of early fetal brain development that are relevant to the pathophysiology behind ASD. We discovered that MIA induces dysregulation of the fetal brain transcriptome in several important ways. MIA downregulates expression of many genes known to be highly penetrant for ASD. Genes with smaller effect size for ASD risk were also downregulated by MIA though to a lesser extent than larger effect size genes. The strength of MIA enrichment tended to follow the strength of evidence for association with ASD. This evidence suggests that MIA may have particular detrimental fetal programming impact for enhancing later risk for ASD by perturbing genes in early periods of fetal development that are of medium-to-high penetrance. MIA also significantly influences transcriptional programs that are downstream to highly penetrant mutations in FMR1 and CHD8 genes, even when expression of these genes themselves may not be MIA-dysregulated. This evidence provides additional proof-of-concept that MIA-induced effects may converge to many of the same or overlapping pathways hit by some highly penetrant ASD mutations, and this can occur without actual dysregulation of the target genes themselves. These findings bolster the intriguing possibility that MIA acts as an environmental etiological factor that disrupts specific key early developmental genomic mechanisms that are risks for ASD. MIA-induced disruption may work in a manner similar in directionality to rare highly penetrant ASD mutations, and may act as ‘priming’27, 28, 29, 30 or fetal programming21, 22 mechanisms that enhance risk of later developing ASD. However, MIA may be different from such mutations in being a temporally transient event since it is restricted to circumscribed windows in fetal development rather than being persistent in disrupting protein synthesis over the entire lifespan as a deleterious mutation would be. Nonetheless, in terms of the sheer number of genes affected by MIA, it could also be that such events may be more potent and/or common in occurrence than highly penetrant germline mutations, which are rare but more commonly found in ASD individuals. These results would further predict that when such environmental and gene mutation effects co-occur, pathology could be amplified and/or lead to more complex heterogeneous phenotypes. Prior work provides some evidence in support of such MIA-gene interactions, such as co-occurrence of MIA with TSC2 haploinsuffiency,92 rare de novo CNVs93 and PTEN mutations.39 Intriguingly, both TSC2 and PTEN are among the genes that are MIA-downregulated within the current dataset, which could increase abnormal neurogenesis. A future extension of our work here along these lines could be to block MIA effects on the mTOR–EIF4E pathway and test the hypothesis whether this ameliorates behavioral phenotypes and cortical pathology linked to MIA.32 If effective, such work would go along with many other therapies tested in animal models of MIA that may be ultimately important for treating ASD-related phenotypes (e.g., Fragile X Syndrome), since such therapies may work on pathways shared with ASD.54, 55, 94, 95, 96 Another future extension of this work is to examine whether the type of immunogen inducing MIA response (i.e. poly(I:C) vs LPS) has similar or different effects on gene expression. While our work utilizes LPS and mimics bacteria-like infections via TLR4 signaling and downstream effects from TNF-α signaling, future work systematically contrasting the effects of immunogens such as LPS or poly(I:C) (mimicking viral infections via TLR3-dependent signaling), will be important. Overall, this work would suggest that MIA itself could constitute a sufficient environmental route through which the transcriptome in fetal brain development could be altered in ways similar to genetic etiologies associated with neural and behavioral phenotypes of ASD.
