

Abstract
A thiol-terminated polyethyleneglycol (PEG)-paclitaxel (PTX) conjugate was synthesized and utilized to construct a novel drug delivery system with thiol-functionalized silica nanoparticles (SLNs) to improve the overall performance of PTX in liver cancer therapy. Drug loading was performed by coating the PTX conjugate on the surface of SLNs. The PTX-PEG/SLNs showed a binary responsive drug release behavior to esterase as well as high concentrations of glutathione. The synergic effects of these cancer cell-specific factors on the release characteristics of PTX-PEG/SLNs resulted in a significantly (P<0.01) elevated anti-cancer efficiency. This included prolonged circulation and passive tumor-targeting properties in vivo due to surface modification of PEG and targeted release of PTX inside tumor cells, which resulted in increased anti-cancer efficiency. Improving the in vitro properties of PTX-PEG/SLNs not only significantly (P<0.01) enhanced its therapeutic efficacy in a murine liver cancer model, but rendered these drug-conjugated SLNs a promising nanoprodrug system for potential use as clinical cancer therapeutics.
Keywords: silica, dual-responsive, paclitaxel, liver cancer
Introduction
A large proportion of the most commonly applied anti-cancer drugs are hydrophobic and have numerous disadvantages, including low solubility and stability as well as severe side effects, which have greatly limited their clinical applications. To solve this dilemma, a wide range of well-designed nanocarriers have been developed with the aim to improve the anti-cancer efficacy of hydrophobic anti-cancer drugs by employing various materials ranging from organic to inorganic ones (1–5). Paclitaxel (PTX) has long been identified as a broad-spectrum anti-cancer drug with higher effectiveness than its competitors; therefore, a variety of PTX formulations have been explored seeking to employ the advantages of PTX while overcoming its disadvantages (6–8). Among them, silica nanoparticle (SLN)-based drug delivery systems (DDS), particularly surface-modified SLNs, are considered to be promising, since they differ from the ordinary drug delivery strategies of encapsulating drug molecules within the hydrophobic core, as SLNs covalently conjugate or physically absorb drug molecules onto their surface (9–11). This requires mono- or multiple layers generated on the surface of SLNs, allowing for surface functionalization of SLNs with a variety of active molecules, such as specific targeting ligands and/or drug payloads with tunable ratios. Moreover, it has been well-documented that the numerous merits of SLNs, such as conjugating reactivity, chemical inertness and low innate cytotoxicity are all desirable properties for their potential usage as drug carriers with favourable biocompatibility (12,13).
In the present study, a well-defined polyethyleneglycol (PEG)-PTX conjugate was employed to construct a novel DDS with thiol-terminated SLNs (PTX-PEG/SLNs) for overcoming the above-mentioned problems. Being different from that used in previous studies, the well-designed PEG-PTX conjugate applied to the surface of SLNs was expected to show certain advantages. First, the PEG shell improved the water solubility of the modified SLNs and granted a ‘stealth effect’ to the PTX-PEG/SLNs in vivo, thereby increasing the circulation time and enhancing the passive targeting effect; Furthermore, the drug loading content of the resulting PTX-PEG/SLNs is freely tunable by adjusting the feeding ratio of PEG vs. PTX. Moreover, the binary responsive drug release characteristics insure the stability of PTX-PEG/SLNs in the circulation system and selective drug release within the tumor cells. As a proof of concept, the present study performed a detailed physicochemical and pharmaceutical characterization of PTX-PEG/SLNs. Moreover, their anti-cancer efficacy in a HepG2 tumor-bearing mouse model was systematically investigated by comparison to Taxol®, the commercial formulation of PTX.
Materials and methods
Materials
Triton X-100, tetraethyl orthosilicate (TEOS) and (3-mercaptopropyl)-trimethoxysilane (MPTMS) obtained from the Shanghai Chemical Regent Company (Shanghai, China). PEG (molecular weight, 1,000 kDa), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazoliumbromide (MTT), fluorescein isothiocyanate (FITC), Hoechst 33342 and pig liver esterase (PLE) were obtained from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). Paclitaxel was obtained from Yew Pharmaceutical Co. (Jiangsu, China). Unless otherwise stated, all starting materials were obtained from commercial suppliers and used without further purification.
Preparation of PTX-PEG/SLNs
Thiol-functionalized SLNs were firstly synthesized in a water-in-oil microemulsion as previously reported (14) with minor modifications. In brief, a water-in-oil microemulsion was prepared by mixing 1.8 ml Triton X-100, 7.5 ml cyclohexane, 1.6 ml n-hexanol and 480 µl water. After stirring for 0.5 h, 180 µl TEOS and 60 µl MPTMS were then added as precursors for silica matrix formation, followed by the addition of 100 µl NH4OH to initiate the polymerization process. The reaction was allowed to continue for 24 h at room temperature. After the reaction was completed, the thiol-functionalized SLNs were precipitated by addition of ethanol and were washed with ethanol and water, respectively, for several times to remove the surfactant and excess dye molecules from the particles. FITC-doped thiol-functionalized SLNs were also prepared in the same way except that the water solution was replaced by a 0.01 mol/l FITC solution.
