

Abstract
P-glycoprotein is an efflux transporter located in the blood–brain barrier. (R)-[11C]Verapamil is widely used as a PET tracer to investigate its function in patients with epilepsy, Alzheimer’s disease, and other neurodegenerative diseases. Currently it is not possible to use this successful tracer in clinics without a cyclotron, because of the short half-life of carbon-11. We developed two new fluorine-18 labeled (R)-verapamil analogs, with the benefit of a longer half-life. The synthesis of (R)-N-[18F]fluoroethylverapamil ([18F]1) and (R)-O-[18F]fluoroethylnorverapamil ([18F]2) has been described. [18F]1 was obtained in reaction of (R)-norverapamil with the volatile [18F]fluoroethyltriflate acquired from bromoethyltosylate and a silver trilate column with a radiochemical yield of 2.7% ± 1.2%. [18F]2 was radiolabeled by direct fluorination of precursor 13 and required final Boc-deprotection with TFA resulting in a radiochemical yield of 17.2% ± 9.9%. Both tracers, [18F]1 and [18F]2, were administered to Wistar rats, and blood plasma and brain samples were analyzed for metabolic stability. Using [18F]1 and [18F]2, PET scans were performed in Wistar rats at baseline and after blocking with tariquidar, showing a 3.6- and 2.4-fold increase in brain uptake in the blocked rats, respectively. In addition, for both [18F]1 and [18F]2, PET scans in Mdr1a/b(−/−), Bcrp1(−/−), and WT mice were acquired, in which [18F]2 showed a more specific brain uptake in Mdr1a/b(−/−) mice and no increased signal in Bcrp1(−/−) mice. [18F]2 was selected as the best performing tracer and should be evaluated further in clinical studies.
Keywords: P-gp, BCRP, positron emission tomography, blood−brain barrier, verapamil, 18F, ABC transporters
Introduction
The blood–brain barrier (BBB) is a diffusion barrier between the central nervous system and the circulation that protects the brain from entrance of neurotoxic substances. Endothelial cells with tight junctions prevent paracellular passage. Transcellular passage is possible by selective transport of, for example, amino acids, vitamins, and sugars via their specific transporters.1 ATP-binding cassette (ABC) transporters constitute an important transporter family at the BBB with the most investigated ones being P-glycoprotein (P-gp/ABCB1) and breast cancer resistance protein (BCRP/ABCG2). Both are ATP dependent efflux transporters, mediating the transport of structurally diverse compounds from brain to blood, and share some substrates. Research supports that P-gp function is diminished in Alzheimer’s disease, causing accumulation of β-amyloid, in the brain.2 On the other hand, several studies have shown increased P-gp function in epilepsy patients, causing drug resistance.3 A number of studies have used positron emission tomography (PET) to investigate the function of P-gp with radiolabeled substrates. The most widely used PET tracer for P-gp is [11C]verapamil, originally a calcium channel blocker, but also a substrate of P-gp.4 In early studies, it was used as a racemic mixture, but later it was shown that the (R)-enantiomer had better in vivo stability than the (S)-isomer and the use of a single isomer is essential for quantification of P-gp function.5 In a clinical PET study using (R)-[11C]verapamil, (mild) AD patients showed 50% reduced P-gp function in brain regions related to AD compared with healthy, age-matched controls.6
Although (R)-[11C]verapamil has shown its usefulness in clinical research, it has several drawbacks. Metabolite studies have shown that N-dealkylation of the parent compound results in the radiolabeled metabolite [11C]D617, which turned out to be a P-gp substrate as well. Metabolite formation could disturb the PET image when the radiolabeled metabolites interact with P-gp in a different manner than the parent compound and makes quantification challenging. To circumvent this problem, [11C]D617 itself was investigated as a P-gp tracer.7In vivo studies, however, showed lower affinity of [11C]D617 for P-gp than (R)-[11C]verapamil, and therefore it was not a satisfactory substitute. The aspect of (R)-[11C]verapamil the we want to tackle is the use of the isotope carbon-11, with the short half-life of 20 min. Although, the rapidly decaying isotope gives the opportunity for multiple PET scans in 1 day with the same object, it is only possible to perform studies with the PET tracer at a facility in close proximity to a cyclotron. A fluorine-18 labeled PET tracer would give the opportunity to investigate P-gp in almost all facilities in possession of a PET scanner. Besides transport, a longer half-life gives the opportunity for longer scan time and multiple patient batches out of one tracer production. The energy of the positron emission of fluorine-18 is lower compared with carbon-11 and results in a lower range and better resolution of the PET image.
