

Summary
Mucopolysaccharidoses (MPS) types I, II and VI are associated with deficiencies in alpha-L-iduronidase, iduronate-2-sulfatase and N-acetylgalactosamine-4-sulfatase, respectively, and generally involve progressive and multi-systemic clinical manifestations. Enzyme replacement therapy (ERT) appears to be reasonably well tolerated. The aim of this study was to examine clinical and diagnostic findings of a series of pediatric and adult MPS patients, and assess the safety and efficacy of ERT in children and adults with MPS type I, II and VI. Pediatric and adult patients were treated weekly with 1 mg/kg recombinant human N-acetylgalactosamine-4-sulphatase (rhASB), 0.45 mg/kg alpha-L-iduronidase, or 0.5 mg/kg iduronate-2-sulfatase. Clinical and biochemical parameters with ERT were evaluated for a mean duration of 5 years. Mantel-Haenszel risk ratios and associated 95% confidence intervals (CIs) were calculated for rates of death among different types of enzyme replacement therapies (ERTs). Twenty-seven patients (mean ages ‒ pediatric: 6.8 years; adult: 29 years) were included. ERT was found to be consistently well tolerated and effective in attenuating symptoms, but did not prevent the progression of the disease or reduce mortality rates. Our findings demonstrated that early diagnosis and initiation of ERT are critical for improvements in patient-important outcomes and quality of life, although disease progression and mortality rates remain high.
Keywords: Lysosomal storage disorders, glycosaminoglycans, treatment, prognosis
1. Introduction
Mucopolysaccharidoses (MPS) are lysosomal storage disorders caused by glycosaminoglycan (GAG) enzymatic catabolism deficiencies, leading to organ and tissue deposition. The clinical manifestations of MPS are generally progressive and multisystemic, and signs and symptoms are variable and may progress rapidly or more slowly after birth. Clinical alterations such as facial infiltration, thick lips, opacity of the cornea, umbilical and/or inguinal hernias, cardiac disease, hepatosplenomegaly, articular stiffness, short stature, intellectual disability and skeletal changes, also known as multiple dysostosis, make this diagnosis challenging (1).
MPS types I, II and VI are associated with deficiencies in alpha-L-iduronidase, iduronate-2-sulfatase and N-acetylgalactosamine-4-sulfatase, respectively (1). Enzyme replacement therapy (ERT) with alpha-L-iduronidase, iduronate-2-sulfatase, and recombinant human N-acetylgalactosamine-4-sulphatase (rhASB) have been shown to be effective in several phase 2 and 3 studies (2–4).
The aim of this case series study was to examine clinical and diagnosis findings of pediatric and adult MPS I (Hurler, Hurler-Scheie, Scheie), MPS II (Hunter) and MPS VI (Marateaux Lamy) patients, and to evaluate the long-term safety and efficacy of ERT for these patients.
2. Materials and Methods
This study was approved and registered under number 1034/08 by the Ethics Committee for the analysis of Research projects (CAPPesq) of the clinical Medical School of the University of São Paulo. This was an open-label, single-center case series study of 27 patients with confirmed diagnoses of MPS I (n = 13), II (n = 8) and VI (n = 6), managed with ERT in three Brazilian referral centers (Genetic Outpatient Clinic of the Children's Institute of Hospital das Clínicas FMUSP, Chemotherapy Division of PUC Campinas, and the Children's Hospital Candido Fontoura) between October 2012 and July 2015.
MPS diagnosis was confirmed by specific enzymatic activity (nmol/h/mg protein) and elevated urinary glycosaminoglycans (GAGs). All patients were evaluated by one geneticist by means of clinical and laboratory protocols, previously described by Giugliani et al. 2007 (5) in their study evaluating the management of patients with MPS with ERT. Laboratory exams included: quantitative and qualitative dosing of urinary GAGs, based on the reference values for age and enzymatic activity in leukocytes; echocardiography; abdominal ultrasonography and/or computed tomography; and magnetic resonance imaging and/or computed tomography of the skull and spine resonance. Genetic mutation research was performed when available. Patients who previously received bone marrow transplantation were excluded.
Treatment consisted of weekly infusions of 0.45 mg/kg alpha-L-iduronidase, 0.5 mg/kg iduronate-2-sulfatase, or 1 mg/kg of rhASB in all patients with MPS I, II, and VI, respectively, except five patients in the alpha-L-iduronidase group who received 0.45 mg/kg every other week.