The synthesis of the PTX-PEG1000-thiol (SH) ligand was performed according to a previously reported method (15).
For the preparation of PTX-PEG/SLNs, PTX-PEG-SH was dissolved in 4 ml methanol. Thiol-functionalized SLNs were added to the solution of PTX-PEG-SH, and the mixture was stirred for 1 h and left to stand overnight to allow for complete reaction. The mixture was dialyzed in a fresh deionized water solution [molecular-weight cut-off (MWCO), 7,500; 3×2l]. After freeze-drying, the resulting substance was stored at −20°C until further use. The single thiol-funcionalized SLNs were indicated as ‘thiol-funcionalized SLNs’ while the PTX-PEG-SH modified thiol-funcionalized SLNs were indicated as ‘PTX-PEG/SLNs’ in the following descriptions.
Drug loading
The prepared PTX-PEG/SLNs were suspended in a solution of ethanol and 0.01 M HCl (1:1) and incubated at 37°C for 48 h. After being filtered through a 0.22-µm membrane, the solution was subjected to high-performance liquid chromatography (HPLC) to calculate the drug loading efficiency (DLE) of PTX-PEG/SLNs, which was calculated according to the following formula:
DLE (% w/w) = (weight of loaded PTX/weight of PTX-PEG/SLNs) × 100%.
Analytical HPLC was performed using an LC-2010 system (Shimadzu Corp., Kyoto, Japan) with ultraviolet detection at 227 nm. The separation of PTX was performed on a Diamonsil™ C18 column (length, 250 mm; inner diameter, 4.6 mm; pore size, 5 µm) with elution with methanol and distilled water (80:20 v/v) at 30°C. The flow rate was set at 1 ml/min (2).
Characterization
The diameter and dispersity of the thiol-functionalized SLNs and PTX-PEG/SLNs were measured using a ZetaPlus Zeta Potential Analyzer (Brookhaven Instruments Corp., Holtsville, NY, USA). For structural observation, a single drop of each solution was deposited on a transmission electron microscopy (TEM) grid and allowed to air-dry. All silica nanoparticle samples and conjugates were imaged using a JEM-200CX TEM (JEOL Ltd., Tokyo, Japan) with an acceleration voltage of 80 kV.
In vitro drug release
The release of PTX and PTX-PEG-SH ligands from PTX-PEG/SLNs was investigated by a standard dialysis method according to a previously described method (15). In brief, the PTX-PEG/SLNs (30 mg) were suspended in 5 ml phosphate-buffered saline (PBS; pH 7.4) and placed in a dialysis bag (MWCO, 7,500). PBS containing Tween-80 (0.1% w/v) was employed as the release medium. Glutathione (GSH; Sigma-Aldrich; Merk KGaA) concentrations in the release medium were set at 2 mM (extracellular level) and 10 mM (intracellular level), and 20 units of PLE units were added. The dialysis bag was immersed in the release medium and the system was agitated at 37°C. At pre-determined time intervals, 200 µl release medium was withdrawn and the same volume of fresh medium was added. The amount of PTX-PEG1000-SH ligand and PTX molecules in the medium were measured by the same.
HPLC analysis as mentioned above. The PTX-PEG/SLNs were designed to achieve a dual-responsive drug release profile: One release is mediated by ligand exchange in response to the high concentration of GSH within tumor cells, and the other is hydrolysis of the ester bond between PTX and the PEG chain by esterase. The drug release profile of PTX-PEG/SLNs meets the requirement that the PTX molecules remain unreleased under normal physiological conditions (i.e., in the blood stream), whereas accelerated PTX release is achieved at tumor sites or within cancer cells. The present study therefore systematically assessed the release properties of PTX-PEG/SLNs under the following conditions: i) PBS solution with a pH of 7.4 (physiological environment); ii) GSH solution with concentrations of 10 and 2 mM (mimicking the intra- and extracellular GSH concentration, respectively); iii) the absence and presence of PLE; and iv) the combination of GSH and PLE (simulates the in vivo environment outside and inside cells, respectively).
Assessment of cytotoxicity
The HepG2 human hepatocellular liver carcinoma cell line was purchased from the China Center for Type Culture Collection (Shanghai, China) and was cultured in Dulbecco's modified Eagle's medium with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) at 37°C in a humidified atmosphere with 5% CO2.
For cytotoxicity assays, HepG2 cells were seeded in a 96-well plate (1×104 cells/well) and incubated to reach 80% confluence. The primary growth medium was removed and replaced with 200 µl fresh medium containing various concentrations of drug-free amine-functionalized SLNs and returned to the incubator for another 48 h. The cells were then subjected to a standard MTT assay (16). Other wells were incubated with different concentrations of PTX-PEG/SLNs, PTX-PEG1000-SH ligand or free PTX for 48 h followed by the same standard MTT assay as mentioned above.