Recently, some new fluorine-18 labeled P-gp PET tracers have been developed. [18F]Gefitinib, originally an epidermal growth factor receptor inhibitor and a substrate for P-gp and BCRP,8 was used in a study to examine drug–drug interactions at the BBB. However, again this is not a specific P-gp tracer, since it is also transported by BCRP.9 In another comprehensive study of Sander et al., fluorine-18 labeled tracers were used in a new approach to metabolically activate a dual P-gp/BCRP substrate. Although the proof-of-concept was successful, slow rates of enzymatic conversion toward the prodrug tracer did not give the anticipated results.10 Another study proposed three new fluorine-18 labeled P-gp tracers. Only one of the three showed P-gp substrate functionality in vivo, although all tracers were evaluated as substrates in vitro.11,12
While these studies are all attempts to label a substrate of P-gp, to image the expression levels of P-gp in relevant brain regions, an inhibitor of binding ligand is needed. Two fluorine-18 labeled P-gp tracers were based on analogs of the P-gp inhibitors tariquidar and elacridar. 1-[18F]Fluoroelacridar showed excessive defluorination in vivo and was not further developed.13 [18F]Fluoroethylelacridar and [18F]fluoroethyltariquidar showed P-gp substrate behavior in vivo, with a small increase in brain uptake for Mdr1a/b(−/−) mice but much higher uptake for Mdr1a/b(−/−)Bcrp1(−/−) mice, indicating that they were substrates for both P-gp and BCRP.14 Therefore, the tracers could not be used for the intended purpose of quantification of P-gp function.
To date, no fluorine-18 labeled P-gp tracer is available for clinical use. Since (R)-[11C]verapamil has shown its value in multiple clinical studies, the purpose of the present study was to investigate two novel fluorine-18 analogs of verapamil, (R)-N-[18F]fluoroethylverapamil ([18F]1) and (R)-O-[18F]fluoroethylnorverapamil ([18F]2), as tracers of P-gp function.
Results and Discussion
Chemistry
The aim of this study was to develop a new fluorine-18 labeled P-gp substrate; therefore two verapamil analogs containing a fluoroethyl group were investigated. The two different positions of the fluoroethyl group were chosen in order to investigate the effect on pharmacokinetics and metabolism. For [18F]1, the fluoroethyl group was placed on the original position of the [11C]methyl group on the amine. To make [18F]2 less prone to metabolism, one of the methoxy groups was replaced with the fluoroethyl group, keeping in mind that the average bond enthalpy between a C–O bond is higher and therefore stronger than a C–N bond.15 The metabolic pathway of [11C]verapamil has been studied thoroughly16 and its main metabolite is norverapamil, with the [11C]methyl group cleaved off resulting in polar radioactive metabolites (Scheme 1). In addition, the formed metabolite [11C]D617 of [11C]verapamil, was proven to be a substrate of P-gp,7 and it is assumed that the 18F-analog of D617 would also act as a substrate of P-gp. The formation of radioactive metabolites could provide a background signal (of unknown amount) to the PET images and, therefore, make quantitative interpretation of PET data difficult if not impossible. To circumvent these issues, the [18F]fluoroethyl group was placed on the phenol that is not part of D617, at the same time removing the methyl group on the amine to avoid the first metabolic step toward norverapamil, since this is a substrate of P-gp as well.17
Scheme 1. Metabolic Pathway of [11C]Verapamil as Adapted from Luurtsema et al.18.

The two proposed P-gp PET tracers with likely positions for metabolic cleavage are depicted at the bottom of the schematic.
It is known that the (R)-enantiomer of verapamil is less prone to metabolism, has lower affinity for the calcium channel, and shows better pharmacokinetics than the (S)-enantiomer.5,19 Therefore, the (R)-enantiomer of the precursor and reference compound of [18F]2 were synthesized. To avoid the use of expensive starting materials or chiral HPLC purification, the method of Gilmore et al. was used20 (Schemes 2 and 3). Using this method, the (R,S/R,R)-diastereomer intermediate 10 was synthesized, which enables stereochemical purification by flash column chromatography in batches up to multiple grams. After purification of the (R,R)-intermediate, acidic hydrolysis, and periodate cleavage of the resulting diol, gave (R)-aldehyde 11. Elongation of aldehyde 11 was executed through a Wittig reaction followed by acidic hydrolysis to (R)-aldehyde 12. The second difficulty occurred during the reductive amination between obtained (R)-aldehyde 12 and amine 7. The secondary amine was formed, but could not be purified. The reaction did not proceed efficiently due to formation of a byproduct by intermolecular substition, with tosylate acting as leaving group to form a dimer, as identified by MS. To prevent this undesirable side reaction, the Boc protection was carried out immediately after workup of the reductive amination, without reducing the volume of solvent. With a yield of 44% over the reductive amination and Boc protection, this is a viable method to synthesize (R)-precursor 13. The reference compound 2 was synthesized from aldehyde 12 and amine 8 containing the fluoroethoxy group.
Scheme 2.

Reagents and conditions: (a) Boc2O, Et3N, MeOH, r.t., 2 h; (b) synthesis of 5, ethylene di(p-toluenesulfonate), K2CO3, KI, DMF, rt, 3 h; synthesis of 6, 1-bromo-2-fluoroethane, K2CO3, KI, DMF, 75 °C, 18 h; (c) TFA, DCM, rt, 1 h.