Primary endpoints were clinical outcomes: coarse facies, cloudy cornea, joint stiffness, claw hands, short neck, macroglossia, macrocephaly, short stature, low weight, hepatosplenomegaly, hernias, intellectual disability, surgery procedures, and clinical and anesthetic complications. Secondary endpoints were laboratory parameters: GAGs, echocardiography, X-ray, MRI of the skull and spine, abdominal ultrasonography, polysomnography, and molecular imaging.
Clinical and biochemical parameters with ERT were evaluated for a mean duration of 5 years. Mantel-Haenszel risk ratios and associated 95% confidence intervals (CIs) were calculated for rates of death among different types of enzyme replacement therapies (ERTs).
3. Results
Twenty-seven patients with confirmed MPS-I (n = 13; 48%), II (n = 8; 30%), and VI (n = 6; 22%) were included (Table 1). No abnormalities were present at birth across included patients; onset of symptoms (i.e., facial anomalies, abdominal volume increase, joint stiffness, growth deficits) were confirmed to have presented from three to six months of age. Frequent findings were joint restraint, claw hands, macrocephaly, short stature, and weight deficit across all types of MPS. Short stature was present in 67% (18/27) of patients: Hurler (1/3), Hurler-Scheie (3/5), Scheie (5/5), MPS II (3/8), and MPS VI (6/6). All MPS patients presented with dysmorphic facial features typical of MPS, with infiltrated face and coarse facies being the most discrete findings in Scheie patients.
Table 1. Enzymatic activity according to their respective enzymes of patients with MPS.
| MPS | Patients | Enzyme activity |
|---|---|---|
| MPS I – Hurler¢ | 1 | 0,073 (32 a 56) |
| 2 | 0,06 (32 a 56) | |
| 3* | 0,0 (32 a 56) | |
| MPS I – Hurler-Scheie¢ | 4 | 0,07 (32 a 56) |
| 5 | 0,0 (32 a 56) | |
| 6** | 0,06 (32 a 56) | |
| 7 | 0,1 (32 a 56) | |
| 8 | 0,1 (32 a 56) | |
| MPS I – Scheie¢ | 9 | 0,5 (32 a 52) |
| 10 | 0,09 (32 a 52) | |
| 11 | 0,0 (32 a 52) | |
| 12 | 0,0 (32 a 52) | |
| 13 | 0,0 (32 a 52) | |
| MPS II – Hunter$ | 14 | 6 (110–370) |
| 15 | 1,1 (31–110) | |
| 16 | 0,73 (31–110) | |
| 17 | 0,0 (31–110) | |
| 18 | 0,35 (122–463) | |
| 19 | 0,4 (110–370) | |
| 20 | 0,0 (30–53) | |
| 21 | 0,4 (122–463) | |
| MPS VI – Marateaux Lamy¥ | 22 | 4 (72–176) |
| 23 | 12 (72–176) | |
| 24 | 8 (72–176) | |
| 25 | 0,0 (5,3–21,8) | |
| 26 | 6 (72–176) | |
| 27 | 1,39 (5,3–21,8) |
α-L-iduronidase;
Idurolnate-2-sulfatase;
N-acetilgalactosamine 4-sulfatase;
Reference value in serum (4,7 a 18,1);
Reference value in leucocites (32 a 56).
In terms of diagnostic findings, initial quantitative GAG was noted to be elevated 2 to 13.3 times the age-specific reference values. As Hunter for GAG chromatography, dermatan sulfate and heparan sulfate were predominant in MPS I and II patients, and dermatan sulfate and chondroitin sulfate in MPS VI (Table 1).
3.1. Mucopolysaccharidosis type I
3.1.1. Clinical findings
Thirteen patients (n = 4 female, n = 9 male) belonging to 11 families with MPS I subtypes (n = 3 Hurler, n = 5 Hurler-Scheie, n = 5 Scheie). Consanguinity was observed in 3/11 (27%) families. One family presented two sisters with MPS VI, and another presented two siblings and one cousin with MPS I (Table 2).
Table 2. Characteristics of mucopolysaccharidosis type I.
