

Abstract
The idea that disturbances occurring early in brain development contribute to the pathogenesis of schizophrenia, often referred to as the neurodevelopmental hypothesis, has become widely accepted. Despite this, the disorder is viewed as being distinct nosologically, and by implication pathophysiologically and clinically, from syndromes such as autism spectrum disorders, attention‐deficit/hyperactivity disorder (ADHD) and intellectual disability, which typically present in childhood and are grouped together as “neurodevelopmental disorders”. An alternative view is that neurodevelopmental disorders, including schizophrenia, rather than being etiologically discrete entities, are better conceptualized as lying on an etiological and neurodevelopmental continuum, with the major clinical syndromes reflecting the severity, timing and predominant pattern of abnormal brain development and resulting functional abnormalities. It has also been suggested that, within the neurodevelopmental continuum, severe mental illnesses occupy a gradient of decreasing neurodevelopmental impairment as follows: intellectual disability, autism spectrum disorders, ADHD, schizophrenia and bipolar disorder. Recent genomic studies have identified large numbers of specific risk DNA changes and offer a direct and robust test of the predictions of the neurodevelopmental continuum model and gradient hypothesis. These findings are reviewed in detail. They not only support the view that schizophrenia is a disorder whose origins lie in disturbances of brain development, but also that it shares genetic risk and pathogenic mechanisms with the early onset neurodevelopmental disorders (intellectual disability, autism spectrum disorders and ADHD). They also support the idea that these disorders lie on a gradient of severity, implying that they differ to some extent quantitatively as well as qualitatively. These findings have important implications for nosology, clinical practice and research.
Keywords: Schizophrenia, neurodevelopment, autism, ADHD, intellectual disability, bipolar disorder, genomics, copy number variants
The neurodevelopmental hypothesis has been the dominant framework within which research on schizophrenia has been conducted since the influential papers of Weinberger1 and Murray and Lewis2 thirty years ago.
The crucial conceptual advance was the proposal that the emergence of schizophrenia in adolescence or early adulthood could be explained by the interaction between an early “lesion” to the developing brain, arising from genetic and environmental factors, and normal developmental processes. According to this view, as the brain develops and takes on new and more complex functions, the impact of early neurodevelopmental pathology can become apparent.
The idea that schizophrenia might have its origins in disturbances of early neurodevelopment was not new, and both Kraepelin and Bleuler were aware that the developmental histories of those with schizophrenia could be abnormal3. However, the neurodevelopmental hypothesis brought together findings implicating early environmental exposures, such as obstetric injury, with those from clinical and basic neuroscience implicating cognitive impairment and cortical dysfunction, and evidence for “premorbid” developmental deviance. Crucially, it provided a framework to explain how early developmental abnormalities might be manifest as psychosis in late adolescence and early adulthood when schizophrenia typically presents, and explained the failure to identify neurodegenerative, traumatic or neurotoxic mechanisms in post mortem studies1.
THE NEURODEVELOPMENTAL CONTINUUM
While the neurodevelopmental hypothesis has been hugely influential within the confines of schizophrenia research, its broader implications for nosology, diagnosis, management, research and prevention remain largely overlooked4.
Despite general acceptance that schizophrenia has a substantial neurodevelopmental basis, the disorder remains widely regarded as being distinct nosologically, and by implication pathophysiologically and clinically, from syndromes such as autism spectrum disorders, attention‐deficit/hyperactivity disorder (ADHD) and intellectual disability, which typically present in childhood and are grouped together as “neurodevelopmental disorders”5.
This separation overlooks several key observations4, 6, 7, 8, 9. First, there are many clinical and other phenotypic similarities between schizophrenia and childhood neurodevelopmental syndromes7, 9. These have tended to be overlooked because of the prominence given to psychotic symptoms in schizophrenia by researchers and clinicians. This focus on symptoms that typically present after childhood has drawn attention from the fact that schizophrenia shares with childhood neurodevelopmental disorders impairments of cognition, which are often present before psychotic breakdown, a greater frequency in males, and associations with varying degrees of developmental delay, neurological soft signs and motor abnormalities. Second, there are no clear diagnostic boundaries between these disorders, and there is a significant comorbidity between them that is obscured by the use of diagnostic hierarchies or exclusions, developmental change in predominant symptom type, and service configurations4. Third, a number of environmental risk factors, particularly those impacting on early brain development, are shared across these disorders4, 9. Finally, and most tellingly, evidence began to emerge about ten years ago, particularly from studies of rare copy number variants, that childhood neurodevelopmental disorders such as intellectual disability, autism spectrum disorders and ADHD share specific genetic risk alleles with each other and with schizophrenia4, 6.
Consideration of these issues led us to reappraise the neurodevelopmental hypothesis of schizophrenia and propose a new model, the neurodevelopmental continuum4, 6, in which neurodevelopmental disorders, including schizophrenia, are seen as representing the diverse range of outcomes that follow from disrupted or deviant brain development. This model was based on the emerging evidence for shared genetic and environmental risk factors and predicts that there are also likely to be overlapping pathogenic mechanisms.
Thus, childhood neurodevelopmental disorders (such as intellectual disability, autism spectrum disorders and ADHD) and adult psychiatric disorders (including both schizophrenia and bipolar disorder), rather than being etiologically discrete entities, could better be conceptualized as lying on an etiological and neurodevelopmental continuum or spectrum, with the major clinical syndromes reflecting the severity, timing and predominant pattern of abnormal brain development and resulting functional abnormalities, as well as the modifying effects of other genetic and environmental factors4, 6.