Scheme 3.

Reagents and conditions: (a) NaH, DMF, 65 °C, 3 h, (R)-2,2-dimethyl-1,3-dioxolan-4-ylmethyl 4-methylbenzenesulfonate, 65 °C, 18 h, flash column chromatography; (b) AcOH, H2O, rt, 18 h; (c) NaIO4, NaHCO3, CH2Cl2, rt, 4 h; (d) MeOCH=PPh3, THF, −80 °C to rt, 4 h; subsequently, p-TSA, i-PrOH, H2O, 80 °C, 3 h; (e) synthesis of 2, 8, NaBH(OAc)3, Na2SO4, MeOH, rt, 18 h; synthesis of 10, (i) 7, NaBH(OAc)3, Na2SO4, DCE, rt, 18 h; (ii) Boc2O, Et3N, EtOAc, rt, 1.5 h.
The precursor of [18F]1, (R)-norverapamil (17), was kindly donated by Abbott Laboratories, and therefore the synthesis of the reference compound of [18F]1 was straightforward. (R)-Norverapamil (17) was alkylated with 1,2-bromofluoroethane to obtain reference compound 1.
Radiochemistry
Although its precursor was readily available, the radiochemical synthesis of [18F]1 was more challenging. Radiolabeling of (R)-norverapamil to obtain [18F]1 was first pursued by fluorinating bromoethyltosylate to give bromo[18F]fluoroethane ([18F]15), which was distilled into a second vial containing a solution with 3 mg of (R)-norverapamil (17) and 1.6 mg of K2CO3 that was heated to 120 °C in MeCN. However, for the second step, only 0.5% conversion was observed on HPLC. Therefore, a silver triflate oven was introduced to conduct volatile bromo[18F]fluoroethane conversion into [18F]fluoroethyltriflate ([18F]16), substituting the bromine for a stronger leaving group, and this intermediate was bubbled through the precursor solution (Scheme 4). This resulted in higher conversion, and when the solution was also stirred to improve solubility of K2CO3, the final reaction conditions were achieved, resulting in an overall yield of 2.4% ± 1.4%, a specific activity of 143 ± 88 GBq/μmol, and a radiochemical purity of >99%. The yield of [18F]1 was still lower than expected, which is mainly caused by poor trapping of activity in the second reaction vial after distillation. Nevertheless, sufficient activity was obtained for preclinical studies. For [18F]2, the precursor synthesis was designed to prevent this problem by addition of a tosyl group for direct fluorination, followed by Boc-deprotection (Scheme 5). No optimization was required for animal experimentation, and the overall yield was 14.3% ± 6.8%, with a specific activity of 151 ± 74 GBq/μmol and a radiochemical purity of >99%.
Scheme 4.

Reagents and conditions: (a) 18F/K2.2.2/K+, DMF, 90 °C, 15 min; (b) AgOTf, 200 °C, 15 min; (c) [18F]fluoroethyltriflate, K2CO3, MeCN, 120 °C, 15 min.
Scheme 5.

Reagents and conditions: (a) 18F/K2.2.2/K+, MeCN, 90 °C, 5 min; (b) TFA, 20 °C, 10 min.
Biodistribution
The biodistribution of [18F]1 in Wistar rats shows a relatively slow washout from the blood pool and other related organs and low brain uptake, presumably caused by the efflux transport of P-gp (Figure 1). Since bone uptake was low and did not increase over time, defluorination of the tracer appears to be absent. Different behavior in rats was observed for [18F]2, with lower brain uptake (compared with [18F]1) and high initial uptake in kidney and lung, which decreases over time. This is similar to the biodistribution of [11C]verapamil.21 In some cases, high lung uptake can be explained by perfusion, which is related to the activity in plasma. However, the blood values for [18F]2 are even lower than [18F]1; therefore this could not be concluded. [18F]2 is not prone to defluorination based on its low bone uptake.
Figure 1.

Biodistribution of [18F]1 and [18F]2 in selected organs. Data are expressed as percentage injected dose per gram tissue (%ID/g).
Metabolite Analysis
Metabolite analyses of both tracers were performed in healthy Wistar rats, 5, 15, and 60 min after tracer injection, and the results are shown in Table 1. For both tracers, the parent tracer was found in the nonpolar fraction and was identified by HPLC. The metabolite analysis showed a rapid rate of metabolism in rats for both PET tracers. The main metabolite fraction was the polar metabolites. Since bone uptake is not observed in the PET scans (vide infra) and biodistributions, the polar metabolites are not products of defluorination. Therefore, the high polar fraction consists of metabolites most likely formed via cleavage of the fluoroethyl group.