We also specifically examined how prenatal MIA-induced transcriptome dysregulation is similar to dysregulation of the ASD cortical transcriptome seen in later life. We found commonality between MIA and ASD in downregulation of synaptic-related processes and upregulation of translation-related processes. Interestingly, many of the genes and enrichment terms we found commonly downregulated are relevant to developmental processes, such as synaptogenesis, that occur at later prenatal and postnatal stages, well after the point of the MIA event (i.e. see Supplementary Figure S1). One possible interpretation of this counterintuitive result could be that these downregulated genes play other important roles at earlier periods of first trimester brain development, and that such roles are not well represented in current gene ontology annotations, especially when compared with their much more well-known canonical roles associated with synaptic processes in later development. This apparent pleiotropy of roles for these ASD-relevant genes is certainly an under-investigated area. For example, although many high-risk genes associated with ASD are commonly interpreted as being involved in later occurring processes such as neurite and synapse development, some research supports the idea that many of these genes also show prominent involvement in very early stages of brain development such as neural induction and early maturation of the neuroblast.97 Thus, our results suggest an interesting new direction for future work that examines much earlier roles for ASD-associated genes, roles beyond those in later synaptic processes. Also important is that our results also show that the vast majority (approximately 94–97%) of MIA down- and upregulated genes that govern numerous early neural development processes (e.g., cell number, cell-type laminar fate, migration, cell growth and differentiation), are not detected by examining differential expression of genes in cortical tissue in older child and adult ASD individuals. Therefore, it is important to consider that gene expression studies of the mature ASD cortex might provide somewhat limited insight into the prenatal functional genomic pathology that may underlie the beginnings of ASD. As such, gene expression data from the mature ASD brain could be prone to false negatives and, as such, should be interpreted with caution. Indeed, a previous study showed age-dependent changes in abnormal cortical gene expression in ASD.53
A specific key dissimilarity between MIA in fetal development and the cortical transcriptome of ASD in children and adults is the presence of strongly upregulated cell cycle expression in MIA. This strong upregulation in fetal development is highly relevant given that the timing of MIA in this study occurs within a prominent time period for neurogenesis and cell differentiation. In the child and adult ASD brain, there is a lack of any dysregulation in such cell cycle processes. However, this developmental time period corresponds to a period where neurogenesis and cell differentiation processes are much less prominent and synaptic plasticity, circuit-dependent activity regulation play more important roles. Thus, a potential major defect underlying early ASD development14, 36, 37, 67 in dysregulated cell proliferation and differentiation processes likely cannot be adequately examined via study of the ASD brain in later development. We argue that MIA upregulation of cell cycle processes and increased neurogenesis, and/or changes in timing of cell differentiation in early fetal development, is likely a shared ASD-relevant aspect of pathophysiology.36, 37 Supporting this inference, we find that the ASD-upregulated M25 co-expression module that is preserved in MIA has strong downstream impact on cell cycle processes (see Supplementary Table S2) that are likely highly relevant in early fetal development, when neurogenesis and early cell differentiation is a highly prominent neurodevelopmental process.
We also found evidence for upregulation of translation processes in both MIA and ASD. Of note, transcriptional alteration of translation regulation was additionally supported by the FMRP target enrichment analysis that showed this is likely one of the strongest convergent signals in our comparative analysis of MIA and ASD effects on the cortical transcriptome. In prior work, we demonstrated upregulation of translation initiation in postnatal blood leukocyte expression in ASD toddlers.98 Furthermore, analysis of how gene co-expression modules interact within the cortical transcriptome of ASD (i.e. via eigengene network analysis of the same ASD transcriptome dataset analyzed here) supports the idea that a MIA-preserved translation initiation module is highly connected with immune/inflammation modules and other synaptic, cell cycle, and neurogenesis processes.99 This systems biology link between atypical translational processes and neural dysregulation at the synapse, immune/inflammation processes and regulation of cell number is important to underscore, as it may suggest a larger more unified systems biological disruption than can be accounted for, by only looking specifically at individual modules. In addition, the fact that dysregulation of translation initiation can be found systemically in blood,98 and is not specific to neural tissue, may allow to further test hypotheses about the relation between this type of dysregulation with other sorts of systemic dysregulation and interaction with immune and inflammation processes.
Translation and protein synthesis mechanisms have been highly important within examination of syndromic forms of ASD. Kelleher and Bear78 suggested a ‘troubled translation’ hypothesis of ASD by linking mutations associated with syndromic forms of ASD to altered translation and disturbance of synaptic processes. This hypothesis has been further elaborated by Santini and Klann and others, with new evidence supporting the crucial role of cap-dependent translation protein EIF4E79, 88, 100, 101 in ASD pathophysiology.77 The current data support these ideas that translation processes are integral to ASD, and that MIA induces substantial early dysregulation of such processes. To investigate possible consequences of upregulated translation in our MIA mouse model experiment we tested whether key regulatory genes such as TSC1/2 and mTOR that are upstream to the EIF4E-complex regulating cap-dependent translation were a reproducible transcriptional phenotype of MIA. We report the novel finding that MIA in rodents during early fetal brain development influences the expression of the TSC–mTOR–EIF4E axis and the regulation of EIF4E-binding proteins. This experimental evidence in a model system plus across species comparison of expression data indicates disrupted cap-dependent translation is common across rat MIA, mouse MIA and human ASD cortex. This finding warrants future examination to identify the specific downstream cellular and molecular phenotypes of abnormal cortical development.