This approach accepts that current diagnostic systems have some utility in defining groups of cases that are more closely related than by chance, but it regards current categorical diagnoses as arbitrary divisions of what is essentially a continuous etiological, pathogenic, developmental and clinical landscape. The implications of this for research and practice are substantial4, 8.
The notion of a spectrum or continuum in childhood neurodevelopmental disorders was not a new one10, 11, but we expanded this further across the hitherto deep nosological divide between childhood neurodevelopmental disorders and psychiatric disorders that present in adulthood, such as schizophrenia and bipolar disorder. Subsequently, others have made a similar suggestion12.
THE NEURODEVELOPMENTAL GRADIENT
We have also proposed a more refined, and testable, conceptualization: the neurodevelopmental gradient hypothesis. This suggests that, within the neurodevelopmental continuum, severe mental illnesses occupy a gradient of decreasing neurodevelopmental impairment as follows: intellectual disability, autism spectrum disorders, ADHD, schizophrenia and bipolar disorder4, 6, 8. The severity of neurodevelopmental impairment is indexed by a number of features. These include typical age at onset (congenital for intellectual disability, early childhood for autism spectrum disorders, adolescence for schizophrenia) as well as the severity of associated cognitive impairment and the persistence of functional impairment (see Figure 1).
Figure 1.
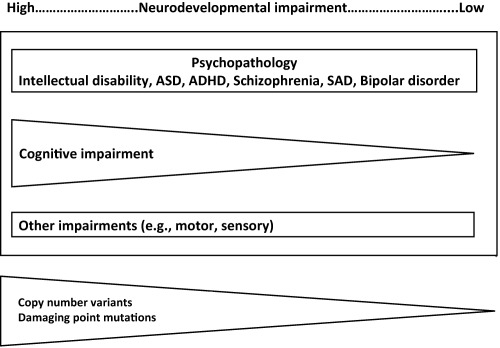
The neurodevelopmental continuum. This shows the different domains of outcome of neurodevelopmental impairment. It also shows the hypothesized relationship between the severity of neurodevelopmental impairment and psychiatric syndromes and degree of associated cognitive impairment. The relative impact of copy number variants and damaging point mutations is also shown. ASD – autism spectrum disorders, ADHD – attention‐deficit/hyperactivity disorder, SAD – schizoaffective disorder
Like all models, that of a neurodevelopmental gradient is certainly an oversimplification. Neurodevelopmental disorders clearly differ along a number of additional clinical dimensions, and presumably there are mechanistic differences as well, but it posits that the degree of neurodevelopmental impairment is currently the most recognizable of these features. It makes clear predictions about the relative importance across the neurodevelopmental spectrum of the most damaging classes of rare mutations, such as large copy number variants and rare coding variants. It also makes predictions about the relative extent of brain dysfunction (number of structures and circuits affected) in the various clinical syndromes and the relationships and likely similarities between disorders according to their relative position on the gradient.
In recent years, there has been increasing evidence from family studies for shared, as well as independent, genetic risk between different adult psychiatric disorders, and between adult disorders and childhood neurodevelopmental disorders7, 13, 14, 15, 16. There has also been an accumulation of evidence that schizophrenia shares environmental risk factors with childhood neurodevelopmental disorders, particularly those likely to index early neurodevelopmental impairment17, 18, 19, 20, 21. At the same time, there has been a profusion of large, increasingly well‐powered genomic studies of childhood neurodevelopmental disorders, particularly autism spectrum disorders and intellectual disability, and of adult psychiatric disorders, in particular schizophrenia.
In contrast to the environmental exposures, which generally are risk indicators rather than factors known to be causal, the identification of large numbers of specific risk DNA changes offers a direct and robust test of the predictions of the continuum model and gradient hypothesis, and for this reason it is considered in detail in this paper.
GENETICS OF SCHIZOPHRENIA
Genetic risk for schizophrenia is conferred by both rare and common alleles distributed across the genome22. The largest published analysis of genome‐wide association study data (up to 36,989 cases and 113,075 controls including replication data) identified a total of 108 conservatively defined loci that contain common risk alleles, and which met genome‐wide significance23.
These robustly implicated loci access only a small fraction of the total number of common alleles involved in conferring risk to schizophrenia, and studies of the en masse effects of common variants have suggested that between a half to a third of the genetic risk of schizophrenia is indexed by common alleles genotyped by current genome‐wide association study arrays24, 25. Recent estimates suggest that there may be many thousands of common risk alleles for schizophrenia, with 71‐100% of 1 Mb regions containing a schizophrenia locus26.
In addition to common alleles, each of which confers only a small increase in individual risk (odds ratio, OR < 1.2), a relatively small number of copy number variants are associated with substantial increases in individual risk, with ORs of 1.5 to >5027, 28. A recent meta‐analysis of previously implicated candidate copy number variants robustly identified eleven specific variants as schizophrenia risk factors28. These schizophrenia‐associated copy number variants are extremely rare, being found in 1 in 200 to 1 in several thousand people with the disorder, and have required large sample sizes to confidently implicate them28.
The genome‐wide burden of >500kb copy number variants has been shown to be significantly increased in schizophrenia compared with controls even after excluding known risk loci29, suggesting the existence of further schizophrenia risk variants. More recently, a genome‐wide investigation applying a centralized analysis pipeline to a schizophrenia cohort of 21,094 cases and 20,227 controls30 reported a global enrichment of copy number variants burden in cases, which persisted after excluding loci implicated in previous studies. Genome‐wide significant evidence was obtained for eight loci, and suggestive support was found for eight additional candidate susceptibility and protective loci.