Table 1. [18F]1 and [18F]2 and Their Radiolabeled Metabolites in Plasma and Brain Tissuea in Wistar rats.
| [18F]1 |
[18F]2 |
||||
|---|---|---|---|---|---|
| min | plasma | brain | plasma | brain | |
| parent tracer | 5 | 46 ± 14 | 41 ± 10 | 20 ± 3 | 26 ± 6 |
| 15 | 19 ± 2 | 14 ± 2 | 8 ± 3 | 17 ± 7 | |
| 60 | 3 ± 1 | 2 ± 0.3 | 4 ± 1 | 6 ± 1 | |
| nonpolar metabolites | 5 | 5 ± 2 | 5 ± 3 | ||
| 15 | 9 ± 3 | 5 ± 1 | |||
| 60 | 5 ± 1 | 3 ± 1 | |||
| polar metabolites | 5 | 49 ± 11 | 75 ± 3 | ||
| 15 | 71 ± 2 | 87 ± 1 | |||
| 60 | 92 ± 1 | 93 ± 2 | |||
| Brain metabolites | 5 | 59 ± 10 | 74 ± 6 | ||
| 15 | 86 ± 2 | 83 ± 7 | |||
| 60 | 98 ± 0.3 | 94 ± 1 | |||
Percentage of total radioactivity, mean ± SD.
For [18F]1, the metabolic pathway is expected to be similar to [11C]verapamil, which gives norverapamil as the main metabolite, next to polar monocarbon labeled ones like [11C]formaldehyde, [11C]formic acid, and [11C]carbon dioxide (Scheme 1). The high polar fraction observed in the metabolite analysis of [18F]1 indicates that the [18F]fluoroethyl group is even more prone to metabolism than the [11C]methyl group, resulting in [18F]fluoroacetaldehyde, and other small oxidized fractions. Furthermore, a peak likely corresponding to [18F]fluoroethyl labeled D617 was observed in the nonpolar fraction on HPLC. This presumably can act as a substrate of P-gp, similar to [11C]D617, which could interfere with the PET signal when it interacts with P-gp in a different matter than the parent tracer.
With this in mind, it was expected that [18F]2 would show a lower rate of metabolism, since this first metabolic step is avoided by eliminating the amine bound methyl group in the designed structure. However, this effect was not observed, and high polar fractions were measured. Possible enzyme cleavage sites are the [18F]fluoroethyl group and the amine bound alkyl groups, with the latter being less sterically hindered in the secondary amine compared with a tertiary amine (Scheme 1). Labeled metabolites will not be analogs of D617, since that part of the molecule was not labeled with fluorine-18. Therefore, it is unlikely that labeled metabolites are substrates of P-gp.
It was shown that metabolism of [11C]verapamil was less rapid in humans compared with rats. While only 28.1% ± 2.7% of the total activity in the rat plasma was parent tracer at 60 min,18 in humans it was 45% ± 9%.22 Therefore, we anticipate that while tracers [18F]1 and [18F]2 are metabolized rapidly in rats, in humans this will likely be slower but must be monitored during the translation of these tracers to humans.
P-gp Blocking Study with Tariquidar
Rats were treated with P-gp inhibitor tariquidar 30 min before injection of [18F]1 or [18F]2, a method in line with previous reports studying P-gp in rats.23 Brain uptake of [18F]1 was 3.6-fold higher than in the baseline scans (Figure 2a). The highest brain uptake was at 5 min. For [18F]2, a 2.4-fold higher brain uptake was observed in tariquidar treated animals compared with baseline brain uptake (Figure 2b). However, a difference was observed in pharmacokinetics between the two tracers. While behavior of [18F]1 was similar to that of verapamil, with an injection peak followed by washout over time, [18F]2 showed no injection peak, while both tracers were injected via a comparable bolus injection. Instead, a more steady time activity curve of [18F]2 resulted in higher activity levels at the end of the scan. It is likely that this difference is caused by the higher polarity of [18F]2, due to the secondary amine, as compared with the tertiary amine of [18F]1. This could lead to more difficult and thus slower passive diffusion of [18F]2 through the BBB and therefore delayed interaction with P-gp. This is supported by the measured log D values of 2.08 and 1.61 for [18F]1 and [18F]2, respectively.
Figure 2.

Whole brain time–activity curves of [18F]1 and [18F]2 in Wistar rats at (●) baseline and (■) after treatment with tariquidar (15 mg/kg).
