

Summary
The calcium/calmodulin-dependent serine protein kinase gene (CASK) mutations are associated with various neurological disorders; a syndrome of intellectual disability (ID) and microcephaly with pontine and cerebellar hypoplasia (MICPCH), FG syndrome, X-linked ID with/without nystagmus, epileptic encephalopathy, and autistic spectrum disorder (ASD). Next generation sequencing was performed to elucidate genetic causes in siblings exhibiting developmental disorders, and a novel CASK mutation, c.1424G>T (p.Ser475Ile), was detected in a male patient with ID, ASD, and microcephaly. Radiological examination of his brain showed no structural abnormality. The identified mutation was shared with the healthy mother and a younger sister exhibiting ASD. Although the mother showed a skewed X-chromosome inactivation (XCI) pattern, the sister showed a paradoxical XCI pattern. This would explain why this sister possessed a normal intellectual level, but showed the same ASD symptoms as the affected brother. A novel CASK mutation was identified in two siblings with ID and/or ASD, suggesting a relationship between the CASK mutation and ASD. Recently performed large molecular cohorts for patients with developmental disorders suggest that CASK is one of the genes related to developmental disorders. For better understanding of genotype-phenotype correlation in ASD cases with CASK mutations, more information should be accumulated.
Keywords: Autism, X-chromosome inactivation, next generation sequencing, obligate carrier, manifesting carrier
1. Introduction
Approximately 1.5–2% and 1–0.7% of children show developmental disorders (DD), including intellectual disability (ID) and autism spectrum disorder (ASD), respectively (1). Etiologies of these disorders are heterogeneous and recent developments in genetic analysis have provided evidence that various types of genomic alterations are related to DD. Especially, many large cohort studies have been performed to elucidate the molecular basis of DD. As a result, the calcium/calmodulin-dependent serine protein kinase gene (CASK) was reported as one of the genes responsible for developmental disorders (2,3).
Human CASK mutations were first identified in female patients with a syndrome of ID and microcephaly with pontine and cerebellar hypoplasia (MICPCH) (4). CASK encodes a calcium/calmodulin-dependent serine protein kinase that belongs to the membrane-associated guanylate kinase (MAGUK) family (4). The proteins included in MAGUK family are scaffold proteins and are located at synapses in the brain. Various neurological disorders have been associated with CASK; FG syndrome (5,6), X-linked ID with/without nystagmus (7), and epileptic encephalopathy (8–10). In this study, a novel CASK mutation, c.1424G>T (p.Ser475Ile), was identified in siblings exhibiting DD with ID and/or ASD.
2. Materials and Methods
This study, which aimed to identify genetic etiologies in patients with DD, was performed in accordance with the Helsinki declaration. We obtained permission from the ethical committee of the institution. Patients were enrolled in this study after receiving written informed consent and thorough genetic counseling regarding appropriate dealing with genetic information and possible incidental findings.
Blood samples were obtained from the patients and their parents. DNA was then extracted using the QIAamp DNA extraction kit (QIAGEN, Hilden, Germany). Next-generation sequencing (NGS) was performed to screen single-nucleotide variants using TruSight One v1.0 sequencing panel (Illumina, San Diego, CA, USA), in accordance with the previously described protocol (11). To confirm SNVs, Sanger sequencing was performed on all samples. The X-chromosome inactivation (XCI) pattern was determined by quantitation of the methylation of the polymorphic human androgen receptor (HUMARA) locus (12).
3. Results
3.1. Patients report
There are two siblings in the indicated family. Both of the siblings, born to healthy parents, are affected. The mother had two spontaneous abortions with unknown etiologies.
A 5-year-old boy (patient 1) was born at 39 weeks of gestational age, with a weight of 2,810 g. There were no remarkable complications during his pregnancy. In early infancy, his mother did not notice any developmental problems. His umbilical herniation was surgically repaired during infancy. He made several motor developmental milestones, with head control at 3 months, crawling at 14 months, and standing with support at 16 months. At 18 months, he could not express speech or walk unassisted. Because of these developmental delays, he was referred to our hospital. There was no history of seizures.