With regard to MIA dysregulation of the TSC1/2–mTOR–EIF4E axis, we predicted that there are potential downstream effects involving genes that regulate cell proliferation, specifically controlling G1–S and G2–M cell cycle phase transition. The link we found between disrupted translation upon MIA or in ASD cortex and cell cycle processes is supported by other studies. MIA alters proliferation of cortical neural progenitor cells, laminar allocation of neurons, increased cortical thickness, increased cell density and patches of cortical dysplasia.32, 38, 42 Increased proliferation of neural progenitor cells associated with brain overgrowth was observed after low-dose LPS treatment and was more pronounced in a Pten haploinsufficient background, demonstrating clear genetic–environmental effects on early brain growth.39 We recently demonstrated that genes frequently found mutated in ASD1 may regulate the downstream expression of genes directly relevant to brain size as well as other regulatory genes with cell cycle functions, particularly those involved in the regulation of the G1–S phase transition.67 Further strengthening this evidence, in vitro iPSC studies have bridged molecular and cellular phenotypes of cell cycle timing during neural progenitor cell division to abnormal cortical development in ASD subjects with enlarged brains.37, 102
Our evidence, showing reproducible MIA-induced upregulation of EIF4E and downregulation of two binding proteins (EIF4EBP1/2), suggest a possible imbalance in the regulation of neurogenic versus neural progenitor divisions during development. In vivo evidence has indeed shown that normal expression and proper binding to EIF4E is required to maintain the correct balance.77 We hypothesize that reduced binding of EIF4E partners may lead, directly or through a compensatory mechanism, to increased production of EIF4E, which in turn is sufficient to abnormally expand the number of neural precursor cells.77
We also find consistent evidence for decreased mTOR expression across both rat and mouse MIA models. Although this finding is in the opposite direction of known hyperactivation of mTOR by some highly ASD-penetrant mutations, it is consistent with other evidence from genetic103 and environmental models104 of ASD as well as evidence showing that mTOR protein levels in post-mortem cortical tissue of ASD patients can be increased105 or decreased.104 Thus, these results further add to the literature supporting the idea that dysregulation of mTOR in either direction is likely important in ASD.104
In addition to underscoring how the results on MIA relate to ASD, it is also important to discuss links between how MIA-induced transcriptome dysregulation maps onto known pathways one would expect to be hit by inflammation/injury in general. In particular, during the acute stage of inflammation/injury, expression of cell cycle and translation processes have been shown to be upregulated, while many of the MIA-downregulated synaptic and neuronal processes implicated here are also known to be downregulated in acute stages of inflammation/injury.106, 107, 108 This similarity is important to underscore since it may point towards the idea that MIA as an environmental risk factor for ASD enhances known biological processes that may mimic effects seen during acute stages of inflammation/injury, and that MIA’s shared pathways with ASD may also overlap with general processes known to occur under these circumstances. A related concept here could also be to link such phenomena with what Naviaux et al.55, 94, 95 refer to as MIA eliciting a ‘cell danger response’. Both of these concepts could be conceptually important for linking MIA-induced effects on the brain with other concepts in molecular biology and immunology.
We initially hypothesized that MIA may result in long-term alterations in transcriptome at later recovery phase. To test this possibility, we investigated DE signals at 24 h post-treatment data-set by comparing with controls in the LPS-injected rat model. Further support for the idea that MIA may result in acute effects can be seen in the absence of much DE signal in the 24 h post-treatment dataset. This evidence of massive transcriptome dysregulation early on, following the MIA-inducing event (i.e. 4 h dataset) followed by relative normalization of most pathological processes (i.e. 24 h dataset), further suggests that MIA’s influence may be most potent during and shortly after the event when immune mediators (e.g. cytokines, chemokines) are also temporarily upregulated. This idea has further impact when hypothesizing about the effects of temporal duration of infections in humans. This evidence may support the idea that temporal duration of maternal infections in pregnancy may be important as longer-lasting MIA events may induce more detrimental effects on the fetus.