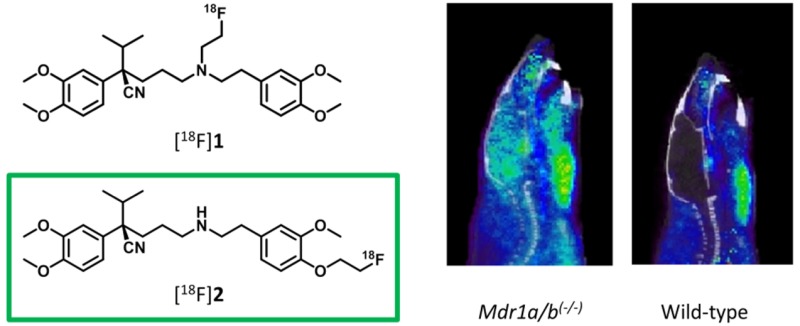
PET Imaging in Mdr1a/b(−/−) and Bcrp1(−/−) Mice
Given that, at a dose of 15 mg/kg, tariquidar is known to be an inhibitor of both P-gp and BCRP,24,25 another PET study using knockout animals was performed to assess the specificity of the two tracers (Figure 3). To enable a direct comparison, a similar study was performed using (R)-[11C]verapamil (Figure 4). Unfortunately, as the HRRT scanner had just been decommissioned, the [18F]2 study in Mdr1a/b(−/−) mice vs wild-type (WT) mice was performed using new PET/CT and PET/MR small animal scanners. This led to slightly different results for the wild-type animals, compared with the Bcrp1(−/−) vs wild-type mice study, probably due to differences in spatial resolution. Therefore, TACs are presented separately in Figure 3a–d. Figure 3e,f represents the ratio of brain uptake in Mdr1a/b(−/−)/WT and Bcrp1(−/−)/WT for both [18F]1 and [18F]2, respectively, to normalize for the scanner differences.
Figure 3.

Time–activity curves of whole brain uptake of [18F]1 and [18F]2 in (●) wild-type (WT) mice, (■) Bcrp1(−/−) mice, or (▲) Mdr1a/b(−/−) mice. [18F]1 in (a) WT vs Mdr1a/b(−/−), (c) WT vs Bcrp1(−/−), (e) ratio of both Mdr1a/b(−/−) and Bcrp1(−/−) over WT; [18F]2 in (b) WT vs Mdr1a/b(−/−), (d) WT vs Bcrp1(−/−), (f) ratio of both Mdr1a/b(−/−) and Bcrp1(−/−) over WT.
Figure 4.

(a) Time–activity curve of whole brain uptake of [11C]verapamil in (●) wild-type (WT) mice (n = 2), (■) Bcrp1(−/−) mice (n = 3), or (▲) Mdr1a/b(−/−) mice (n = 2). (b) Ratio of both Mdr1a/b(−/−) and Bcrp1(−/−) over WT.
For both tracers, no significant differences in brain uptake were observed between wild-type and Bcrp1(−/−) mice, indicating that neither are BCRP substrates (Figures 3c,d). Both tracers showed increased brain uptake in Mdr1a/b(−/−) mice (Figures 3a,b). A smaller difference in brain uptake was noticed for [18F]1, with only a 1.4-fold higher brain uptake during the first 10 min compared with wild-type mice. This is mainly due to high uptake in the wild-type animals, which suggests that [18F]1 is a poorer tracer of P-gp (Figure 3e) than [18F]2. For [18F]2, uptake in wild-type mice was similar to that of [11C]verapamil. Although metabolism of [18F]2 was even more rapid than that of [18F]1, [18F]2 in Mdr1a/b(−/−) mice showed steady brain uptake up to a 6.4-fold higher level than that in wild-type mice, reaching a SUV plateau of about 0.9 at the end of the scan. The slower increase in brain uptake could be due to the lower log D value and therefore difficulty in crossing the BBB. The irreversible kinetics of [18F]2 are in general not ideal for PET analysis. In this study, no blood samples were collected from mice; therefore kinetic modeling was not possible. To obtain more insight in the relationship between the activity concentration in the blood and total brain uptake, blood data was collected from the scans.
As shown in Table 2, both [18F]1 and [18F]2 showed a higher brain-to-blood AUC ratio in Mdr1a/b(−/−) mice compared with WT animals. However, [18F]1 also showed an increased brain-to-blood ratio in Bcrp1(−/−) mice. Since both compounds are close analogs of (R)-[11C]verapamil, which is a P-gp specific PET tracer, the dual P-gp/Bcrp PET tracer behavior of [18F]1 is an unexpected observation.
Table 2. Brain-to-Blood AUC Ratios.
| groups | brain-to-blood AUC ratios | |
|---|---|---|
| [18F]1 | WT | 0.52 ± 0.05 |
| Mdr1a/b(−/−) | 0.98 ± 0.07 | |
| Bcrp1(−/−) | 0.88 ± 0.06 | |
| [18F]2 | WT | 0.18 ± 0.04 |
| Mdr1a/b(−/−) | 0.75 ± 0.14 | |
| Bcrp1(−/−) | 0.20 ± 0.02 |
In the case of [18F]2, brain-to-blood AUC ratios were statistically significant (P < 0.05, Student’s t test for paired data) for Mdr1a/b(−/−) vs WT and Mdr1a/b(−/−) vs Bcrp(−/−) animals. This was not the case for [18F]1 where the difference between Mdr1a/b(−/−) vs WT mice and Bcrp(−/−) vs WT mice were significant but not that between Mdr1a/b(−/−) vs Bcrp(−/−) animals.