At 26 months, a behavioral assessment indicated the following symptoms of ASD: i) poor verbal communication (lack of vocabulary, poor pronunciation, and dysarthria), ii) qualitative impairment in social communication (he is not empathetic and only shows a superficial interest in things), and iii) limited stereotypical interests and behaviors (when distressed, he strikes his head on the wall repeatedly or will break magazines). He shows a special interest in some marks and signs. The pervasive developmental disorders autism society Japan rating scale (PARS) was scaled at 9, indicating a high possibility of ASD (13). Brain magnetic resonance imaging showed no structural abnormality (Figure 1). Conventional chromosomal analysis showed a normal male karyotype of 46, XY and chromosomal microarray testing showed no abnormality.
Figure 1.
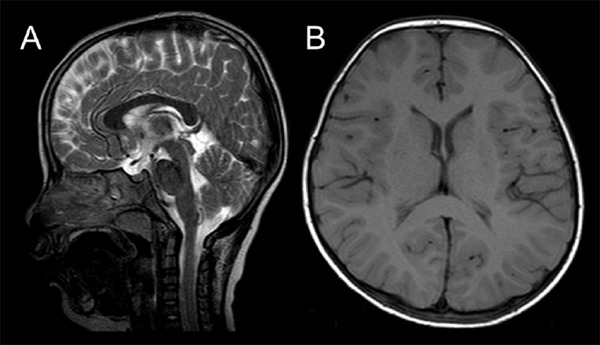
Results of brain magnetic resonance imaging. There is no abnormality, including the volume of cerebellum. T2-sagittal (A) and T1-axial images (B) examined at 5 years of age.
At present, his height is 99.5 cm (3rd-10th centile), weight is 13.4 kg (< 3rd centile), and occipitofrontal circumference (OFC) is 47 cm (< 3rd centile), indicating microcephaly. He requires assistance in eating, bathing, and getting dressed. There is no history of sleep disturbance. The Kyoto Scale of Psychological Development (KSPD) indicated an intelligence quotient (IQ) of 61, indicating mild developmental delay (14).
A younger sister of patient 1 (patient 2) also visited our hospital. She is currently 3 years old. She was crawling at 8 months and walking unassisted at 18 months, indicating mild motor developmental delay. She misbehaves/acts out when a situation does not go her way. Based on further inputs regarding her behavior and developmental history, she was diagnosed with ASD. She showed typical autistic behavior, like her brother (patient 1). However, she did not demonstrate short stature or microcephaly (OFC 46 cm). The KSPD indicated an IQ of 84, which is a normal level. The PARS score was 9, which was the same as her elder brother (patient 1), indicating a high possibility of ASD.
3.2. Genetic analysis
NGS for patient 1 extracted 2,561 Gb of targeted aligned sequences and a mean coverage depth of 105.3 was obtained (target coverage at 20× was 95.8%). After filtering, only a missense variant (chrX:41437672C>A, NM_003688.3(CASK):c.1424G>T [NP_003679.2:p. Ser475Ile]) remained (Figure 2A). Integrative Genomics Viewer (IGV; http://www.broadinstitute.org/igv/) showed the same alteration in all reads mapped at this position, indicating a 100% detection ratio and denying mosaicism (Figure 2B). This codon is conserved among species (Figure 2D). The prediction scores were calculated through wANNOVAR (http://wannovar.wglab.org/) (Table 1) and the scores by LRT_pred, MutationTaster_pred, and CADD_phred suggested “damaging”. Previously reported CASK variants are summarized in Table 2. As shown, p.Ser475Ile has never been reported previously and is supposedly a novel mutation. This variant was reconfirmed by Sanger sequencing and identified in the mother and patient 2 (the younger sister of patient 1) (Figure 2C), suggesting segregation by the X-linked recessive trait. Although patient 2 did not show ID, she shared ASD symptoms with her brother (patient 1). Because CASK is located on the X chromosome, XCI patterns were analyzed in the mother and patient 2. The mother showed an almost completely skewed XCI pattern; however, patient 2 showed a paradoxical XCI pattern, i.e., the paternally derived allele (supposed normal allele) was predominantly inactivated (Figure 3). This may indicate that the mother is an obligate carrier and patient 2 is a manifesting carrier.