In summary, we show that many genes that are strongly dysregulated in early fetal brain development by MIA highly overlap with known ASD-associated genes and gene targets of two key ASD genes, FMR1 and CHD8. At the same time, we show MIA additionally dysregulates large numbers of other genes that impact a multitude of early pivotal fetal programs that govern cell number, type, migration, laminar organization, axon guidance, growth and differentiation, and these early functional genomic aberrances are largely not detectable at later ages in the mature ASD cortex. Increased awareness and knowledge about the impact of maternal infections during pregnancy on later risk for neuropsychiatric disorders like autism are particularly important given that such events are potentially preventable or could be largely reduced by changing practices.27, 28 In addition, MIA represents a potential etiology that could be more amenable to novel treatments.54, 55 Our work has highlighted the particular pathways related to translation initiation that could help to potentially explain the links between MIA and ASD, and more work is needed to explore dysregulation of these processes and how potentially one could intervene and reshape such dysregulation. Finally, this work explains why MIA is a prominent risk factor for ASD and suggests that interactions between such risk and gene risk factors may enhance ASD risk.
Acknowledgments
We thank Nathan Lewis and Anthony Wynshaw-Boris for helpful discussion of this work. This work is supported by grants (KL2TR00099 and 1KL2TR001444) from the University of California, San Diego Clinical and Translational Research Institute to Dr Pramparo and grants from the Simons Foundation Autism Research Initiative awarded to Prof. Courchesne (SFARI #176540) and Prof. Theo D Palmer (SFARI #323220), by a grant from the National Institute of Health awarded to Prof. Courchesne (R01MH110558), and a postdoctoral fellowship from Child Health Research Institute at Stanford University School of Medicine awarded to Dr Moon.
Author contributions
MVL, EC and TP conceived the idea for the study. MVL and TP conceived and implemented all data analyses. HMM, JS, TDP and TP conceived the idea for MIA mouse model validation study and performed qRT-PCR experiment/data analysis. MVL, EC and TP interpreted the results and wrote the manuscript. All authors read and approved the final manuscript.
Footnotes
Supplementary Information accompanies the paper on the Molecular Psychiatry website (http://www.nature.com/mp)
The authors declare no conflict of interest.
Supplementary Material
References
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014; 515: 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH, State MW. Gene hunting in autism spectrum disorder: on the path to precision medicine. Lancet Neurol 2015; 14: 1109–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014; 515: 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumm N, Turner TN, Baker C, Vives L, Mohajeri K, Witherspoon K et al. Excess of rare, inherited truncating mutations in autism. Nat Genet 2015; 47: 582–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova EL, Sharp JL, Chakraborty H, Sumi NS, Casanova MF. Genes with high penetrance for syndromic and non-syndromic autism typically function within the nucleus and regulate gene expression. Mol Autism 2016; 7: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012; 485: 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klei L, Sanders SJ, Murtha MT, Hus V, Lowe JK, Willsey AJ et al. Common genetic variants, acting additively, are a major source of risk for autism. Mol Autism 2012; 3: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB et al. Most genetic risk for autism resides with common variation. Nat Genet 2014; 46: 881–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandin S, Lichtenstein P, Kuja-Halkola R, Larsson H, Hultman CM, Reichenberg A. The familial risk of autism. JAMA 2014; 311: 1770–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry 2011; 68: 1095–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoner R, Chow ML, Boyle MP, Sunkin SM, Mouton PR, Roy S et al. Patches of disorganization in the neocortex of children with autism. N Engl J Med 2014; 370: 1209–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum R, Jaffe AE, Hyde TM, Kleinman JE, Weinberger DR. Prenatal expression patterns of genes associated with neuropsychiatric disorders. Am J Psychiatry 2014; 171: 758–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 2013; 155: 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courchesne E, Mouton PR, Calhoun ME, Semendeferi K, Ahrens-Barbeau C, Hallet MJ et al. Neuron number and size in prefrontal cortex of children with autism. JAMA 2011; 306: 2001–2010. [DOI] [PubMed] [Google Scholar]
- Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 2013; 155: 1008–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brimberg L, Sadiq A, Gregersen PK, Diamond B. Brain-reactive IgG correlates with autoimmunity in mothers of a child with an autism spectrum disorder. Mol Psychiatry 2013; 18: 1171–1177. [DOI] [PubMed] [Google Scholar]
- Atladottir HO, Thorsen P, Ostergaard L, Schendel DE, Lemcke S, Abdallah M et al. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J Autism Dev Disord 2010; 40: 1423–1430. [DOI] [PubMed] [Google Scholar]
- Jiang HY, Xu LL, Shao L, Xia RM, Yu ZH, Ling ZX et al. Maternal infection during pregnancy and risk of autism spectrum disorders: a systematic review and meta-analysis. Brain Behav Immun 2016; 58: 165–172. [DOI] [PubMed] [Google Scholar]
- Lee BK, Magnusson C, Gardner RM, Blomstrom A, Newschaffer CJ, Burstyn I et al. Maternal hospitalization with infection during pregnancy and risk of autism spectrum disorders. Brain Behav Immun 2015; 44: 100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron-Cohen S, Auyeung B, Norgaard-Pedersen B, Hougaard DM, Abdallah MW, Melgaard L et al. Elevated fetal steroidogenic activity in autism. Mol Psychiatry 2015; 20: 369–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ. In utero programming of chronic disease. Clin Sci (Lond) 1998; 95: 115–128. [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med 2008; 359: 61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chess S. Autism in children with congenital rubella. J Autism Child Schizophr 1971; 1: 33–47. [DOI] [PubMed] [Google Scholar]
- Brown AS, Sourander A, Hinkka-Yli-Salomaki S, McKeague IW, Sundvall J, Surcel HM. Elevated maternal C-reactive protein and autism in a national birth cohort. Mol Psychiatry 2014; 19: 259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbo O, Qian Y, Yoshida C, Grether JK, Van de Water J, Croen LA. Maternal infection during pregnancy and autism spectrum disorders. J Autism Dev Disord 2015; 45: 4015–4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goines PE, Croen LA, Braunschweig D, Yoshida CK, Grether J, Hansen R et al. Increased midgestational IFN-γ IL-4 and IL-5 in women bearing a child with autism: a case-control study. Mol Autism 2011; 2: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuesel I, Chicha L, Britschgi M, Schobel SA, Bodmer M, Hellings JA et al. Maternal immune activation and abnormal brain development across CNS disorders. Nat Rev Neurol 2014; 10: 643–660. [DOI] [PubMed] [Google Scholar]
- Patterson PH. Maternal infection and immune involvement in autism. Trends Mol Med 2011; 17: 389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes ML, McAllister AK. Maternal immune activation: implications for neuropsychiatric disorders. Science (New York, NY) 2016; 353: 772–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U. Prenatal poly(i:C) exposure and other developmental immune activation models in rodent systems. Biol Psychiatry 2014; 75: 307–315. [DOI] [PubMed] [Google Scholar]
- Oskvig DB, Elkahloun AG, Johnson KR, Phillips TM, Herkenham M. Maternal immune activation by LPS selectively alters specific gene expression profiles of interneuron migration and oxidative stress in the fetus without triggering a fetal immune response. Brain Behav Immun 2012; 26: 623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science (New York, NY) 2016; 351: 933–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci 2003; 23: 297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkova NV, Yu CZ, Hsiao EY, Moore MJ, Patterson PH. Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav Immun 2012; 26: 607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci 2007; 27: 10695–10702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer A. Neocortical neurogenesis and the etiology of autism spectrum disorder. Neurosci Biobehav Rev 2016; 64: 185–195. [DOI] [PubMed] [Google Scholar]
- Marchetto MC, Belinson H, Tian Y, Freitas BC, Fu C, Vadodaria KC et al. Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol Psychiatry 2016; doi: 10.1038/mp.2016.95. [DOI] [PMC free article] [PubMed]