The high Mdr1a/b(−/−)/WT ratio of [18F]2 over 60 min (Figure 3f) shows a specific P-gp substrate at the same ratio of (R)-[11C]verapamil, which is confirmed by the significant differences of the brain-to-blood AUC ratios, despite the rapid rate of metabolism from the previous analysis (Table 1). Possible explanations are differences in expression of P-gp between species and the rate of metabolism in rats vs mice.18,26,27 Another explanation of the stable TAC pattern of [18F]2 is found in the similarity with [11C]dLop, another P-gp PET tracer, which was shown to be caused by acidic lysosomal trapping in the brain. This is possible for more basic radiotracers, like [18F]2 with the secondary amine, that have the ability to diffuse into lysosome and subsequently be protonated in the acid lysosomal interior.28 A third possibility is the effect of anesthesia on the different species and the different time frames (repeated anesthesia in rats vs one time anesthesia in mice), which was not examined. However, in a paper of Wanek et al., mice were tested with (R)-[11C]verapamil under long and short periods of time (160 and 5 min, respectively) under anesthesia, and no significant effect was observed.27 While this behavior is not fully explained for [18F]2, it does show specific brain uptake in Mdr1a/b(−/−) mice supporting its potential use as P-gp PET tracer.
Conclusion
Both PET tracers showed substrate behavior in tariquidar treated rats with different patterns over time. While [18F]1 showed higher initial uptake followed by faster washout, [18F]2 showed slower brain uptake. As tariquidar is an inhibitor of both BCRP and P-gp, the PET study in knockout mice showed that [18F]2 was more specific for P-gp, despite its fast rate of metabolism. As the rate of metabolism is species dependent, further studies in humans are needed to assess the potential of [18F]2 as a clinical PET tracer.
Methods
General
Chemicals and solvents were purchased from commercial sources, Sigma-Aldrich (Zwijndrecht, the Netherlands), Fluorochem (Hadfield Derbyshire, UK), ABCr GmbH (Karlsruhe, Germany), and Biosolve (Valkenswaard, the Netherlands), without further purification unless stated otherwise. (R)-Desmethyl-verapamil was kindly donated by Abbott Laboratories (IL, USA). Dichloromethane (DCM), dichloroethane (DCE), methanol (MeOH), and dimethylformamide (DMF) were dried over 3 Å molecular sieves for at least 24 h prior to use. Tetrahydrofuran (THF) was first distilled from LiAlH4 and then dried over 3 Å molecular sieves. Thin layer chromatography (TLC) was performed on Merck (Darmstadt, Germany) precoated silica gel 60 F254 plates. Spots were visualized by UV quenching or ninhydrin. Column chromatography was carried out either manually by using silica gel 60 Å (Sigma-Aldrich) or on a Buchi (Flawil, Switzerland) sepacore system (comprising a C-620 control unit, a C-660 fraction collector, 2 C601 gradient pumps, and a C640 UV detector) equipped with Buchi sepacore prepacked flash columns. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on a Bruker (Billerica, USA) Avance 500 (500.23 and 125.78 MHz, respectively) with chemical shifts (δ) reported in ppm relative to the solvent. Electrospray ionization mass spectrometry (ESI-MS) was carried out using a Bruker microTOF-Q instrument in positive ion mode (capillary potential of 4500 V). QC analysis was performed using an HPLC system of Jasco (Easton, MD, USA) containing a PU-2089 pump station equipped with a Varian Kromasil C18 column (10 μm, 250 mm × 4.6 mm, EKA Chemicals AB or AkzoNobel, Sweden) using H2O/MeCN/diisopropanolamine (DIPA) (40:60:0.2, v/v/v, method A) as eluent or Grace Alltima C18 column (5 μm, 250 mm × 4.6 mm, Grace, Columbia, USA) using H2O/MeCN/DIPA (40:60:0.2, v/v/v, method B) or H2O/MeCN/TFA (60:40:0.2, v/v/v, method C) or H2O/MeCN/TFA (30:70:0.2, v/v/v, method D (intermediate)) as eluent at a flow rate of 1 mL·min–1, with a Jasco UV-2075 UV detector (λ = 232 nm) and a NaI radioactivity detector (Raytest, Straubenhardt, Germany). Chromatograms were acquired using Raytest GINA Star software (version 5.01). Semipreparative HPLC was performed on a Jasco PU-2089 pump station equipped with Luna C18(2) column (10 μm, 250 mm × 10 mm, Phenomenex, California, USA) using 5 mM K3PO4/MeCN (28:72, v/v, pH = 10.0 method E) or H2O/MeCN/TFA (60:40:0.2, v/v/v, method F) as eluent at a flow rate of 4 mL·min–1, a Jasco UV-1575 Plus UV detector (λ = 254 nm), a custom-made radioactivity detector, and Jasco ChromNAV CFR software (version 1.14.01) for data acquisition. Metabolite analysis was performed on Dionex (Sunnyvale, CA, USA) UltiMate 3000 HPLC equipment with Chromeleon software (version 6.8). A LUNA C8 (5 μm, 250 mm × 10 mm, Phenomenex) column was used (method F) using 5 mM NH4OAc/MeCN (1:1, v/v, pH = 4.2) as eluent at a flow rate of 3.5 mL/min.