Cellular uptake studies
FITC-doped thiol-functionalized SLNs were employed to construct PTX-PEG/SLNs, which were incubated with HepG2 cells to determine the cellular uptake profile of PTX-PEG/SLNs. HepG2 cells were seeded in 6-well plates at a density of 4×105 cells/well and incubated overnight to reach 70% confluence. The cells were then incubated for an additional 2 or 4 h with thiol-functionalized SLNs or PTX-PEG/SLNs at a PTX dose of 2 µg/ml. At different time points, cells were treated with Hoechst 33342 (10 µg/ml) for 15 min and rinsed three times with PBS. The intracellular trafficking of polyplexes was observed using a Leica TCS SP5 confocal laser scanning microscope (Leica Microsystems, Wetzlar, Germany). To quantitatively analyze the cellular uptake profile, cells were washed with PBS subsequent to incubation, trypsinized and subjected to flow cytometric analysis (BD Biosciences, Franklin Lakes, NJ, USA).
In vivo antitumor efficacy and histological assays
The in vivo antitumor efficacy of different PTX formulations was evaluated using animal tumor models after inoculation of HepG2 cells. All procedures were performed in compliance with the US National Institute of Health's Guide for the Care and Use of Laboratory Animals. The protocol was approved by the Committee on the Ethics of Animal Experiments of the HeiLongJiang BaYi Agricultural University. Healthy male BALB/c nude mice (18–20 g) were purchased from Zhejiang Experimental Animal Center (Hangzhou, China), housed under pathogen-free conditions and allowed free access to food and water with 12 h light/dark, temperature of 25°C and humidity of 55%. To establish the tumor-bearing mouse model, HepG2 cell suspensions containing 106 cells in 0.1 ml saline solution were subcutaneously implanted into the axillary space. The tumor-bearing mice were subjected to antitumor activity studies once the tumor volume reached 100 mm3.
The mice were randomly divided into three groups: Saline, Taxol® and PTX-PEG/SLNs. Each treatment group was comprised of six tumor-bearing mice. All formulations were injected through the tail vein with a PTX dosage of 20 mg/kg in 2-day intervals. Tumor volume and body weight of all tested mice were recorded every 2 days using a caliper prior to administration of the formulations.
At the end of the treatment (after 14 days), mice were sacrificed by decaptitation following anesthesia with 100 µl/per mouse of 10% (v/v) chloral hydrate (Shanghai Aladdin Bio-Chem Technology Co., Ltd., Shanghai, China) and their tumor tissues were excised and fixed with 4% paraformaldehyde. The samples were processed, sectioned and stained with hematoxylin and eosin (H&E) according to a standard procedure. The tissue sections were subjected to histological observation with a microscope (Olympus BX51-Pol; Olympus Optical Co., Ltd., Tokyo, Japan).
Statistical analysis
Values are expressed as the mean ± standard deviation. P<0.05 was considered to indicate a statistically significant difference. Statistical significance was determined using two-tailed Student's t-test or one-way analysis of variance using Origin 8.0 (OriginLab, Northampton, MA, USA.).
Results and Discussion
Particle size, dispersity, morphology and drug loading of PTX-PEG/SLNs
The particle size and dispersity of thiol-functionalized SLNs and PTX-PEG/SLNs were first measured by a dynamic light scattering technique. As shown in Fig. 1A, the thiol-functionalized SLNs obtained via the water-in-oil microemulsion method were spherical, with a diameter of ~90 nm. This is in accordance with a previous study, which prepared thiol-functionalized SLNs using the same method (17). Moreover, the as prepared thiol-functionalized SLNs displayed a relatively small polydispersion index (PDI) of 0.214. This was attributed to the well-formed water pools in the microemulsion, which are thought to have an important role in controlling the size and dispersity of the generated particles (18,19). Compared with the particle size of the thiol-functionalized SLNs, the PTX-PEG/SLNs demonstrated a slightly increased size of ~110 nm, which may be due to the anchoring of PTX-PEG1000-SH ligand on the surface of SLNs. At the same time, it was observed that the PDI of the obtained PTX-PEG/SLNs (0.083; Fig. 1B) was lower than that of thiol-functionalized SLNs, indicating that surface modification may be beneficial for the dispersion of PTX-PEG/SLNs. This was mainly due to the high hydrophilic nature of PEG, which is able to reduce the aggregation of the modified nanoparticles (20). To further confirm the successful modification achieved by the PTX-PEG1000-SH ligand on the surface of thiol-functionalized SLNs, TEM was applied to observe the structure of thiol-functionalized SLNs and PTX-PEG/SLNs. Fig. 1A shows that thiol-functionalized SLNs were individual particles with evident boundaries to one another. However, it was also observed that certain neighbouring thiol-functionalized SLNs were connected with each other, which may be due to the reaction of surface thiol groups among thiol-functionalized SLNs that are in close proximity. By contrast, the interparticle distance of PTX-PEG/SLNs increased compared with that of the thiol-functionalized SLNs, which was in accordance with the decreased PDI in the dispersity test. Furthermore, the edges of the PTX-PEG/SLNs were relatively vague, with an apparent core-shell core-structure being observed, which may indicate the presence of organic ligands (15). All of these results confirmed the successful modification of the surface of thiol-functionalized SLNs achieved by the PTX-PEG1000-SH ligand, resulting in the formation of well-shaped PTX-PEG/SLNs.