Most of the specific copy number variants definitively associated with schizophrenia impact on multiple genes. The exception to this is deletions of NRXN128, 31, the gene that encodes the presynaptic cell adhesion protein neurexin1. In order to infer the biological mechanism(s) through which multigenic copy number variants contribute to disease, researchers have sought to determine whether the genes impacted by schizophrenia‐related variants are enriched for functionally related sets of genes. This is often termed pathway analysis. Studies using this approach have yielded remarkably consistent findings. Schizophrenia‐related variants are enriched for synaptic genes30, 32, 33, 34, 35, 36, and particularly those encoding members of N‐methyl‐D‐aspartate receptor and neuronal activity regulated cytoskeleton‐associated protein complexes, both of which are known to be important for glutamatergic signaling and synaptic plasticity30, 34, 36. A recent large case‐control study showed that case copy number variants are also enriched for genes involved in GABAergic neurotransmission36.
Finally, recent large‐scale work using new generation sequencing approaches, predominantly exome sequencing to date, has shown that rare coding variants that change the DNA sequence at one or a few nucleotides are enriched in specific gene pathways, particularly those involved in synaptic function, including many of those implicated in studies of copy number variants37, 38, 39, and that ultra‐rare, gene‐disruptive and putatively protein damaging variants are more abundant in schizophrenia than among controls39. Finally, loss‐of‐function rare coding variants in a gene that encodes the histone methyltransferase SETD1A have been shown to be associated with schizophrenia40. This is the first gene to be implicated in schizophrenia by exome sequencing at Bonferroni corrected genome‐wide levels of statistical significance and, when combined with previous common variant evidence41, points to chromatin remodelling, specifically histone H3K4 methylation, as an important mechanism in the pathogenesis of schizophrenia.
COMPARATIVE GENETIC ARCHITECTURE OF SCHIZOPHRENIA AND OTHER NEURODEVELOPMENTAL DISORDERS
Copy number variants
A major impetus for the continuum model and gradient hypothesis came from the observation that specific rare copy number variants that are significantly associated with schizophrenia are also associated with a range of other neurodevelopmental disorders, such as autism spectrum disorders, ADHD and intellectual disability31, 42, 43.
Although there have been no unbiased population studies conducted to date, it is apparent that the severity of the neurodevelopmental outcome associated with such copy number variants is highly variable, with phenotypes ranging from mild cognitive impairment in some individuals44, 45 through to schizophrenia, autism, ADHD or intellectual disability in others42, 46. Moreover, the evidence suggests that this reflects true pleiotropy rather than heterogeneity resulting from the multigenic nature of most copy number variants47.
Support for the neurodevelopmental gradient hypothesis has come from a number of observations. First, Girirajan et al48 showed that, in children, the burden of large copy number variants is positively correlated with the severity of childhood neurodevelopmental disorders, being greater in intellectual disability than in autism spectrum disorders, and greater in autism spectrum disorders with intellectual disability than in those without. Second, Kirov et al46 found that the burden of large rare copy number variants implicated in neurodevelopmental disorders is greater in cases with developmental delay, autism or congenital malformations than in schizophrenia. For most variants, penetrance for the early onset developmental disorders was greater than for schizophrenia; importantly, this was not only true for variants robustly identified first in the childhood disorders, but also for variants identified initially in schizophrenia, thus minimizing bias.
Furthermore, studies of patients with autism spectrum disorders, intellectual disability and congenital neurodevelopmental disorders referred to clinical genetics clinics for chromosomal microarray analysis have highlighted ninety loci enriched for copy number variants in these disorders, albeit not all are definitively implicated. Emphasizing the overlap between these disorders and schizophrenia, every schizophrenia‐associated variant is in this set of ninety childhood neurodevelopmental disorder copy number variants. Moreover, in a recent study of over 20,000 cases of schizophrenia, even after excluding known schizophrenia loci, copy number variations associated with intellectual disability were en masse significantly enriched in patients in schizophrenia49, supporting the view that many additional intellectual disability‐related variants also confer risk to schizophrenia, but at reduced penetrance.
The evidence suggests that large copy number variants are less strongly associated with bipolar disorder than schizophrenia50 and, where direct comparisons have been made, large rare variants were indeed found to be significantly less common in bipolar disorder than schizophrenia51, 52, 53, 54. These findings do not exclude the involvement of copy number variants at specific loci in susceptibility to bipolar disorder53: actually, there is strong evidence that duplications of 16p11.2 that are associated with schizophrenia are also associated with bipolar disorder53. However, it is now clear that relatively large copy number variants contributing to childhood neurodevelopmental disorders, and to impaired cognition in non‐clinical populations, contribute less to susceptibility for bipolar disorder than they do for schizophrenia. This is in keeping with the generally higher level of cognitive function and less persistent impairment seen in bipolar disorder, and supports the view that this disorder lies between schizophrenia and controls on the neurodevelopmental gradient (see Figure 1).
The neurodevelopmental gradient hypothesis further predicts that, among bipolar cases, those with cognitive impairments or earlier onsets would show a higher burden of large copy number variants. There is already some evidence, albeit not definitive, to support this55, 56.
Neurodevelopmental disorders, including schizophrenia, are associated with reduced fecundity57. Mutations that confer very high risk for those disorders should, therefore, be rare in the population due to strong negative selection, and the frequency in the population should, hypothetically, be a function of that selection pressure versus the rate of replacement by de novo mutation. Such a postulated relationship between selection pressure and de novo mutation rate has recently been empirically demonstrated for neurodevelopmental disorder‐associated copy number variants46. Assuming neurodevelopmental impairment to be a major driver of loss of fecundity, this leads to the prediction that the relative contribution of de novo mutations to different neurodevelopmental disorders should correlate with their position on the proposed neurodevelopmental gradient.