| Patients | Sex | Cons | Syndromes | Age of Symptoms | Age of Diagnosis | Follow-up | Current age at the time of this publication |
|---|---|---|---|---|---|---|---|
| 1 | M | + | Hurler | 8m | 2y | Death at 5 y and 6 m) | - |
| 2 | F | - | Hurler | 6m | 1y | BMT performed, ongoing | 4y 10m |
| 3 | F | - | Hurler | 8m | 1y 8m | Ongoing | 6y 6m |
| 4 | M | - | Hurler-Scheie | 1y | 1y 5m | Ongoing | 13y 5m |
| 5 | M | + | Hurler-Scheie | 3y | 4y | Death at 13y 10m | - |
| 6 | F | - | Hurler-Scheie | 2y 6m | 7y 9m | Death at 18y 5m | - |
| 7 | M | - | Hurler-Scheie | 3y | 6y | Ongoing | 20y 5m |
| 8 | M | - | Hurler-Scheie | 2y | 4y 1m | Ongoing | 18y 10m |
| 9 | F | - | Scheie | 7y | 17y | Ongoing | 42y 8m |
| 10 | M | - | Scheie | 5y | 24a | Ongoing | 41y 6m |
| 11* | M | + | Scheie | 6y | 12y | Ongoing | 40y 2m |
| 12* | M | + | Scheie | 8y | 7y | Ongoing | 35y 4m |
| 13* | M | + | Scheie | 8y | 8y | Ongoing | 37y |
Same family;
Cons: Consanguinity; +: Yes; −: No; m: months; y.: years
The onset of symptoms for Hurler patients varied from 6 to 8 months of age, with a mean of 7 months; for Hurler-Scheie and Scheie, patients ranged from 1 year to 3 years (mean 2 years), and from 5 years to 8 years (mean 6 years 9 months), respectively.
The first symptoms for Hurler patients were spinal deformity, ocular alteration (i.e. glaucoma) and infiltrated face, while for Hurler-Scheie, patients presented with increased abdominal volume, infiltrated face and macrocephaly. Joint stiffness and short stature were the only symptoms observed in Scheie patients.
3.1.2. Diagnostic findings
The enzymatic activity of α-L-iduronidase in leukocytes and/or plasma was undetectable in 1/3, 1/5, and 3/5 of patients with Hurler, Hurler-Scheie, and Scheie, respectively (Table 1).
3.2. Mucopolysaccharidosis type II
3.2.1. Clinical findings
Eight male patients with MPS II, belonging to 8 families, were included. The first symptoms noted were neuropsychomotor development delay, joint restriction and infiltrated face, which appeared between 2 and 5 years of age (mean: 3 years 6 months). Age at diagnosis was 2 to 10 years (mean: 5 years) (Table 3).
Table 3. Characteristics of mucopolysaccharidosis type II (Hunter).
| Patients | Sex | Cons | Age of Symptoms | Age of Diagnosis | Follow-up | Current age at the time of this publication |
|---|---|---|---|---|---|---|
| 14 | M | N | 2y | 4y 6m | Ongoing | 23y 9m |
| 15 | M | N | 2y 6m | 3y 3m | Ongoing | 7y 10m |
| 16 | M | N | 4y | 6y | Ongoing | 15y 5m |
| 17 | M | N | 5y | 5y | Ongoing | 18y |
| 18 | M | N | 2y 6m | 5y | Death at 13y 9m | - |
| 19 | M | N | 3y | 10y | Death at 23y | - |
| 20 | M | N | 2y | 5y 6m | Ongoing | 10y |
| 21 | M | N | 2y | 2y 6m | Ongoing | 11y |
M: male; N: no; Cons: Consanguinity; y: years; m: month.
3.2.2. Diagnosis findings
The enzymatic activity of α-L-iduronidase in leukocytes and/or plasma was undetectable in 2/8 patients (Table 1).
3.3. Mucopolysaccharidosis type VI
3.3.1. Clinical findings
Six patients (n = 4 female, n = 2 male) with MPS VI, belonging to five families, were included. The first symptoms to present were infiltrated face, spinal deformity and macrocephaly. The onset of symptoms varied from 3 months to 3 years of age (mean 1 year). Age at diagnosis ranged from 8 months to 10 years (mean 5 years) (Table 4).
Table 4. Characteristics of mucopolysaccharidosis type VI (Marateaux Lamy).
| Patients | Sex | Cons | Age of Symptoms | Age of Diagnosis | Follow-up | Current age at the time of this publication |
|---|---|---|---|---|---|---|
| 22 | F | + | 3m | 8y | Ongoing | 18y |
| 23 | F | + | 7m | 8m | Ongoing | 10y 8m |
| 24 | M | + | 6m | 3y 8m | Death at 13y 5m | - |
| 25 | F | - | 3y | 10y | Ongoing | 16y 8m |
| 26 | M | + | 1y | 3y | Ongoing | 11y 8m |
| 27 | F | - | 1y | 5y | Ongoing | 10y 5m |
*Same family; M: Male; F: female; Cons: Consanguinity; y: years; m: months.
3.3.2. Diagnosis findings
The enzymatic activity of α-L-iduronidase in leukocytes and/or plasma was undetectable in only 1 patient (Table 1).