Figure 1.
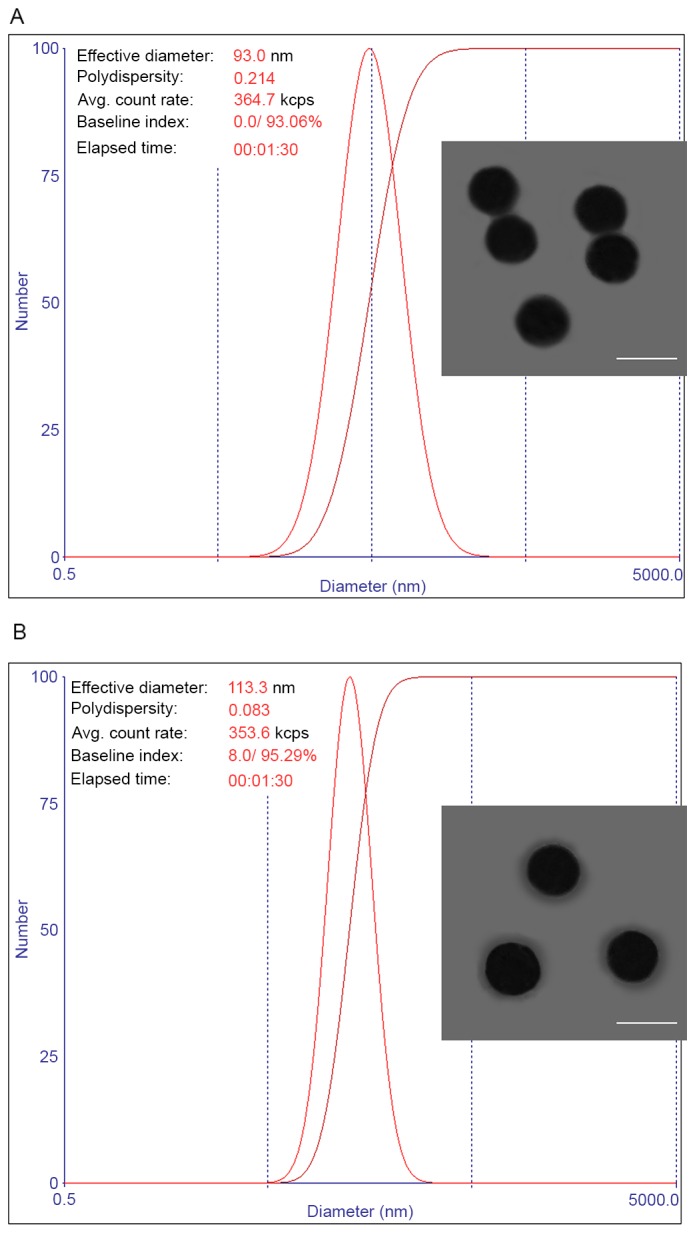
Particle size, size dispersion and morphology of (A) thiol-functionalized SLNs and (B) PTX-PEG/SLNs (scale bar, 100 nm). PTX, paclitaxel; PEG, polyethyleneglycol; SLN, silica nanoparticles.
The DLE calculated by the HPLC method was 10.7% (data not shown), which is sufficient for both in vitro and in vivo applications.
In vitro drug release
One fatal drawback of certain currently existing DDS is that they are not capable of achieving selective drug release inside tumor cells, which leads to severe side effects in vivo. The drug release behaviors of PTX-PEG/SLNs, including the release of the PTX-PEG1000-SH ligand and free PTX, were detected using HPLC and the results are summarized in Fig. 2. It was concluded that PTX-PEG/SLNs are stable under normal physiological conditions, as supported by the fact that the release of PTX-PEG1000-SH ligand and PTX in PBS solution was only 3.56 and 2.15%, respectively, within 120 h (Fig. 2A). This was further confirmed by the results displayed in Fig. 2B, showing that PTX-PEG/SLNs also remained stable in the presence of 2 mM GSH (extracellular GSH concentration). However, increased release was obtained for PTX-PEG1000-SH ligand and PTX in the presence of a high concentration of GSH (10 mM) at 120 h post-incubation (P<0.01). The mechanism of GSH-induced drug release from PTX-PEG/SLNs was associated with the thiol group exchange reaction, in which GSH functions as a reducing agent to replace the PTX-PEG1000-SH ligand on the surface of the SLNs. However, Fig. 2C demonstrated that PLE also had an important role in the drug release from PTX-PEG/SLNs in a site-specific manner: It was observed that only PTX release was accelerated, while the release profile of PTX-PEG1000-SH ligand remained unaffected. This was possibly due to the selective hydrolysis of the ester bond between PTX and the PEG chain by PLE. Finally, the combined effect of GSH and PLE was assessed to demonstrate the dual-responsive drug release properties of PTX-PEG/SLNs. As shown in Fig. 2D, PTX-PEG/SLNs treated with GSH (10 mM) combined with PLE demonstrated the highest release rate with >80% of the loaded PTX being dissociated after 120 h. All of the abovementioned results indicated that GSH and PLE achieve targeted drug release from PTX-PEG/SLNs in a synergistic manner. The drug release mechanism is involves ligand exchange with GSH at a high concentration and enzymatic hydrolysis reactions. The combination of the two drug release mechanisms permits the rapid transformation of PTX-PEG/SLNs into free PTX inside tumor cells, which is beneficial for tumor cell inhibition. As a proof of concept, in vitro cytotoxicity assays were then performed to further demonstrate the intracellular targeted drug release of the PTX-PEG/SLNs.