Unfortunately, precise comparisons of the de novo mutation rate between diagnoses are difficult, because there have been no direct tests based on identical arrays, mutation size cut‐offs, and epidemiologically ascertained samples fully representative of each diagnosis. However, the findings to date are broadly in line with the predictions of the neurodevelopmental gradient hypothesis. For example, it has been reported58 that the frequency of large (>100kb) de novo mutations in bipolar disorder (2.2%) is intermediate between schizophrenia (4.3%) and controls (1.5%). A recent large study of autism59 found a de novo mutation rate of 5.2% in cases and 1.6% in unaffected siblings. Finally, a recent large study of intellectual disability reported a de novo rate of 11.5% for rare mutations60.
Rare coding variants
As we have seen, specific mutations conferring high individual risk to neurodevelopmental disorders are likely to be rare, and large samples will be required to implicate them in case‐control studies. However, as is the case for copy number variants, in people with neurodevelopmental disorders, very high risk rare coding variants are likely to be enriched among mutations occurring de novo.
A higher than expected burden of mutations predicted to be functionally deleterious, loss of function and missense de novo mutations predicted by algorithms to be damaging, is clearly seen in intellectual disability and autism spectrum disorders40, 59, 61. The de novo burden in schizophrenia is much less pronounced, but it is nevertheless clearly present with respect to loss of function mutations40, especially in genes that are highly constrained by natural selection and in which loss of function mutations are more likely to be damaging62. When the relative enrichment of de novo mutations is compared across disorders, the rates are higher in intellectual disability than autism spectrum disorders, and higher in autism spectrum disorders than schizophrenia37, 40, 61, 62, in line with the predictions of the gradient hypothesis.
Moreover, there is evidence that schizophrenia patients with intellectual disability have a greater enrichment of rare damaging variants in highly constrained genes and developmental disorder genes, but that a weaker but significant enrichment exists throughout the larger schizophrenia population62. Also, even amongst those with schizophrenia who do not have intellectual disability, the rate of de novo loss of function mutations is higher in those with poorer educational attainment37. These findings are consistent with those in autism spectrum disorders, in which the burden of de novo mutations is positively correlated with the degree of cognitive impairment63.
We can also explore whether the same genes, or sets of functionally related genes, tend to be implicated across neurodevelopmental disorders, and this would appear to be the case. Genes affected by loss of functioning de novo mutations in schizophrenia are enriched for those affected by this same class of mutation in people with autism spectrum disorders and intellectual disability37. Genes and mutation sites were most highly conserved in intellectual disability, then autism spectrum disorders, with those in schizophrenia least conserved. When loss of function mutations in highly constrained genes are considered, a similar pattern is seen, with enrichment in schizophrenia concentrated in known autism spectrum disorder and intellectual disability genes62. At an even finer level of resolution, the same loss of function mutation in SETD1A gene that contributes high risk for schizophrenia also does so for severe intellectual disability and developmental delay40.
Finally, there is also evidence that the burden of rare variation found in schizophrenia, autism and intellectual disability is concentrated in functionally related sets of genes, particularly those involved in synaptic function and histone remodelling and other neurodevelopmental gene sets37, 62, 64, 65. These findings all converge on the conclusion that at least some of the risk to schizophrenia conferred by rare mutations of large effect is shared with childhood neurodevelopmental disorders and impacts on synaptic development and function. They also support the prediction of the neurodevelopmental gradient hypothesis that the burden of such mutations is greatest in intellectual disability, then in autism spectrum disorders and then in schizophrenia.
Bipolar disorder has been much less extensively studied by exome sequencing. Consistent with the picture that is more clearly emerging from studies of intellectual disability, autism spectrum disorders and schizophrenia, one small study found an excess of de novo loss of function and protein altering variants in mutation intolerant genes, and an association with early onset66, while a second study found that damaging variants were enriched for genes previously found to contain de novo mutations in autism and schizophrenia67.
Common variants
The evidence for shared genetic risk across psychiatric disorders arising from common alleles detected by genome‐wide association studies is strong. This was first demonstrated by the International Schizophrenia Consortium24 using a polygenic risk score approach. A highly robust evidence for genetic overlap between schizophrenia and bipolar disorder was found. Subsequent work has shown that common alleles that confer risk for schizophrenia also do so for major depressive disorder and to a lesser extent autism spectrum disorders, ADHD, anorexia nervosa and obsessive‐compulsive disorder47, 68.
A note of caution should be sounded here, in that the sample sizes subjected to genome‐wide association studies for a number of these disorders, including autism spectrum disorders and ADHD, are relatively small compared to those studied in schizophrenia, and the estimates of shared risk may well change as larger samples are studied69.
At the level of individual loci, there is evidence that those implicated in schizophrenia genome‐wide association studies are enriched for genes in which de novo non‐synonymous mutations have been observed in schizophrenia, autism spectrum disorders and intellectual disability, pointing to shared biological mechanisms across the common and rare variant signals and between disorders23. There is also emerging evidence that some of the genes and gene pathways implicated by common variants overlap with those enriched for rare variants in autism spectrum disorders and intellectual disability70.
That there is at least a partial convergence of the common and rare variant signals is also supported by the observation that carriers of pathogenic copy number variants who develop schizophrenia have a higher load of common risk variants than carriers who do not71, suggesting that the outcome of rare variants is to some extent determined by the complement of common risk alleles present in the carrier and supporting the liability threshold model of schizophrenia.