Figure 2.

Results of genetic studies. (A) Filtering steps and the number of filtered variants are shown in inverted pyramids. The Human Genetic Variation Database (HGVD) (http://www.genome.med.kyoto-u.ac.jp/SnpDB) provided from Kyoto University was used for the filtering. (B) The Integrative Genomics Viewer (IGV) shows the identified CASK variant in 100% of reads. (C) Electropherograms of Sanger sequencing. The father shows the wild type sequence, whereas the mother and patient 2 exhibit heterozygous status. Patient 1 is completely hemizygous for the mutation. (D) The neighboring amino-acid sequences are compared among species. The affected codon “S” is conserved among species.
Table 1. Results of prediction scores.
| SIFT_score | 0.19 |
| SIFT_pred | T |
| Polyphen2_HDIV_score | 0.914 |
| Polyphen2_HDIV_pred | P |
| Polyphen2_HVAR_score | 0.887 |
| Polyphen2_HVAR_pred | P |
| LRT_score | 0 |
| LRT_pred | D* |
| MutationTaster_score | 1 |
| MutationTaster_pred | D* |
| MutationAssessor_score | 0.255 |
| MutationAssessor_pred | N |
| FATHMM_score | 0.64 |
| FATHMM_pred | T |
| RadialSVM_score | −0.843 |
| RadialSVM_pred | T |
| LR_score | 0.162 |
| LR_pred | T |
| VEST3_score | 0.936 |
| CADD_raw | 3.177 |
| CADD_phred | 16.63* |
| GERP++_RS | 5.44 |
| phyloP46way_placental | 2.41 |
| phyloP100way_vertebrate | 7.429 |
| SiPhy_29way_logOdds | 18.55 |
T, tolerate; P, possibly damaging;
damaging is suggested.
Table 2. Summary of the intragenic nucleotide changes in CASK and phenotypic features of the patients.

M; male, F; female, NA; not available, DN; de novo, XLR; X-linked recessive trait, MICPCH; microcephaly with pontine and cerebellar hypoplasia, MCA; multiple congenital anomalies, DD; developmental disorder, ASD; autism spectrum disorder, ID; intellectual disability, DDDS; Deciphering Developmental Disorders Study.
Figure 3.
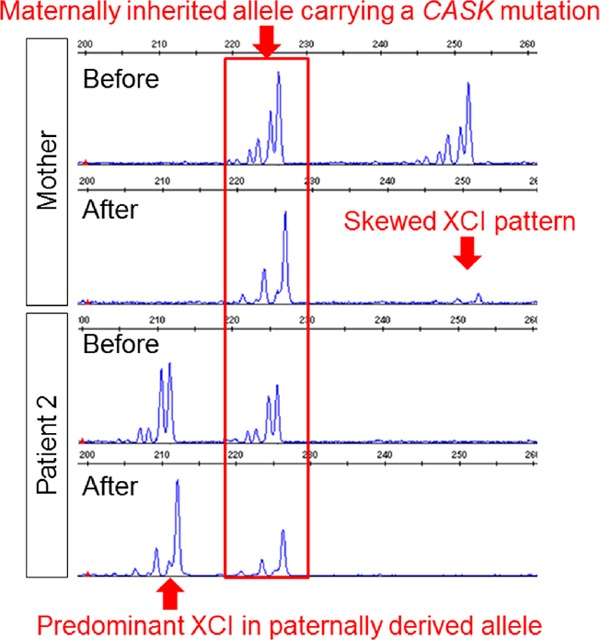
Results of X-chromosome inactivation (XCI) patterns. The mother shows a skewed XCI pattern, because one of the X-chromosomes (not shared with patient 2) is almost completely missing after digestion by a methylation sensitive restriction enzyme. Patient 2 shows a paradoxical pattern with predominant XCI in the paternally derived allele.