Figure 2.

Drug release profiles of PTX and the therapeutic ligand from the PTX-PEG/SLNs (A) in the release media (PBS at pH 7.4), (B) release media with intracellular and extracellular concentration of GSH (10 and 2 mM, respectively), (C) release media containing pig liver esterase (20 units), and (D) the combination of release media with GSH and in the presence of pig liver esterase. **P<0.01 vs. ligand. Values are expressed as the mean ± standard deviation (n=3). PTX, paclitaxel; PEG, polyethyleneglycol; GSH, glutathione.
Cytotoxicity assays and cellular uptake
The cytotoxicity of thiol-functionalized SLNs was first measured to seek its safety profile by a standard MTT assay. As shown in Fig. 3A, thiol-functionalized SLNs exhibited low cytotoxicity at all the tested concentrations. At the highest SLN concentration of 1,000 µg/ml, of cell viability remained >85% after 48 h of incubation, suggesting that thiol-functionalized SLNs were a safe material without any marked cytotoxic effect. This was beneficial for PTX-PEG/SLNs to act as a DDS to be applied in vivo.
Figure 3.
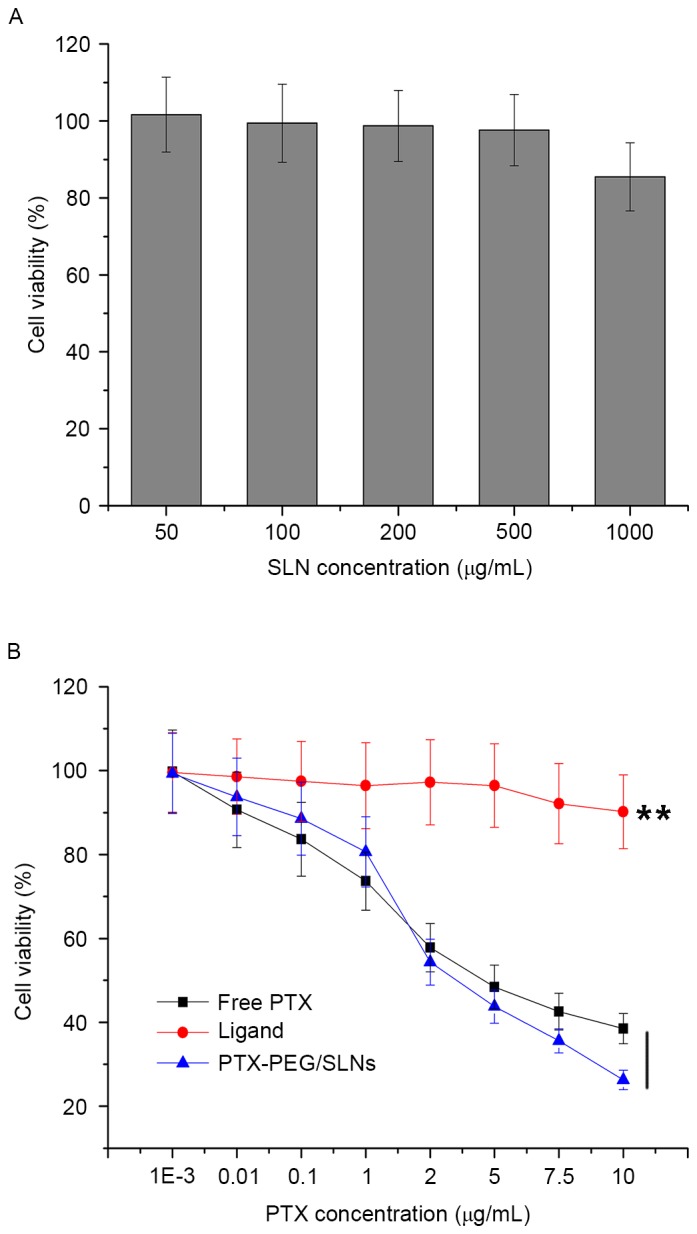
(A) Viability of HepG2 cells incubated with thiol-functionalized SLNs at various concentrations for 48 h. (B) Cytotoxicity of free PTX, PTX-PEG1000-thiol ligand and PTX-PEG/SLNs with varying PTX concentrations for 48 h on HepG2 cells. Values are expressed as the mean ± standard deviation (n=5). **P<0.01 vs. ligand. SLN, silica nanoparticles; PTX, paclitaxel; PEG, polyethyleneglycol.