- Smith SE, Elliott RM, Anderson MP. Maternal immune activation increases neonatal mouse cortex thickness and cell density. J Neuroimmune Pharmacol 2012; 7: 529–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Belle JE, Sperry J, Ngo A, Ghochani Y, Laks DR, Lopez-Aranda M et al. Maternal inflammation contributes to brain overgrowth and autism-associated behaviors through altered redox signaling in stem and progenitor cells. Stem Cell Rep 2014; 3: 725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann CM, Bloss CS, Barnes CC, Wideman GM, Carper RA, Akshoomoff N et al. Longitudinal magnetic resonance imaging study of cortical development through early childhood in autism. J Neurosci 2010; 30: 4419–4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen MD, Nordahl CW, Young GS, Wootton-Gorges SL, Lee A, Liston SE et al. Early brain enlargement and elevated extra-axial fluid in infants who develop autism spectrum disorder. Brain 2013; 136(Pt 9): 2825–2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soumiya H, Fukumitsu H, Furukawa S. Prenatal immune challenge compromises the normal course of neurogenesis during development of the mouse cerebral cortex. J Neurosci Res 2011; 89: 1575–1585. [DOI] [PubMed] [Google Scholar]
- Patrich E, Piontkewitz Y, Peretz A, Weiner I, Attali B. Maternal immune activation produces neonatal excitability defects in offspring hippocampal neurons from pregnant rats treated with poly I:C. Sci Rep 2016; 6: 19106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovanoli S, Engler H, Engler A, Richetto J, Voget M, Willi R et al. Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. Science (New York, NY) 2013; 339: 1095–1099. [DOI] [PubMed] [Google Scholar]
- Morgan JT, Chana G, Pardo CA, Achim C, Semendeferi K, Buckwalter J et al. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol Psychiatry 2010; 68: 368–376. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Sugihara G, Ouchi Y, Nakamura K, Futatsubashi M, Takebayashi K et al. Microglial activation in young adults with autism spectrum disorder. JAMA Psychiatry 2013; 70: 49–58. [DOI] [PubMed] [Google Scholar]
- Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol 2005; 57: 67–81. [DOI] [PubMed] [Google Scholar]
- Richetto J, Calabrese F, Riva MA, Meyer U. Prenatal immune activation induces maturation-dependent alterations in the prefrontal GABAergic transcriptome. Schizophr Bull 2014; 40: 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Smith SE, Malkova N, Tse D, Su Y, Patterson PH. Activation of the maternal immune system alters cerebellar development in the offspring. Brain Behav Immun 2009; 23: 116–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir RK, Forghany R, Smith SE, Patterson PH, McAllister AK, Schumann CM et al. Preliminary evidence of neuropathology in nonhuman primates prenatally exposed to maternal immune activation. Brain Behav Immun 2015; 48: 139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Ellis SE, Ashar FN, Moes A, Bader JS, Zhan J et al. Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat Commun 2014; 5: 5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011; 474: 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow ML, Pramparo T, Winn ME, Barnes CC, Li HR, Weiss L et al. Age-dependent brain gene expression and copy number anomalies in autism suggest distinct pathological processes at young versus mature ages. PLoS Genet 2012; 8: e1002592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013; 155: 1451–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naviaux JC, Schuchbauer MA, Li K, Wang L, Risbrough VB, Powell SB et al. Reversal of autism-like behaviors and metabolism in adult mice with single-dose antipurinergic therapy. Transl Psychiatry 2014; 4: e400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012; 28: 882–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet 2007; 3: 1724–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015; 43: e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD. A direct approach to false discovery rates. J R Statist Soc B 2002; 64: 479–498. [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA 2003; 100: 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008; 9: 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JA, Cai C, Langfelder P, Geschwind DH, Kurian SM, Salomon DR et al. Strategies for aggregating gene expression data: the collapseRows R function. BMC Bioinformatics 2011; 12: 322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Zhang B, Horvath S. Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics 2008; 24: 719–720. [DOI] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. Eigengene networks for studying the relationships between co-expression modules. BMC Syst Biol 2007; 1: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhagen J, Wagenmakers EJ. Bayesian tests to quantify the result of a replication attempt. J Exp Psychol Gen 2014; 143: 1457–1475. [DOI] [PubMed] [Google Scholar]