A number of studies have used a polygenic risk score or similar approaches to show overlap in common genetic variation between schizophrenia and developmental outcomes in the general population, and have shown that alleles which increase risk for schizophrenia also associate with, for example, poorer cognitive function and impaired social and communication difficulties, similar to those seen in people with autism spectrum disorders72, 73, 74. While the overlaps are not large, neither are they trivial (genetic correlations between 0.18 and 0.37) and support the involvement of alleles that increase risk for schizophrenia in a wider set of developmental traits.
PLEIOTROPY, PSYCHOPATHOLOGY AND COGNITION
We have seen that a large amount of recent genomic data point to shared genetic risk across childhood neurodevelopmental and adult psychiatric disorders. But do the findings allow us to be more specific about the relationship between shared risk and variable outcome? The term “pleiotropy” is used to describe the phenomenon of an individual gene influencing two or more distinct traits47. Genetic pleiotropy is said to occur when the altered function of a gene influences multiple traits, whereas allelic pleiotropy, a subtype of genetic pleiotropy, occurs when the same gene variant influences multiple traits. It should also be noted that “pseudo‐pleiotropy” can arise as a result of imprecision in gene mapping, whereby two phenotypes are influenced by different genes in close proximity, but it can also arise from poor study design, or associations that are due to chance or publication bias47.
The evidence in relation to pleiotropy in psychiatric disorders has been reviewed in detail elsewhere47. It suggests that, in the majority of instances, the pleiotropy observed between different psychiatric diagnoses and between psychiatric disorders and cognitive impairment is a true allelic pleiotropy rather than a pseudo‐pleiotropy47. The data from rare variants (copy number variations and rare coding variants) are also largely inconsistent with the view that the findings reflect mediated pleiotropy, in which an allele influences two traits, but its effects on one are secondary to more direct effects on the other47. In other words, the findings suggest that intellectual disability, autism spectrum disorders, ADHD and schizophrenia represent direct outcomes of the same rare pathogenic mutations. Moreover, the risk of psychiatric disorders does not appear to be mediated by cognitive impairment, which itself seems to be an additional pleiotropic outcome of the same genetic risk47.
However, the concept of pleiotropy requires a phenotype to be linked directly to a particular gene or mutation, and this is not an easy test to perform for psychiatric disorders, for a number of reasons. First, these are highly polygenic disorders, and the relationship between risk alleles and specific phenotypic outcomes is complex and combinatorial. One clear example of this is that an individual's burden of common risk alleles can influence psychiatric outcome in copy number variants carriers71. Second, despite our use of categorical diagnoses, the boundaries between disorders are not clear‐cut, and comorbidity frequently occurs.
While more work is needed, considering all these elements together leads to the conclusion that what we perceive as pleiotropic manifestations of a particular mutation, such as a copy number variant, likely represent the net effects of an individual's polygenic and environmental background on multiple traits representing various domains of brain function47. Thus, psychiatric, cognitive and motor phenotypes tend to co‐occur in clinical populations because they share underlying etiological and pathogenic mechanisms, but the mix of outcomes in any individual case will reflect that individual's particular genetic complement and environmental history.
CONCLUSIONS AND IMPLICATIONS
Findings from genomic studies have implicated large, rare copy number variants in conferring risk to schizophrenia and shown that the same variants also confer risk to intellectual disability, autism spectrum disorders and ADHD. Similarly, there is emerging evidence that rare coding variants also confer risk of schizophrenia and for overlap between the genes impacted by damaging variants found in schizophrenia and those seen in autism spectrum disorders and intellectual disability.
The enrichment of large, rare copy number variants is highest in intellectual disability, then autism spectrum disorders, then schizophrenia, then bipolar disorder. There is also evidence that the enrichment of damaging rare coding variants is greatest in intellectual disability, then autism spectrum disorders and then schizophrenia, with insufficient data to date for ADHD and bipolar disorder.
The enrichment of rare mutations appears to be correlated with the degree of cognitive impairment both across and within diagnostic groups, but pathogenic copy number variants and rare coding variants are found in autism spectrum disorders and schizophrenia in the absence of gross cognitive impairment, and pathogenic copy number variants are present in individuals with subtle impairments of cognition but who do not have a psychiatric diagnosis.
There is also evidence for shared common allele genetic risk across schizophrenia and other neurodevelopmental disorders, and evidence that this overlaps with the genes and pathways implicated by rare variant studies. Indeed, the fact that, regardless of the specific diagnoses, rare de novo and damaging rare coding variants tend to implicate broadly similar processes (synaptic plasticity, chromatin modifiers and targets of fragile X mental retardation protein) suggests that individual mutations are likely to influence the same pathogenic mechanisms across disorders.
These findings not only support the view that schizophrenia is a disorder whose origins lie in disturbances of brain development, but also that it shares genetic risk and pathogenic mechanisms with the early onset neurodevelopmental disorders (intellectual disability, autism spectrum disorders and ADHD). They also support the view that these disorders lie on a gradient of severity, implying that they differ to some extent quantitatively as well as qualitatively.
There are a number of important implications of these findings for nosology, research and clinical practice. First, they suggest that we should widen the nosological concept of neurodevelopmental disorders to include the functional psychoses. Further work will be required to establish the extent to which genomic data support the inclusion of bipolar disorder and ADHD as well as other neurodevelopmental disorders, such as dyslexia and coordination disorder, not discussed in this paper. But there is compelling genomic evidence for the existence of a group of neurodevelopmental disorders that includes what are generally considered to be adult onset disorders and that is associated with pleiotropic effects on cognitive impairment. The pleiotropic nature of the relationship between psychopathology and cognition predicts that the severity of cognitive impairment in individuals with psychopathology who meet diagnostic criteria for one of these disorders will be variable and sometimes subtle and may possibly only be detected by comparison with parental cognitive function12, 75.