The cytotoxicity of PTX-PEG/SLNs, PTX-PEG1000-SH ligand and free PTX toward HepG2 cells was then assessed at various concentrations (Fig. 3B). It was observed that at all tested PTX concentrations, PTX-PEG1000-SH ligand showed inferior cytotoxicity compared to free PTX. It may be speculated that the PTX-PEG1000-SH ligand, as a high-molecular-weight polymer with a shielding effect of the PEG space, has a significantly (P<0.01) impaired biological activity and cellular uptake. By contrast, free PTX as a relatively small molecule with hydrophobic properties is readily taken up by cells. As for PTX-PEG/SLNs, it was noted that its competitive cytotoxicity towards free PTX is concentration-dependent. The cytotoxicity of PTX-PEG/SLNs with the same dose of PTX was slightly lower than that of free PTX at low concentrations (0–2 µg/ml PTX). However, a reversed phenomenon was observed at PTX concentrations >2 µg/ml, as PTX-PEG/SLNs showed significantly higher cytotoxicity than free paclitaxel. As the results proved that thiol-functionalized SLNs were not cytotoxic towards HepG2 cells at the concentration of 1,000 µg/ml (corresponding PTX concentration was 10 µg/ml), the cytotoxicity of PTX-PEG/SLNs towards HepG2 cells is fully attributed to the release of free PTX molecules from PTX-PEG/SLNs. Therefore, the high cytotoxicity of drug-loaded nanoparticles was due to increased cellular uptake via endocytosis, and intracellular paclitaxel was released via the synergistic mechanism described above.
The cellular uptake properties of PTX-PEG/SLNs compared with those of thiol-functionalized SLNs were investigated to verify that surface modification by using the PTX-PEG1000-SH ligand is beneficial. FITC-doped thiol-functionalized SLNs were employed to construct PTX-PEG/SLNs and HepG2 cells were incubated with the thiol-functionalized SLNs and PTX-PEG/SLNs (equivalent to 2 µg/ml PTX) for 2 or 4 h. As shown in Fig. 4A, PTX-PEG/SLNs were present in the cytoplasm of HepG2 cells after 2 h of incubation and more nanoparticles were localized in the cytoplasm after 4 h of incubation compared to those at 2 h, indicating that the cellular uptake of PTX-PEG/SLNs was time-dependent. Moreover, it was noted that, compared with thiol-functionalized SLNs at the two designated time intervals, the extent of PTX-PEG/SLNs was superior. This may be due to the enhanced dispersion profile induced by modification with the PTX-PEG1000-SH ligand. Quantitative analysis using flow cytometry (Fig. 4B) further confirmed that above-mentioned results. The results indicated that particle endocytosis-induced drug accumulation in tumor cells was likely to be responsible for the differences between the therapeutic ligand and drug-conjugated SLNs in the cytotoxicity studies.
Figure 4.
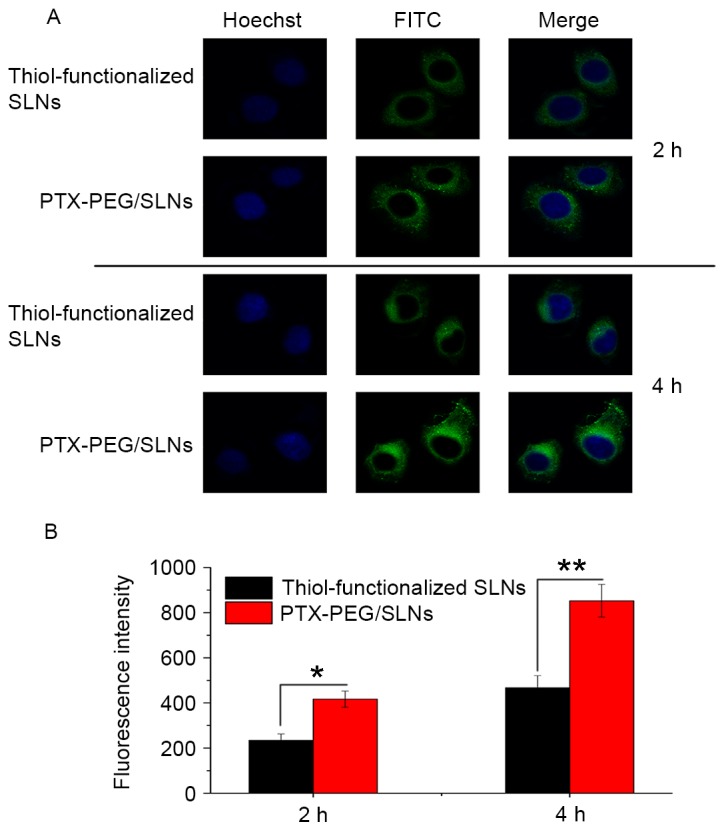
(A) Cellular uptake images of fluorescein isothiocyanate-labeled, thiol-functionalized SLNs and PTX-PEG/SLNs incubated with HepG2 cells for 2 or 4 h (magnification, ×600). (B) Flow cytometric analysis of the uptake fluorescence intensity of thiol-functionalized SLNs and PTX-PEG/SLNs at 2 and 4 h. Values are expressed as the mean ± standard deviation (n=3). *P<0.05; **P<0.01. SLN, silica nanoparticles; PTX, paclitaxel; PEG, polyethyleneglycol.