Chemistry
(R)-2-(3,4-Dimethoxyphenyl)-2-(((R)-2,2-dimethyl-1,3-dioxolan-4-yl)methyl)-3-methylbutanenitrile (10)
To a stirred solution of 2-(3,4-dimethoxyphenyl)-3-methylbutanenitrile (9, 3.83 g, 17.5 mmol) in 60 mL of dry DCM was added sodium hydride (60% dispersion in mineral oil; 1.40 g, 34.9 mmol), and the mixture was heated to 65 °C. After 3 h, the reaction mixture was cooled to room temperature, (R)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl 4-methylbenzenesulfonate (5.00 g, 17.5 mmol) dissolved in 5 mL of dry THF was added, and the reaction mixture was stirred at 65 °C overnight. The dark brown mixture was quenched with water and extracted into diethyl ether, organic layers were washed with water and brine and dried over Na2SO4, and the solvent was evaporated in vacuo. The crude product was purified by flash column chromatography (10–30% EtOAc/hexane) to yield the R,R-diastereomer 10 (3.96 g, 11.9 mmol, 68.1% yield) as a light brown oil. 1H NMR (CDCl3) δ 6.95–6.83 [3H, m, CHAR], 4.03–3.92 [1H, m, CHO], 3.89 [3H, s, CH3O], 3.90 [3H, s, CH3O], 3.19 [1H, dd, J = 5.6 and 8.3 Hz, CH2O], 2.98 [1H, t, J = 7.9 Hz, CH2O], 2.65 [1H, dd, J = 4.6 and 13.8 Hz, CH2CHO], 2.15 [1H, sept, J = 6.7 Hz, CH(CH3)2], 1.90 [1H, dd, J = 7.7 and 13.6 Hz, CH2CHO], 1.36 and 1.26 [3H each, s, C(CH3)2], 1.23 and 0.79 [3H each, d, J = 6.7 Hz, CH(CH3)2]; 13C NMR (CDCl3) δ 149.12, 148.71, 129.9, 120.47, 118.6, 111.19, 109.53, 108.14, 73.7, 69.34, 56.06, 55.92, 51.1, 42.38, 37.92, 26.78, 25.72, 18.6, 18.52; ESI-HRMS calculated for C19H27NO4 333.1940, found 334.2058 [M + H]+, 356.1898 [M + Na]+.
(R)-2-(3,4-Dimethoxyphenyl)-2-isopropyl-4-oxobutanenitrile (11)
(R)-2-(3,4-Dimethoxyphenyl)-2-(((R)-2,2-dimethyl-1,3-dioxolan-4-yl)methyl)-3-methylbutanenitrile (10, 847 mg, 2.54 mmol) was dissolved in water (4 mL) and acetic acid (12 mL) and stirred at room temperature overnight. The volume was reduced by evaporation, giving the crude diol as a colorless oil. The crude reaction mixture was dissolved in DCM (35 mL), and 1 M NaHCO3 (16.9 mL, 16.9 mmol) was added. To the stirred biphasic system was added a solution of sodium periodate (2.20 g, 10.3 mmol) in water (11 mL) dropwise over 2 h. The white suspension was stirred for another 2 h. The suspension was diluted with DCM, and the organic phase was separated, washed with water, and dried over Na2SO4, and the solvent was evaporated in vacuo. The resulting brown oil was purified by flash column chromatography (25% EtOAc/Hex) to obtain (R)-2-(3,4-dimethoxyphenyl)-2-isopropyl-4-oxobutanenitrile, 11 (434 mg, 1.66 mmol, 65.4% yield), as a colorless oil. 1H NMR (CDCl3) δ 6.93–6.84 [3H, m, CHAR], 3.90 [3H, s, OCH3], 3.88 [3H, s, OCH3], 3.14–2.87 [2H, m, CH2CHO], 2.14 [1H, sept, J = 6.7 Hz, CH(CH3)2], 1.18 and 0.89 [3H each, d, J = 6.7 Hz, CH(CH3)2]; 13C NMR (CDCl3) δ 149.35, 148.93, 128.75, 120.45, 118.81, 111.35, 109.70, 56.09, 55.95, 49.80, 48.66, 38.19, 18.43, 18.22; ESI-HRMS calculated for C15H19NO3 261.1365, found 262.1458 [M + H]+ and 284.1285 [M + Na]+.