3.4. Enzyme replacement therapy
3.4.1. Initiation of ERT
The age at onset of ERT ranged from 1 to 31 years (mean 14 years). Duration between time of diagnosis and initiation of ERT ranged from 2 months to 9 years, and was primarily associated with difficulty obtaining the recombinant enzyme. Duration of the infusion ranged from 40 weeks to 556 weeks (mean 259 weeks).
3.4.2. Non-compliance with ERT
The proportion of patients lacking infusions ranged from 0% to 32% (mean 17.8%), and were primarily due to the following reasons: lack of enzyme (MPS I, 40%; MPS II, 47%; MPS VI, 29%), holidays (MPS I, 26%; MPS II, 20%; MPS VI 29%), situational reasons such as family illness and public transport strikes (MPS I, 18%; MPS II, 19%; MPS VI 25%), and illnesses (e.g. upper respiratory infections, hospitalizations, surgical procedures) (MPS I, 16%; MPS II, 14%; MPS VI, 17%).
3.4.3. Adverse reactions to ERT
Adverse reactions to infusions were observed in 55% (n = 15) of patients, mostly occurring during the first few weeks of treatment. Common adverse reactions included skin rash (Figure 1a–1d), subarachnoid hemorrhage, fever and bronchospasm; symptoms were generally responsive to antihistamines, antipyretics and reduction of infusion speed.
Figure 1.
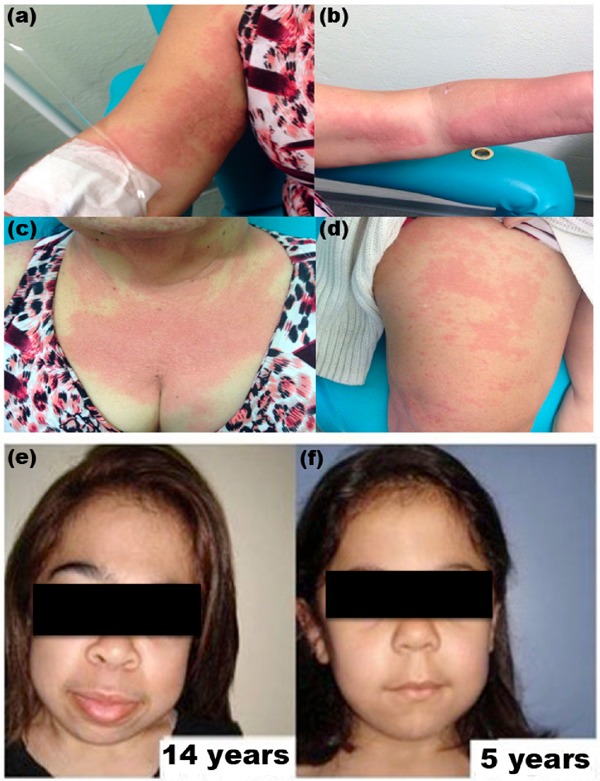
(a–d) Patient with allergic skin reaction during ERT; (e–f) Facial appearance from two sisters with MPS VI.
Serious adverse reactions were observed in only 2 (7.4%) patients. One patient with MPS VI, aged 4 years 5 months, presented with shortness of breath, facial flushing, tachypnea, tachycardia and respiratory failure requiring hospitalization and intensive care unit admission for 30 days under mechanical ventilation, during week 48 of ERT. ERT was suspended and pulmonary hypertension was diagnosed. When ERT was subsequently re-introduced, the patient presented with milder reactions responsive to antihistamines and antipyretics, in which these symptoms lasted 8 months until its resolution. The second patient was a MPS I Scheie patient, aged 29 years, who presented with arterial hypertension at the beginning of the ERT therapy (between weeks 1 and 2); her symptoms were responsive to antihypertensive medications. She subsequently presented with severe shortness of breath, tachycardia, urticarial, reddish and itchy plaques with all infusions from week 320 of her therapy, unresponsive to antihistamines and corticosteroids and requiring the initiation of a rapid desensitization protocol. The protocol consisted of reducing the infusion rate and administering drugs such as diphenhydramine, ranitidine, montelukaste, acetaminophen, methylprednisolone and benzodiazepines. Due to limited infrastructure, the patient was transferred to another private infusion center.
3.4.4. Clinical outcomes: Dysmorphic features
Most patients continued to present with infiltrated facies even with ERT. However, we observed a less pronounced facial appearance in one of the two sisters who started ERT earlier (age 1 year 5 months) when compared to her sister, who started ERT at age 8 years 9 months (Figure 1e–1f).