Antitumor activity in vivo
The antitumor efficacy was evaluated in BALB/c nude mice bearing HepG2 tumors. Changes in tumor volume and body weight of mice treated with saline, Taxol® and PTX-PEG/SLNs are presented in Fig. 5A and B. It was revealed that tumor growth was significantly suppressed in the group treated with PTX-PEG/SLNs as compared to that in the saline and Taxol® groups. It was noted that the mice in the PTX-PEG/SLNs group not only had the smallest tumor size, but that their body weight (23.23±0.61) and increment of body weight (1.86 g) also exceeded that of the other groups. These results indicated that the quality of life of mice treated with the PTX-PEG/SLNs was significantly (P<0.01) improved. By contrast, mice in the Taxol® group demonstrated the lowest body weight and with a negative body weight increment.
Figure 5.

In vivo antitumor efficacy of saline, Taxol® and PTX-PEG/SLNs in HepG2 tumor xenograft BALB/c nude mouse models. Changes in (A) tumor volume and (B) body weight. Values are expressed as the mean ± standard deviation (n=6). (C) Hematoxylin and eosin-stained tumor tissue sections at 14 days post-injection (magnification, ×200). **P<0.01 vs. saline. SLN, silica nanoparticles; PTX, paclitaxel; PEG, polyethyleneglycol.
To determine the extent of the anti-cancer effect in response to the concentrated distribution and cancer-specific drug release of PTX-PEG/SLNs, histological examination of H&E-stained tumor tissue sections was performed after 14 days of treatment (Fig. 5C). Almost no damage was observed in tumors from the saline-treated group, as tumor tissue cells retained the characteristics of tumor cells with a compacted cell matrix. By contrast, significant necrosis was present in the Taxol® and PTX-PEG/SLNs-treated groups. Moreover, a higher necrosis ratio was observed in tumors from the PTX-PEG/SLNs group compared to that in the Taxol® group. These results further confirmed the therapeutic effect of the nanoprodrug delivery system of PTX-PEG/SLNs, further confirming that enhanced accumulation and controlled drug release in tumor tissues from PTX-PEG/SLNs ensured higher amounts of PTX being delivered into cancer cells, resulting in increased therapeutic efficacy and antitumor efficiency.
In conclusion, with the aim to improve the overall performance of SLNs, PEG was conjugated with PTX on one end and a thiol group on the other end and employed to construct a nanoprodrug system (PTX-PEG/SLNs). The resulting hybrid structure of PTX-PEG/SLNs had superior properties compared with previous DDSs, including enhanced dispersity and targeted drug release inside tumor cells, resulting in enhanced anti-cancer activity in vitro and tumor therapeutic efficacy in vivo. The PTX-PEG/SLNs prepared in the present study overcame certain problems associated with the practical application of PTX and further improved SLNs as a DDS. The advantages of this system, such as superior biological activity and therapeutic efficiency, suggested that PTX-PEG/SLNs are a promising approach for in vivo tumor therapy.
References
- 1.Abbad S, Wang C, Waddad AY, Lv H, Zhou J. Preparation, in vitro and in vivo evaluation of polymeric nanoparticles based on hyaluronic acid-poly (butyl cyanoacrylate) and D-alpha-tocopheryl polyethylene glycol 1000 succinate for tumor-targeted delivery of morin hydrate. Int J Nanomedicine. 2015;10:305–320. doi: 10.2147/IJN.S73971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guo Q, Guan D, Dong B, Nan F, Zhang Y. Charge-conversional binary drug delivery polymeric micelles for combined chemotherapy of cervical cancer. Int J Polym Mate Polym Biomater. 2015;64:978–987. doi: 10.1080/00914037.2015.1038819. [DOI] [Google Scholar]
- 3.Li M, Li Y, Huang X, Lu X. Captopril-polyethyleneimine conjugate modified gold nanoparticles for co-delivery of drug and gene in anti-angiogenesis breast cancer therapy. J Biomater Sci Polym Ed. 2015;26:813–827. doi: 10.1080/09205063.2015.1057991. [DOI] [PubMed] [Google Scholar]
- 4.Liu E, Zhou Y, Liu Z, Li J, Zhang D, Chen J, Cai Z. Cisplatin loaded hyaluronic acid modified TiO 2 nanoparticles for neoadjuvant chemotherapy of ovarian cancer. J Nanomaterials. 2015;2015:390358. doi: 10.1155/2015/390358. [DOI] [Google Scholar]