4. Discussion
Most of the CASK mutations found in patients with MICPCH showed loss-of-function effects and were related to an X-linked dominant trait with reduced male viability or even in utero lethality; however, mild and hypomorphic mutations such as missense mutations would be compatible with live birth in affected males (4). Thus, CASK mutations found in male patients with clinically mild neurological impairments (compared to MICPCH) would be related to an X-linked recessive trait. Although phenotypic features were recognized as a clinical continuum, Moog et al. reviewed CASK mutations in males and classified them into three categories (15); i) MICPCH with severe epileptic encephalopathy, associated with loss-of-function mutation, ii) MICPCH associated with inactivating alterations in the mosaic state, and iii) syndromic/nonsyndromic mild to severe ID, with or without nystagmus caused by CASK missense/splicing mutations that leave the CASK protein intact but likely alter its function.
In this study, we identified a novel CASK mutation in a family associated with clinical characteristics of ASD/DD and microcephaly. Recently published large cohort studies for ASD or DD suggested CASK as one of the responsible genes for ASD/DD (3,16). Thus, it is reasonable to conclude that the identified CASK variant is related to ASD/DD in this family.
In this family, the mother, exhibiting a skewed XCI pattern, was considered an obligate carrier. On the contrary, the younger sister of the proband (patient 2) having the same CASK mutation showed a paradoxical XCI pattern (the maternally inherited allele was predominantly activated). This would explain why the sister (patient 2), a female carrier, showed a mild ASD phenotype.
The previously reported CASK mutations are summarized in Table 2 (17–20). The mutation identified in this study was the first missense mutation located on the PDZ domain. This may be related to the phenotypic consequence. Owing to identification of CASK mutations in patients with atypical clinical features, the clinical entity of CASK related disorder is expanding beyond the typical and classical three categories. It is important to accumulate individual cases with CASK mutations that have been analyzed using multidisciplinary approaches including genetic analysis and periodic developmental assessments and neuroimaging.
Acknowledgements
We would like to express our gratitude to the patients and their families for their cooperation.
Funding
This research was supported by the Practical Research Project for Rare/Intractable Diseases from Japan Agency for Medical Research and Development (AMED) and Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Number 15K09631 (TY).
Disclosure
We certify that we have read the journal's position regarding issues pertaining to ethical publication, and affirm that this report is consistent with the guidelines. The authors have no conflicts of interest to declare.
References
- 1. Mefford HC, Batshaw ML, Hoffman EP. Genomics, intellectual disability, and autism. N Engl J Med. 2012; 366:733-743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Neale BM, Kou Y, Liu L, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012; 485:242-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017; 542:433-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Najm J, Horn D, Wimplinger I, et al. Mutations of CASK cause an X-linked brain malformation phenotype with microcephaly and hypoplasia of the brainstem and cerebellum. Nat Genet. 2008; 40:1065-1067. [DOI] [PubMed] [Google Scholar]