- Clancy B, Darlington RB, Finlay BL. Translating developmental time across mammalian species. Neuroscience 2001; 105: 7–17. [DOI] [PubMed] [Google Scholar]
- Pramparo T, Lombardo MV, Campbell K, Carter Barnes C, Marinero S, Solso S et al. Cell cycle networks link gene expression dysregulation, mutation, and brain maldevelopment in autistic toddlers. Mol Syst Biol 2015; 11: 841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu SN, Kollu R, Banerjee-Basu S. AutDB: a gene reference resource for autism research. Nucleic Acids Res 2009; 37: D832–D836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahams BS, Arking DE, Campbell DB, Mefford HC, Morrow EM, Weiss LA et al. SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol Autism 2013; 4: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernier R, Golzio C, Xiong B, Stessman HA, Coe BP, Penn O et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell 2014; 158: 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JD, Bassell GJ, Klann E. Dysregulation and restoration of translational homeostasis in fragile X syndrome. Nat Rev 2015; 16: 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascano MJr., Mukherjee N, Bandaru P, Miller JB, Nusbaum JD, Corcoran DL et al. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 2012; 492: 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 2011; 146: 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotney J, Muhle RA, Sanders SJ, Liu L, Willsey AJ, Niu W et al. The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nat Commun 2015; 6: 6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugathan A, Biagioli M, Golzio C, Erdin S, Blumenthal I, Manavalan P et al. CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc Natl Acad Sci USA 2014; 111: E4468–E4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Luo R, Oldham MC, Horvath S. Is my network module preserved and reproducible? PLoS Comput Biol 2011; 7: e1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Smibert CA, Kaplan DR, Miller FD. An eIF4E1/4E-T complex determines the genesis of neurons from precursors by translationally repressing a proneurogenic transcription program. Neuron 2014; 84: 723–739. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ3rd, Bear MF. The autistic neuron: troubled translation? Cell 2008; 135: 401–406. [DOI] [PubMed] [Google Scholar]
- Santini E, Klann E. Reciprocal signaling between translational control pathways and synaptic proteins in autism spectrum disorders. Sci Signal 2014; 7: re10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Alberts I, Li X. Dysregulation of the IGF-I/PI3K/AKT/mTOR signaling pathway in autism spectrum disorders. Int J Dev Neurosci 2014; 35: 35–41. [DOI] [PubMed] [Google Scholar]
- Gao X, Zhang Y, Arrazola P, Hino O, Kobayashi T, Yeung RS et al. Tsc tumour suppressor proteins antagonize amino-acid-TOR signalling. Nat Cell Biol 2002; 4: 699–704. [DOI] [PubMed] [Google Scholar]
- Smalley SL. Autism and tuberous sclerosis. J Autism Dev Disord 1998; 28: 407–414. [DOI] [PubMed] [Google Scholar]
- Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 2003; 17: 1829–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Klann E, Costa-Mattioli M, Zukin RS. Dysregulation of mammalian target of rapamycin signaling in mouse models of autism. J Neurosci 2015; 35: 13836–13842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Hoeffer CA, Takayasu Y, Miyawaki T, McBride SM, Klann E et al. Dysregulation of mTOR signaling in fragile X syndrome. J Neurosci 2010; 30: 694–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi JA, Collins KA, Hays SA, Tsai NP, Guo W, Birnbaum SG et al. Disrupted Homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nat Neurosci 2012; 15: 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Klann E. The translation of translational control by FMRP: therapeutic targets for FXS. Nat Neurosci 2013; 16: 1530–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves-Pereira M, Muller B, Massie D, Williams JH, O'Brien PC, Hughes A et al. Deregulation of EIF4E: a novel mechanism for autism. J Med Genet 2009; 46: 759–765. [DOI] [PubMed] [Google Scholar]