- 5.Wang C, Li M, Yang T, Ding X, Bao X, Ding Y, Xiong H, Wu Y, Wang W, Zhou J. A self-assembled system for tumor-targeted co-delivery of drug and gene. Mater Sci Eng C Mater Biol Appl. 2015;56:280–285. doi: 10.1016/j.msec.2015.06.034. [DOI] [PubMed] [Google Scholar]
- 6.Shao W, Paul A, Rodes L, Prakash S. A new carbon nanotube-based breast cancer drug delivery system: Preparation and in vitro analysis using paclitaxel. Cell Biochem Biophys. 2015;71:1405–1414. doi: 10.1007/s12013-014-0363-0. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Z, Mei L, Feng SS. Paclitaxel drug delivery systems. Expert Opin Drug Deliv. 2013;10:325–340. doi: 10.1517/17425247.2013.752354. [DOI] [PubMed] [Google Scholar]
- 8.Yang S, Gursoy RN, Lambert G, Benita S. Enhanced oral absorption of paclitaxel in a novel self-microemulsifying drug delivery system with or without concomitant use of P-glycoprotein inhibitors. Pharma Res. 2004;21:261–270. doi: 10.1023/B:PHAM.0000016238.44452.f1. [DOI] [PubMed] [Google Scholar]
- 9.Tang F, Li L, Chen D. Mesoporous silica nanoparticles: Synthesis, biocompatibility and drug delivery. Adv Mater. 2012;24:1504–1534. doi: 10.1002/adma.201104763. [DOI] [PubMed] [Google Scholar]
- 10.Vivero-Escoto JL, Slowing II, Wu CW, Lin VS. Photoinduced intracellular controlled release drug delivery in human cells by gold-capped mesoporous silica nanosphere. J Am Chem Soc. 2009;131:3462–3463. doi: 10.1021/ja900025f. [DOI] [PubMed] [Google Scholar]
- 11.Lu J, Liong M, Sherman S, Xia T, Kovochich M, Nel AE, Zink JI, Tamanoi F. Mesoporous silica nanoparticles for cancer therapy: Energy-dependent cellular uptake and delivery of paclitaxel to cancer cells. Nanobiotechnology. 2007;3:89–95. doi: 10.1007/s12030-008-9003-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang X, Li L, Liu T, Hao N, Liu H, Chen D, Tang F. The shape effect of mesoporous silica nanoparticles on biodistribution, clearance, and biocompatibility in vivo. ACS Nano. 2011;5:5390–5399. doi: 10.1021/nn200365a. [DOI] [PubMed] [Google Scholar]
- 13.He Q, Shi J. Mesoporous silica nanoparticle based nano drug delivery systems: Synthesis, controlled drug release and delivery, pharmacokinetics and biocompatibility. J Materials Chem. 2011;21:5845–5855. doi: 10.1039/c0jm03851b. [DOI] [Google Scholar]
- 14.Wu H, Zhao Y, Mu X, Wu H, Chen L, Liu W. A silica-polymer composite nano system for tumor-targeted imaging and p53 gene therapy of lung cancer. J Biomat Sci Polymer Edition. 2015;26:384–400. doi: 10.1080/09205063.2015.1012035. [DOI] [PubMed] [Google Scholar]
- 15.Ding Y, Zhou YY, Chen H, Geng DD, Wu DY, Hong J, Shen WB, Hang TJ, Zhang C. The performance of thiol-terminated PEG-paclitaxel-conjugated gold nanoparticles. Biomaterials. 2013;34:10217–10227. doi: 10.1016/j.biomaterials.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 16.Wang C, Bao X, Ding X, Dang Y, Abbad S, Wang Y, Li M, Su Y, Wang W, Zhou J. A multifunctional self-dissociative polyethyleneimine derivative coating polymer for enhancing the gene transfection efficiency of DNA/polyethyleneimine polyplexes in vitro and in vivo. Polymer Chem. 2015;6:780–796. doi: 10.1039/C4PY01135J. [DOI] [Google Scholar]
- 17.Wang JK, Zhou YY, Guo SJ, Wang YY, Nie CJ, Wang HL, Wang JL, Zhao Y, Li XY, Chen XJ. Cetuximab conjugated and doxorubicin loaded silica nanoparticles for tumor-targeting and tumor microenvironment responsive binary drug delivery of liver cancer therapy. Materials Science and Engineering: C. 2017;76:944–950. doi: 10.1016/j.msec.2017.03.131. [DOI] [PubMed] [Google Scholar]
- 18.Antonietti M, Basten R, Lohmann S. Polymerization in microemulsions-a new approach to ultrafine, highly functionalized polymer dispersions. Macromol Chem Phys. 1995;196:441–466. doi: 10.1002/macp.1995.021960201. [DOI] [Google Scholar]
- 19.Zarur AJ, Ying JY. Reverse microemulsion synthesis of nanostructured complex oxides for catalytic combustion. Nature. 2000;403:65–67. doi: 10.1038/47450. [DOI] [PubMed] [Google Scholar]
- 20.Wang C, Chen S, Yu Q, Hu F, Yuan H. Taking advantage of the disadvantage: Employing the high aqueous instability of amorphous calcium carbonate to realize burst drug release within cancer cells. J Material Chem B. 2017;5:2068–2073. doi: 10.1039/C6TB02826H. [DOI] [PubMed] [Google Scholar]