As far as research is concerned, the neurodevelopmental continuum underscores the need for new and flexible approaches to patient stratification8, 76. First, it suggests that such approaches, rather than being categorical, will need to be multidimensional, accessing multiple different domains of brain function. Second, it indicates that etiological and mechanistic research should not be constrained by current diagnostic or age‐related silos. In particular, there needs to be much greater communication and integration between the communities researching childhood neurodevelopmental disorders such as ADHD and autism spectrum disorders and those studying adult psychiatric disorders such as schizophrenia and bipolar disorder. Third, the pleiotropic effects of genetic risk factors have clear implications for mechanistic research using endophenotypes in human studies or animal models: researchers should be cautious when attempting to chart causal pathways that mediate the effects of genetic risk on clinical phenotypes8, 47.
Fourth, the range of outcomes of rare mutations such as copy number variants and some rare coding variants suggests that the brain is to some extent able to compensate for the disruptive effects of such mutations, and this, together with the identification of protective mutations30, 77, suggests that some of the biology may be tractable. A focus on what factors influence outcome in specific mutation carriers might be a fruitful area for future research71. Indeed, it is possible that a component of the common variant signal in schizophrenia detected by polygenic risk score and similar approaches relates to mechanisms that mitigate the consequences of neurodevelopmental disruption by damaging mutations or early environmental exposures.
Finally, the findings reviewed above have implications for understanding the potential role of psychosocial risk factors, a number of which have been implicated in schizophrenia9. One possibility is that the presence of pre‐existing neurodevelopmental impairment increases susceptibility to these risk factors. Another is that there is a degree of etiological heterogeneity, and that both psychosocial and neurodevelopmental risk factors can result in similar syndromic outcomes. However, it is also possible that associations with psychosocial risk factors reflect confounding, pleiotropy or reverse causation rather than true causation, and we must await the application of study designs that allow these possibilities to be distinguished9.
There are also implications of the neurodevelopmental continuum for clinical practice. There should be a high expectation of comorbidity, and greater emphasis on developmental history and on multi‐domain assessment (psychopathological, cognitive, sensorimotor). Clinicians should increasingly take a developmental life‐course approach ensuring that patients are effectively managed across the transition from childhood to adulthood, and developmental change over time should be expected and anticipated. The various agencies that currently assess and manage childhood neurodevelopmental and adult psychiatric disorders will need to build up shared language, classification and methods of assessment.
It will be challenging to treat underlying neurodevelopmental mechanisms, and therapeutic approaches, at least in the short and medium term, might need to focus upon symptomatic management of the particular domains (psychopathological, cognitive, sensorimotor) affected in an individual. For the medium and long term, recent genomic findings offer many opportunities for mechanistic research78. Moreover, there is evidence from genomics for tractable biology, and the high degree of pleiotropy suggests that therapeutic approaches might be successful across current diagnostic boundaries47.
ACKNOWLEDGEMENTS
This work was funded by Medical Research Council centre grant MR/L010305/1 and program grant G0800509.
REFERENCES
- 1. Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry 1987;44:660‐9. [DOI] [PubMed] [Google Scholar]
- 2. Murray RM, Lewis SW. Is schizophrenia a neurodevelopmental disorder? BMJ ( Clin Res Ed) 1988;296:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Weinberger DR, Levitt P. Neurodevelopmental origins of schizophrenia. In: Weinberger DR, Harrison PJ. (eds). Schizophrenia, 3rd ed. Oxford: Wiley‐Blackwell, 2011:393‐412. [Google Scholar]
- 4. Owen MJ, O'Donovan MC, Thapar A et al. Neurodevelopmental hypothesis of schizophrenia. Br J Psychiatry 2011;198:173‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rutter M, Moffitt TE, Caspi A. Gene‐environment interplay and psychopathology: multiple varieties but real effects. J Child Psychol Psychiatry 2006;47:226‐61. [DOI] [PubMed] [Google Scholar]
- 6. Craddock N, Owen MJ. The Kraepelinian dichotomy ‐ Going, going… but still not gone. Br J Psychiatry 2010;196:92‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Doherty JL, Owen MJ. Genomic insights into the overlap between psychiatric disorders: implications for research and clinical practice. Genome Med 2014;6:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Owen MJ. New approaches to psychiatric diagnostic classification. Neuron 2014;84:564‐71. [DOI] [PubMed] [Google Scholar]
- 9. Owen MJ, Sawa A, Mortensen PB. Schizophrenia. Lancet 2016;388:86‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lilienfeld AM, Pasamanick B, Rogers M. Relationship between pregnancy experience and the development of certain neuropsychiatric disorders in childhood. Am J Publ Health Nations Health 1955;45:637‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Capute AJ, Palmer FB. A pediatric overview of the spectrum of developmental disabilities. J Dev Behav Pediatr 1980;1:66‐9. [PubMed] [Google Scholar]
- 12. Moreno‐De‐Luca A, Myers SM, Challman TD et al. Developmental brain dysfunction: revival and expansion of old concepts based on new genetic evidence. Lancet Neurol 2013;12:406‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lichtenstein P, Yip BH, Björk C et al. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population‐based study. Lancet 2009;373:234‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sullivan PF, Magnusson C, Reichenberg A et al. Family history of schizophrenia and bipolar disorder as risk factors for autism. Arch Gen Psychiatry 2012;69:1099‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sanchez‐Gistau V, Romero S, Moreno D et al. Psychiatric disorders in child and adolescent offspring of patients with schizophrenia and bipolar disorder: a controlled study. Schizophr Res 2015;168:197‐203. [DOI] [PubMed] [Google Scholar]