3.4.5. Clinical outcomes: Cardiovascular and respiratory changes
Before ERT, 24/26 (92%) patients presented with echocardiographic alterations, primarily involving thickening and/or insufficiency of the mitral or aortic valve (21/24, 88%) and right and/or left ventricular hypertrophy (14/24, 58%). A Scheie patient presenting with ventricular dysfunction with mild reduction of left ventricular contractile function and mitral and aortic valve alterations showed no improvement with ERT. Two patients (n = 1 Hurler, n = 1 Hurler-Scheie) did not present echocardiographic alterations before ERT, but developed valve alterations and ventricular hypertrophy during therapy.
Pulmonary hypertension was observed before ERT in three patients (n = 2 Hurler-Scheie, n = 1 MPS VI). During ERT, three patients presented with pulmonary hypertension, two of whom died (n = 1 Hurler Scheie, n = 1 MPS VI). Among the 6 patients who presented with pulmonary hypertension across the study, four evolved with ventricular dysfunction (n = 1 Hurler, n = 1 Hurler-Scheie, n = 1 Scheie, n = 1 MPS VI), while two did not develop echocardiographically-diagnosed cardiac abnormalties.
There was a difference in the progression of cardiac lesions in two sisters with MPS VI. The older sister began ERT at 8 years 10 months of age and required surgical repair of the mitral valve three months prior to ERT initiation (8 years 7 months). The younger sister who initiated ERT at 1 year 7 months of age showed a partial reversal of ventricular hypertrophy after 3 years of therapy, thereby not warranting cardiac surgery.
3.4.6. Clinical outcomes: surgical procedures
Twenty-two patients (81%) from a total of 27 underwent a total of 63 surgical procedures (mean 2.9 surgeries per patient). Inguinal and/or umbilical herniorrhaphy accounted for 50% of the interventions.
Mitral valve replacement with metal prosthesis, patent ductus arteriosus repair and aortic valve repair were conducted in one MPS VI patient at 8 years 7 months of age, prior to ERT. Two Scheie patients underwent carpal tunnel corrective surgery prior to ERT at 22 and 25 years of age. Eight patients (30%) also underwent adenotonsillectomies prior to ERT (Table 5).
Table 5. Surgical procedures before (B) and after (A) ERT.
| Item | MPS I — | MPS I — | MPS I — | MPS II — | MPS VI — | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Hurler |
Hurler-Scheie |
Scheie |
Hunter |
Marateaux Lamy |
||||||
| B | A | B | A | B | A | B | A | B | A | |
| Adenotonsillectomy | 1/3 | 1/3 | 3/5 | 1/5 | 1/5 | - | 6/7 | - | - | - |
| Hernia | 1/3 | 1/3 | 2/5 | 1/5 | 4/5 | - | 5/7 | - | 2/6 | - |
| Tympanostomy | - | - | 1/5 | 1/5 | - | - | 1/7 | - | - | - |
| Glaucoma | 1/3 | - | - | - | - | - | - | - | - | - |
| Carpal tunnel | - | - | 1/5 | - | 2/5 | - | - | - | - | - |
| Aquiles tendon | - | - | 1/5 | - | - | - | - | 1/7 | - | - |
| Scoliosis | - | - | 1/5 | - | - | - | - | - | - | - |
| Peritoneal Ventricular | - | 2/3 | 1/5 | - | - | - | 1/7 | - | 1/6 | - |
| Bypass | - | - | 1/5 | - | - | - | - | - | - | - |
| Cervical Mielopathy | - | - | - | - | - | - | - | - | 1/6 | - |
| Mitral valve replacement | - | - | - | - | - | - | - | - | 1/6 | - |
One patient with Hurler-Scheie underwent surgical correction of her lumbar kyphoscoliosis at 4 years of age prior to ERT initiation. However, her condition evolved from age 10 years onwards despite ERT therapy, eventually leading to her death at 18 years of age due to pneumonia, pulmonary hypertension, and sepsis (Table 5).
During ERT, one Hurler-Scheie patient underwent bilateral surgical carpal tunnel repair at age 8 years 6 months after 150 weeks of ERT. Two patients (n = 1 Hurler; n = 1 Hurler-Scheie) underwent adenotonsillectomies, and another Hurler-Scheie patient underwent adenotonsillectomy, three tympanostomies, two shunt ventricular peritoneal and surgery for cervical myelopathy and herniorrhaphy. One Hurler-Scheie patient underwent two orthopedic surgeries (Table 5).
3.4.7. Clinical outcomes: mortality
No significant difference in mortality rates was identified across MPS types: MPS I versus MPS II (RR: 0.92, 95% CI: 0.19-4.38; p = 0.92); MPS I versus MPS VI (RR: 1.38, 95% CI: 0.18-10.71; p = 0.76); and MPS II versus MPS VI (RR: 1.50, 95% CI: 0.17-12.94; p = 0.71) (Figure 2, Table S1, http://www.irdrjournal.com/action/getSupplementalData.php?ID=12).