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev 2004; 18: 1926–1945. [DOI] [PubMed] [Google Scholar]
- Carpentier PA, Dingman AL, Palmer TD. Placental TNF-α signaling in illness-induced complications of pregnancy. Am J Pathol 2011; 178: 2802–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentier PA, Haditsch U, Braun AE, Cantu AV, Moon HM, Price RO et al. Stereotypical alterations in cortical patterning are associated with maternal illness-induced placental dysfunction. J Neurosci 2013; 33: 16874–16888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehninger D, Sano Y, de Vries PJ, Dies K, Franz D, Geschwind DH et al. Gestational immune activation and Tsc2 haploinsufficiency cooperate to disrupt fetal survival and may perturb social behavior in adult mice. Mol Psychiatry 2012; 17: 62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazina V, Gerdts J, Trinh S, Ankenman K, Ward T, Dennis MY et al. Epigenetics of autism-related impairment: copy number variation and maternal infection. J Dev Behav Pediatr 2015; 36: 61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naviaux JC, Wang L, Li K, Bright AT, Alaynick WA, Williams KR et al. Antipurinergic therapy corrects the autism-like features in the Fragile X (Fmr1 knockout) mouse model. Mol Autism 2015; 6: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naviaux RK, Zolkipli Z, Wang L, Nakayama T, Naviaux JC, Le TP et al. Antipurinergic therapy corrects the autism-like features in the poly(IC) mouse model. PLoS ONE 2013; 8: e57380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker-Athill E, Luo D, Bailey A, Giunta B, Tian J, Shytle RD et al. Flavonoids, a prenatal prophylaxis via targeting JAK2/STAT3 signaling to oppose IL-6/MIA associated autism. J Neuroimmunol 2009; 217: 20–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova EL, Casanova MF. Genetics studies indicate that neural induction and early neuronal maturation are disturbed in autism. Front Cell Neurosci 2014; 8: 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramparo T, Pierce K, Lombardo MV, Carter Barnes C, Marinero S, Ahrens-Barbeau C et al. Prediction of autism by translation and immune/inflammation coexpressed genes in toddlers from pediatric community practices. JAMA Psychiatry 2015; 72: 386–394. [DOI] [PubMed] [Google Scholar]
- Lombardo MV, Courchesne E, Lewis NE, Pramparo T. Hierarchical cortical transcriptome disorganization in autism. bioRxiv, 2016; Available at: http://biorxiv.org/. bioRxiv 042937; doi: https://doi.org/10.1101/042937. [DOI] [PMC free article] [PubMed]
- Gkogkas CG, Khoutorsky A, Ran I, Rampakakis E, Nevarko T, Weatherill DB et al. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 2013; 493: 371–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Huynh TN, MacAskill AF, Carter AG, Pierre P, Ruggero D et al. Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature 2013; 493: 411–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani J, Coppola G, Zhang P, Abyzov A, Provini L, Tomasini L et al. FOXG1-dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell 2015; 162: 375–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Lehtinen MK, Sessa A, Zappaterra MW, Cho SH, Gonzalez D et al. The apical complex couples cell fate and cell survival to cerebral cortical development. Neuron 2010; 66: 69–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolini C, Ahn Y, Michalski B, Rho JM, Fahnestock M. Decreased mTOR signaling pathway in human idiopathic autism and in rats exposed to valproic acid. Acta Neuropathol Commun 2015; 3: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Gudsnuk K, Kuo SH, Cotrina ML, Rosoklija G, Sosunov A et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 2014; 83: 1131–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu XC, Williams AJ, Yao C, Berti R, Hartings JA, Whipple R et al. Microarray analysis of acute and delayed gene expression profile in rats after focal ischemic brain injury and reperfusion. J Neurosci Res 2004; 77: 843–857. [DOI] [PubMed] [Google Scholar]
- Matzilevich DA, Rall JM, Moore AN, Grill RJ, Dash PK. High-density microarray analysis of hippocampal gene expression following experimental brain injury. J Neurosci Res 2002; 67: 646–663. [DOI] [PubMed] [Google Scholar]
- Schreiber G, Howlett G, Nagashima M, Millership A, Martin H, Urban J et al. The acute phase response of plasma protein synthesis during experimental inflammation. J Biol Chem 1982; 257: 10271–10277. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.