- 5. Piluso G, D'Amico F, Saccone V, Bismuto E, Rotundo IL, DiDomenico M, Aurino S, Schwartz CE, Neri G, Nigro V. A missense mutation in CASK causes FG syndrome in an Italian family. Am J Hum Genet. 2009; 84:162-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dunn P, Prigatano GP, Szelinger S, et al. A de novo splice site mutation in CASK causes FG syndrome-4 and congenital nystagmus. Am J Med Genet A. 2017; 173:611-617. [DOI] [PubMed] [Google Scholar]
- 7. Hackett A, Tarpey PS, Licata A, et al. CASK mutations are frequent in males and cause X-linked nystagmus and variable XLMR phenotypes. Eur J Hum Genet. 2010; 18:544-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Saitsu H, Kato M, Osaka H, Moriyama N, Horita H, Nishiyama K, Yoneda Y, Kondo Y, Tsurusaki Y, Doi H, Miyake N, Hayasaka K, Matsumoto N. CASK aberrations in male patients with Ohtahara syndrome and cerebellar hypoplasia. Epilepsia. 2012; 53:1441-1449. [DOI] [PubMed] [Google Scholar]
- 9. Nakamura K, Nishiyama K, Kodera H, Nakashima M, Tsurusaki Y, Miyake N, Matsumoto N, Saitsu H, Jinnou H, Ohki S, Yokochi K, Okanishi T, Enoki H. A de novo CASK mutation in pontocerebellar hypoplasia type 3 with early myoclonic epilepsy and tetralogy of Fallot. Brain Dev. 2014; 36:272-273. [DOI] [PubMed] [Google Scholar]
- 10. Nakajiri T, Kobayashi K, Okamoto N, Oka M, Miya F, Kosaki K, Yoshinaga H. Late-onset epileptic spasms in a female patient with a CASK mutation. Brain Dev. 2015; 37:919-923. [DOI] [PubMed] [Google Scholar]
- 11. Shimojima K, Okamoto N, Yamamoto T. A novel TUBB3 mutation in a sporadic patient with asymmetric cortical dysplasia. Am J Med Genet A. 2016; 170:1076-1079. [DOI] [PubMed] [Google Scholar]
- 12. Shimada S, Okamoto N, Ito M, Arai Y, Momosaki K, Togawa M, Maegaki Y, Sugawara M, Shimojima K, Osawa M, Yamamoto T. MECP2 duplication syndrome in both genders. Brain Dev. 2013; 35:411-419. [DOI] [PubMed] [Google Scholar]
- 13. Ito H, Tani I, Yukihiro R, Adachi J, Hara K, Ogasawara M, Inoue M, Kamio Y, Nakamura K, Uchiyama T, Ichikawa H, Sugiyama T, Hagiwara T, Tsujii M. Validation of an interview-based rating scale developed in Japan for pervasive developmental disorders. Res Autism Spectr Disord. 2012; 6:1265-1272. [Google Scholar]
- 14. Aoki S, Hashimoto K, Ikeda N, Takekoh M, Fujiwara T, Morisaki N, Mezawa H, Tachibana Y, Ohya Y. Comparison of the Kyoto Scale of Psychological Development 2001 with the parent-rated Kinder Infant Development Scale (KIDS). Brain Dev. 2016; 38:481-490. [DOI] [PubMed] [Google Scholar]
- 15. Moog U, Bierhals T, Brand K, et al. Phenotypic and molecular insights into CASK-related disorders in males. Orphanet J Rare Dis. 2015;10:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Iossifov I, O'Roak BJ, Sanders SJ, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014; 515:216-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moog U, Kutsche K, Kortum F, et al. Phenotypic spectrum associated with CASK loss-of-function mutations. J Med Genet. 2011; 48:741-751. [DOI] [PubMed] [Google Scholar]
- 18. Burglen L, Chantot-Bastaraud S, Garel C, et al. Spectrum of pontocerebellar hypoplasia in 13 girls and boys with CASK mutations: Confirmation of a recognizable phenotype and first description of a male mosaic patient. Orphanet J Rare Dis. 2012;7:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hayashi S, Okamoto N, Chinen Y, Takanashi J, Makita Y, Hata A, Imoto I, Inazawa J. Novel intragenic duplications and mutations of CASK in patients with mental retardation and microcephaly with pontine and cerebellar hypoplasia (MICPCH). Hum Genet. 2012; 131:99-110. [DOI] [PubMed] [Google Scholar]
- 20. Valayannopoulos V, Michot C, Rodriguez D, et al. Mutations of TSEN and CASK genes are prevalent in pontocerebellar hypoplasias type 2 and 4. Brain. 2012; 135:e199; author reply e200. [DOI] [PubMed] [Google Scholar]