Figure 2.
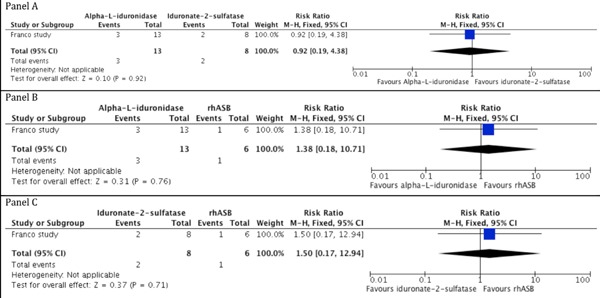
Death rates among studied groups with the use of ERT.
4. Discussion
Our findings regarding age of onset and primary symptoms associated with MPS I, II and VI are consistent with the literature, (1,6–8) except for one MPS I H patient, who presented with glaucoma at 8 months of age. There are few reports of glaucoma with such an early onset (9).
Mean age at time of diagnosis of MPS I in our case series was approximately 7 years of age. In contrast, a multicenter study involving 891 patients with MPS I from 24 countries reported a mean age at time of diagnosis was approximately 5 years (10). Similarly, mean age at time of diagnosis of MPS II in our case series was approximately 5 years of age, while the Hunter Outcome Survey, involving 16 countries with a total of 263 patients, reported a mean age at diagnosis of 3.5 years (11). Mean age at diagnosis for MPS VI patients in our case series was 5 years; the literature generally reports ages of diagnosis ranging from 2 years to 12 years (1,6,12). The discrepancy between age of diagnosis in our study and the literature may be explained by the care of a large number of patients being limited to few specialized in Brazil, delaying time of detection.
In terms of clinical presentations, 6 of 8 MPS I patients (n = 3 Hurler; n = 3 Hurler-Scheie) and 4 of 8 MPS II patients presented with intellectual disability; in contrast, no patients with MPS VI presented with such disabilities. These findings are supported by previous reports in the literature (1,6,13).
The main presenting features such as facial appearance (Figure 1e–1f), joint restriction, and short stature for patients with MPS VI in our case series were similar to those presented in the literature (5,12,14,15).
Current methods for dosing enzyme activity are not sensitive enough to measure the complete absence or presence of residual enzymatic activity (16). Thus, it is difficult to correlate the undetectable activity of the enzyme to severe phenotypes. In terms of GAG chromatography, dermatan sulfate and heparan sulfate were predominant in MPS I and II patients and dermatan sulfate and chondroitin sulfate in MPS VI.
Regarding the adverse effects, we found that 55% of patients experienced at least one mild adverse effect, most frequently erythema, urticaria (as noted in Figure 1a–1d), and redness/flushing. Such events have been reported in 36% of studied populations as per previous reports in the literature (5,17).
One adult female MPS I Scheie patient in our case series specifically presented with a severe infusion reaction after five years of ERT. She presented with shortness of breath, tachycardia, urticaria, and reddish and itchy plaques in all subsequent infusions. As there was no improvement with standard medication (anti-histamines, and corticosteroids), a rapid desensitization adapted protocol was performed, in line with previous reports. The protocol consisted of a reduction of infusion speed (lasting up to 10 hours using three saline bags with different laronidase dilutions) and administration of diphenhydramine, ranitidine, montelukaste, acetaminophen, methylprednisolone and benzodiazepines with infusions, to which the patient was responsive.
Our findings support a strategy that involves the following steps in addition to pre-medication for symptom relief in response to an infusion reaction: i) temporary decrease of the infusion; ii) additional administration of antipyretics and antihistamines; and iii) discontinuation of the infusion and initiation of appropriate supportive measures immediately if a severe hypersensitivity reaction (14).
The most serious adverse reactions observed in our study involved anaphylaxis and impaired respiratory function, warranting further monitoring; these have been previously evidenced in the literature as life-threatening as well (18,19).
Regarding genetic mutations (Table S2, http://www.irdrjournal.com/action/getSupplementalData. php?ID=12), our data aligns with the literature for MPS I, showing the most frequent mutations are W402X and P533R (20) and presence of both alleles is linked to a severe phenotype of the disease. Genotypic determination may be useful in providing individual parameters of the patient's therapeutic response to therapies such as ERT and BMT as well. While the literature shows either large or small deletions being associated with MPS II, our study did not identify any of these findings; contrarily, we noticed only punctual deletions (16,21). In terms of MPS VI, the IVS-8T>G intronic mutation was found in two sisters with the condition, both of whom presented with a more serious phenotype of the disease including medullary compression, short stature and need for cardiac surgery. A multicenter study involving 105 patients of different nationalities observed the IVS-8T>G mutation only in Brazilian patients (22).