- 16. Chou I‐J, Kuo C‐F, Huang Y‐S et al. Familial aggregation and heritability of schizophrenia and co‐aggregation of psychiatric illnesses in affected families. Schizophr Bull 2016;460:744‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thapar A, Cooper M, Eyre O et al. Practitioner review: what have we learnt about the causes of ADHD? J Child Psychol Psychiatry Allied Discip 2013;54:3‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mandy W, Lai M‐C. Annual research review: the role of the environment in the developmental psychopathology of autism spectrum condition. J Child Psychol Psychiatry 2016;57:271‐92. [DOI] [PubMed] [Google Scholar]
- 19. Nosarti C, Reichenberg A, Murray RM et al. Preterm birth and psychiatric disorders in young adult life. Arch Gen Psychiatry 2012;69:610‐7. [DOI] [PubMed] [Google Scholar]
- 20. Abel KM, Wicks S, Susser ES et al. Birth weight, schizophrenia, and adult mental disorder: is risk confined to the smallest babies? Arch Gen Psychiatry 2010;67:923‐30. [DOI] [PubMed] [Google Scholar]
- 21. Mathewson KJ, Chow CHT, Dobson K et al. Mental health of extremely low birth weight survivors: a systematic review and meta‐analysis. Psychol Bull 2017;143:347‐83. [DOI] [PubMed] [Google Scholar]
- 22. Sullivan PF, Daly MJ, O'Donovan M. Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat Rev Genet 2012;13:537‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ripke S, Neale BM, Corvin A et al. Biological insights from 108 schizophrenia‐associated genetic loci. Nature 2014;511:421‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Purcell SM, Wray NR, Stone JL et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009;10:8192‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ripke S, O'Dushlaine C, Chambert K et al. Genome‐wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet 2013;45:1150‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Loh P‐R, Bhatia G, Gusev A et al. Contrasting genetic architectures of schizophrenia and other complex diseases using fast variance‐components analysis. Nat Genet 2015;47:1385‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Malhotra D, Sebat J. CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell 2012;148:1223‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rees E, Walters JTR, Georgieva L et al. Analysis of copy number variations at 15 schizophrenia‐associated loci. Br J Psychiatry 2014;204:108‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rees E, Walters JTR, Chambert KD et al. CNV analysis in a large schizophrenia sample implicates deletions at 16p12.1 and SLC1A1 and duplications at 1p36.33 and CGNL1. Hum Mol Genet 2014;23:1669‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marshall CR, Howrigan DP, Merico D et al. Contribution of copy number variants to schizophrenia from a genome‐wide study of 41,321 subjects. Nat Genet 2016;49:27‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kirov G, Gumus D, Chen W et al. Comparative genome hybridization suggests a role for NRXN1 and APBA2 in schizophrenia. Hum Mol Genet 2008;17:458‐65. [DOI] [PubMed] [Google Scholar]
- 32. Walsh T, McClellan JM, McCarthy SE et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 2008;320:539‐43. [DOI] [PubMed] [Google Scholar]
- 33. Glessner JT, Reilly MP, Kim CE et al. Strong synaptic transmission impact by copy number variations in schizophrenia. Proc Natl Acad Sci USA 2010;107:10584‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kirov G, Pocklington AJ, Holmans P et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol Psychiatry 2012;17:142‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Szatkiewicz JP, O'Dushlaine C, Chen G et al. Copy number variation in schizophrenia in Sweden. Mol Psychiatry 2014;19:762‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pocklington AJ, Rees E, Walters JTR et al. Novel findings from CNVs implicate inhibitory and excitatory signaling complexes in schizophrenia. Neuron 2015;86:1203‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fromer M, Pocklington AJ, Kavanagh DH et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 2014;506:179‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Purcell SM, Moran JL, Fromer M et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 2014;506:185‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Genovese G, Fromer M, Stahl EA et al. Increased burden of ultra‐rare protein‐altering variants among 4,877 individuals with schizophrenia. Nat Neurosci 2016;19:1433‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Singh T, Kurki MI, Curtis D et al. Rare loss‐of‐function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat Neurosci 2016;19:571‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. O'Dushlaine C, Rossin L, Lee PH et al. Psychiatric genome‐wide association study analyses implicate neuronal, immune and histone pathways. Nat Neurosci 2015;18:199‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sebat J, Levy DL, McCarthy SE. Rare structural variants in schizophrenia: one disorder, multiple mutations; one mutation, multiple disorders. Trends Genet 2009;25:528‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Williams NM, Zaharieva I, Martin A et al. Rare chromosomal deletions and duplications in attention‐deficit hyperactivity disorder: a genome‐wide analysis. Lancet 2010;376:1401‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stefansson H, Meyer‐Lindenberg A, Steinberg S et al. CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature 2013;505:361‐6. [DOI] [PubMed] [Google Scholar]
- 45. Kendall KM, Rees E, Escott‐Price V et al. Cognitive performance among carriers of pathogenic copy number variants: analysis of 152,000 UK biobank subjects. Biol Psychiatry 2017;82:103‐10. [DOI] [PubMed] [Google Scholar]