In 26 MPS patients (I, II, III, IV, VI and VII), Leal et al. (23), observed changes in the mitral valve (60.3%); Of the aortic valve (35.8%); ventricular hypertrophy (43%); and pulmonary hypertension (36%).
High mortality rates were found in our patients across all three types of MPS and regardless of ERT use. A study with 24 MPS I patients (24), also reported deaths in 7 patients during ERT. The main causes of death in our case series were respiratory problems such as pneumonia, complications of anesthesia, and pulmonary hypertension, in line with previous reports (23,24).
5. Conclusion
Our case series demonstrated that clinical heterogeneity exists across MPS-affected patients by inter- and intrafamilial variability in terms of phenotype. Our findings suggested that while ERT was well tolerated and able to attenuate symptoms effectively, it did not seem to prevent disease progression and reduce mortality. This study emphasizes that early diagnosis and use of ERT are critical for better outcomes and for enhancing the quality of life of these patients, despite mortality and morbidity remaining high. Further well-designed robust studies with larger sample sizes and longer follow-ups are warranted to further examine the effects of ERT in these patient populations in terms of symptom and disease progression control.
Acknowledgements
José Francisco da Silva Franco received grants from Biomarin for lectures as speaker. Regina El Dib received a Brazilian Research Council (CNPq) scholarship (CNPq 310953/2015-4).
Supplementary PDF file supplied by authors.
References
- 1. Neufield E, Muenzer J. The mucopolysaccharidoses. In: The metabolic and molecular bases of inherited disease (Scriver C, Beaudet A, Valle D, Sly W, eds). The McGraw-Hill Companies, New York, USA, 2001, pp.3421-52. [Google Scholar]
- 2. Jameson E, Jones S, Remmington T. Enzyme replacement therapy with laronidase (Aldurazyme®) for treating mucopolysaccharidosis type I. Cochrane Database Syst Rev. 2016; 4:CD009354. [DOI] [PubMed] [Google Scholar]
- 3. Tomanin R, Zanetti A, D'Avanzo F, Rampazzo A, Gasparotto N, Parini R, Pascarella A, Concolino D, Procopio E, Fiumara A, Borgo A, Frigo AC, Scarpa M. Clinical efficacy of Enzyme Replacement Therapy in paediatric Hunter patients, an independent study of 3.5 years. Orphanet J Rare Dis. 2014; 9:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brunelli MJ, Atallah AN, da Silva EM. Enzyme replacement therapy with galsulfase for mucopoly-saccharidosis type VI. Cochrane database Syst Rev. 2016; 3:CD009806. [DOI] [PubMed] [Google Scholar]
- 5. Giugliani R, Harmatz P, Wraith JE. Management guidelines for mucopolysaccharidosis VI. Pediatrics. 2007; 120:405-418. [DOI] [PubMed] [Google Scholar]
- 6. Harmatz P, Whitley CB, Waber L, Pais R, Steiner R, Plecko B, Kaplan P, Simon J, Butensky E, Hopwood JJ. Enzyme replacement therapy in mucopolysaccharidosis VI (Maroteaux-Lamy syndrome). J Pediatr. 2004; 144:574- 580. [DOI] [PubMed] [Google Scholar]
- 7. Beck M, Arn P, Giugliani R, Muenzer J, Okuyama T, Taylor J, Fallet S. The natural history of MPS I: Global perspectives from the MPS I Registry. Genet Med. 2014; 16:759-765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Muenzer J, Hendriksz CJ, Fan Z, Vijayaraghavan S, Perry V, Santra S, Solanki GA, Mascelli MA, Pan L, Wang N, Sciarappa K, Barbier AJ. A phase I/II study of intrathecal idursulfase-IT in children with severe mucopolysaccharidosis II. Genet Med. 2016; 18:73-81. [DOI] [PubMed] [Google Scholar]
- 9. Nowaczyk MJ, Clarke JT, Morin JD. Glaucoma as an early complication of Hurler's disease. Arch Dis Child. 1988; 63:1091-1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pastores GM, Arn P, Beck M, Clarke JT, Guffon N, Kaplan P, Muenzer J, Norato DYJ, Shapiro E, Thomas J, Viskochil D, Wraith JE. The MPS I registry: Design, methodology, and early findings of a global disease registry for monitoring patients with Mucopolysaccharidosis Type I. Mol Genet Metab. 2007; 91:37-47. [DOI] [PubMed] [Google Scholar]
- 11. Wraith JE, Beck M, Lane R, van der Ploeg A, Shapiro E, Xue Y, Kakkis ED, Guffon N. Enzyme replacement therapy in patients who have mucopolysaccharidosis I and are younger than 5 years: Results of a multinational study of recombinant human alpha-L-iduronidase (laronidase). Pediatrics. 2007; 120:e37-46. [DOI] [PubMed] [Google Scholar]
- 12. Valayannopoulos V, Nicely H, Harmatz P, Turbeville S. Mucopolysaccharidosis VI. Orphanet J Rare Dis. 2010; 5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Muenzer J. The mucopolysaccharidoses: A heterogeneous group of disorders with variable pediatric presentations. J Pediatr. 2004; 144:S27-34. [DOI] [PubMed] [Google Scholar]
- 14. Giugliani R, Federhen A, Muñoz Rojas MV, et al. Terapia de reposição enzimática para as mucopolissacaridoses I, II e VI: Recomendações de um grupo de especialistas brasileiros. Rev Assoc Med Bras. 2010; 56:271-7. [DOI] [PubMed] [Google Scholar]
- 15. Hendriksz CJ, Giugliani R, Harmatz P, Lampe C, Martins AM, Pastores GM, Steiner RD, Leão Teles E, Valayannopoulos V, CSP Stdudy Group Design, baseline characteristics, and early findings of the MPS VI (mucopolysaccharidosis VI) Clinical Surveillance Program (CSP). J Inherit Metab Dis. 2013; 36:373-384. [DOI] [PubMed] [Google Scholar]
- 16. Froissart R, Da Silva IM, Maire I. Mucopolysaccharidosis type II: An update on mutation spectrum. Acta Paediatr. 2007; 96:71-77. [DOI] [PubMed] [Google Scholar]
- 17. Muenzer J, Beck M, Giugliani R, Suzuki Y, Tylki-Szymanska A, Valayannopoulos V, Vellodi A, Wraith JE. Idursulfase treatment of Hunter syndrome in children younger than 6 years: Results from the Hunter Outcome Survey. Genet Med. 2011; 13:102-109. [DOI] [PubMed] [Google Scholar]
- 18. Wraith JE, Clarke LA, Beck M, Kolodny EH, Pastores GM, Muenzer J, Rapoport DM, Berger KI, Swiedler SJ, Kakkis ED, Braakman T, Chadbourne E, Walton-Bowen K, Cox GF. Enzyme replacement therapy for mucopolysaccharidosis I: A randomized, double-blinded, placebo-controlled, multinational study of recombinant human alpha-L-iduronidase (laronidase). J Pediatr. 2004; 144:581-588. [DOI] [PubMed] [Google Scholar]
- 19. Muenzer J. Early initiation of enzyme replacement therapy for the mucopolysaccharidoses. Mol Genet Metab. 2014; 111:63-72. [DOI] [PubMed] [Google Scholar]
- 20. Matte U, Yogalingam G, Brooks D, Leistner S, Schwartz I, Lima L, Norato DY, Brum JM, Beesley C, Winchester B, Giugliani R, Hopwood JJ. Identification and characterization of 13 new mutations in mucopoly-saccharidosis type I patients. Mol Genet Metab. 2003; 78:37-43. [DOI] [PubMed] [Google Scholar]
- 21. Hopwood JJ, Bunge S, Morris CP, Wilson PJ, Steglich C, Beck M, Schwinger E, Gal A. Molecular basis of mucopolysaccharidosis type II: Mutations in the iduronate-2-sulphatase gene. Hum Mutat. 1993; 2:435-442. [DOI] [PubMed] [Google Scholar]
- 22. Karageorgos L, Brooks DA, Pollard A, et al. Mutational analysis of 105 mucopolysaccharidosis type VI patients. Hum Mutat. 2007; 28:897-903. [DOI] [PubMed] [Google Scholar]
- 23. Leal GN. Echocardiographic study of pediatric patients with mucopolysaccharidosis (dissertation). São Paulo: University of São Paulo, Faculty of Medicine; 2009. http://www.teses.usp.br/teses/disponiveis/5/5141/tde-08122009-172753/pt-br.php (accessed August 02, 2017). [Google Scholar]
- 24. Dornelles AD, de Camargo Pinto LL, de Paula AC, et al. Enzyme replacement therapy for Mucopolysaccharidosis Type I among patients followed within the MPS Brazil Network. Genet Mol Biol. 2014; 37:23-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary PDF file supplied by authors.