- 46. Kirov G, Rees E, Walters JTR et al. The penetrance of copy number variations for schizophrenia and developmental delay. Biol Psychiatry 2014;75:378‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. O'Donovan MC, Owen MJ. The implications of the shared genetics of psychiatric disorders. Nat Med 2016;22:1214‐9. [DOI] [PubMed] [Google Scholar]
- 48. Girirajan S, Brkanac Z, Coe BP et al. Relative burden of large CNVs on a range of neurodevelopmental phenotypes. PLoS Genet 2011;7:e1002334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rees E, Kendall K, Pardiñas AF et al. Analysis of intellectual disability copy number variants for association with schizophrenia. JAMA Psychiatry 2016;73:963‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Craddock N, Sklar P. Genetics of bipolar disorder. Lancet 2013;381:1654‐62. [DOI] [PubMed] [Google Scholar]
- 51. Grozeva D. Rare copy number variants: a point of rarity in genetic risk for bipolar disorder and schizophrenia. Arch Gen Psychiatry 2010;67:318‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bergen SE, O'Dushlaine CT, Ripke S et al. Genome‐wide association study in a Swedish population yields support for greater CNV and MHC involvement in schizophrenia compared with bipolar disorder. Mol Psychiatry 2012;17:880‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Green EK, Rees E, Walters JTR et al. Copy number variation in bipolar disorder. Mol Psychiatry 2016;21:89‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Grozeva D, Kirov G, Conrad DF et al. Reduced burden of very large and rare CNVs in bipolar affective disorder. Bipolar Disord 2013;15:893‐8. [DOI] [PubMed] [Google Scholar]
- 55. Priebe L, Degenhardt FA, Herms S et al. Genome‐wide survey implicates the influence of copy number variants (CNVs) in the development of early‐onset bipolar disorder. Mol Psychiatry 2012;17:421‐32. [DOI] [PubMed] [Google Scholar]
- 56. Malhotra D, McCarthy S, Michaelson JJ et al. High frequencies of de novo CNVs in bipolar disorder and schizophrenia. Neuron 2011;72:951‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Power RA, Kyaga S, Uher R et al. Fecundity of patients with schizophrenia, autism, bipolar disorder, depression, anorexia nervosa, or substance abuse vs their unaffected siblings. JAMA Psychiatry 2013;70:22‐30. [DOI] [PubMed] [Google Scholar]
- 58. Georgieva L, Rees E, Moran JL et al. De novo CNVs in bipolar affective disorder and schizophrenia. Hum Mol Genet 2014;23:6677‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sanders SJ, He X, Willsey AJ et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 2015;87:1215‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vulto‐van Silfhout AT, Hehir‐Kwa JY, van Bon BWM et al. Clinical significance of de novo and inherited copy‐number variation. Hum Mutat 2013;34:1679‐87. [DOI] [PubMed] [Google Scholar]
- 61. McRae JF, Clayton S, Fitzgerald TW et al. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017;542:433‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Singh T, Walters JTR, Johnstone M et al. Rare schizophrenia risk variants are enriched in genes shared with neurodevelopmental disorders. bioRxiv 2016:69344. [Google Scholar]
- 63. Robinson EB, St Pourcain B, Anttila V et al. Genetic risk for autism spectrum disorders and neuropsychiatric variation in the general population. Nat Genet 2016;48:552‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rauch A, Wieczorek D, Graf E et al. Range of genetic mutations associated with severe non‐syndromic sporadic intellectual disability: an exome sequencing study. Lancet 2012;380:1674‐82. [DOI] [PubMed] [Google Scholar]
- 65. De Rubeis S, He X, Goldberg AP et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014;515:209‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kataoka M, Matoba N, Sawada T et al. Exome sequencing for bipolar disorder points to roles of de novo loss‐of‐function and protein‐altering mutations. Mol Psychiatry 2016;21:885‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Goes FS, Pirooznia M, Parla JS et al. Exome sequencing of familial bipolar disorder. JAMA Psychiatry 2016;73:590‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Anttila V, Bulik‐Sullivan B, Finucane HK et al. Analysis of shared heritability in common disorders of the brain. bioRxiv 2016:48991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. O'Donovan MC. What have we learned from the Psychiatric Genomics Consortium. World Psychiatry 2015;14:291‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pardiñas AF, Holmans P, Pocklington AJ et al. Common schizophrenia alleles are enriched in mutation‐intolerant genes and maintained by background selection. bioRxiv 2016:68593. [Google Scholar]
- 71. Tansey KE, Rees E, Linden DE et al. Common alleles contribute to schizophrenia in CNV carriers. Mol Psychiatry 2016;21:1085‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hubbard L, Tansey KE, Rai D et al. Evidence of common genetic overlap between schizophrenia and cognition. Schizophr Bull 2016;42:832‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Riglin L, Collishaw S, Richards A et al. Schizophrenia risk alleles and neurodevelopmental outcomes in childhood: a population‐based cohort study. Lancet Psychiatry 2017;4:57‐62. [DOI] [PubMed] [Google Scholar]
- 74. St Pourcain B, Robinson EB, Anttila V et al. ASD and schizophrenia show distinct developmental profiles in common genetic overlap with population‐based social communication difficulties. Mol Psychiatry (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. D'Angelo D, Lebon S, Chen Q et al. Defining the effect of the 16p11.2 duplication on cognition, behavior, and medical comorbidities. JAMA Psychiatry 2016;73:20‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Owen MJ, Doherty JL. What can we learn from the high rates of schizophrenia in people with 22q11.2 deletion syndrome? World Psychiatry 2016;15:23‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Rees E, Kirov G, Sanders A et al. Evidence that duplications of 22q11.2 protect against schizophrenia. Mol Psychiatry 2014;19:37‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. McCarroll SA, Hyman SE, Han Y et al. Progress in the genetics of polygenic brain disorders: significant new challenges for neurobiology. Neuron 2013;80:578‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]