(R)-2-(3,4-Dimethoxyphenyl)-2-isopropyl-5-oxopentanenitrile (12)
(Methoxymethyl)triphenylphosphonium chloride (3.33 g, 9.71 mmol) was suspended in THF (50 mL) and brought to −50 °C, and n-butyllithium (1.6 M in hexanes; 12.1 mL, 19.4 mmol) was added dropwise via a dropping funnel over 30 min, stirring for 1 h at −80 °C. To the resulting dark red solution, (R)-2-(3,4-dimethoxyphenyl)-2-isopropyl-4-oxobutanenitrile (11, 705 mg, 2.70 mmol) dissolved in 4 mL of THF was added dropwise over 15 min, and the mixture was stirred for 2 h at −80 °C and slowly brought to rt. The reaction was quenched with water and extracted with Et2O. The organic layers were washed with water and brine and dried over Na2SO4, and the solvent was evaporated in vacuo. The crude oil was purified by flash column chromatography (5–20% EtOAc/Hex) to obtain a mixture of the E/Z-isomers of the desired intermediate (356 mg, 1.23 mmol, 45.6% yield). To a solution of (R)-2-(3,4-dimethoxyphenyl)-2-isopropyl-5-methoxypent-4-enenitrile (356 mg, 1.23 mmol) in 2-propanol (3 mL) and water (3 mL), 4-methylbenzenesulfonic acid hydrate (14.0 mg, 0.074 mmol) was added, and the mixture was stirred to reflux for 3 h. The reaction was quenched with water and extracted with Et2O. The organic layers were washed with 1 M NaHCO3 and brine and dried over Na2SO4, and the solvent was evaporated in vacuo. The crude oil was purified with flash column chromatography (10% EA in hexane) obtaining the preferred product 12 (95 mg, 0.35 mmol, 28% yield). 1H NMR (CDCl3) δ 6.94–6.82 [3H, m, CHAR], 3.89 [6H, m, (OCH3)2], 2.70–2.40 [2H, m, CH2CH2CHO], 2.24–2.06 [3H, m, CH(CH3)2 and CH2CHO], 1.22 and 0.81 [3H each, d, J = 6.7 Hz, CH(CH3)2]; 13C NMR (CDCl3) δ 200.44, 149.22, 148.58, 129.58, 120.80, 118.69, 111.19, 109.21, 56.03, 55.93, 52.67, 40.60, 38.00, 29.92, 19.06, 18.60; ESI-HRMS calculated for C16H21NO3 275.1521, impossible to ionize.
tert-Butyl 4-Hydroxy-3-methoxyphenethylcarbamate (4)
Triethylamine (0.42 mL, 3.0 mmol) and di-tert-butyl dicarbonate (0.69 mL, 3.0 mmol) dissolved in 3 mL of methanol were added to a solution of 4-(2-aminoethyl)-2-methoxyphenol (3) (500 mg, 2.99 mmol) in 10 mL of methanol and stirred for 2 h at 50 °C. The solvent was evaporated in vacuo, and the crude product was extracted with EtOAc, washed with water and brine, and dried over Na2SO4, and the solvent was evaporated in vacuo. The white crystallized solid obtained was purified using flash column chromatography (1–5% MeOH/DCM) resulting in the desired product as a clear oil (460 mg, 1.72 mmol, 57.5% yield). 1H NMR (CDCl3) δ 6.87–6.66 [3H, m, CHAR], 4.54 [1H, bs, OH], 3.88 [3H, s, OCH3], 3.34 [2H, q, J = 6.4 Hz, NHCH2], 2.72 [2H, t, J = 7.1 Hz, NHCH2CH2], 1.44 [9H, s, C(CH3)3]; 13C NMR (CDCl3) δ (ppm) 158.49, 148.9, 145.96, 132.15, 122.32, 116.21, 113.47, 80.02, 56.39, 43.4, 36.89, 28.92; ESI-HRMS calculated for C14H21NO4 267.1471, found 290.1328 [M + Na]+.
tert-Butyl 4-(2-Fluoroethoxy)-3-methoxyphenethylcarbamate (6)
tert-Butyl 4-hydroxy-3-methoxyphenethylcarbamate (4, 309 mg, 1.16 mmol), 1-bromo-2-fluoroethane (0.345 mL, 4.62 mmol), K2CO3 (639 mg, 4.62 mmol), and potassium iodide (768 mg, 4.62 mmol) were stirred overnight at 75 °C in 8 mL of dry DMF. The reaction was quenched with water, extracted twice with ethyl acetate, washed with brine, and dried over Na2SO4, and the solvent was evaporated in vacuo. The crude yellow oil was purified using flash column chromatography (15–25% EtOAc/hex), and the desired product was obtained (170 mg, 0.542 mmol, 46.9% yield) as a yellow oil. 1H NMR (CDCl3) δ 6.87–6.69 [3H, m, CHAR], 4.76 [2H, dt, J = 47 and 4.1 Hz, (OCH2CH2F)], 4.20 [2H, dt, J = 27 and 4.3 Hz, (OCH2CH2F)], 3.86 [3H, s, OCH3], 3.35 [2H, q, J = 6.4 Hz, NHCH2], 2.74 [2H, t, J = 7.1 Hz, NHCH2CH2], 1.43 [9H, s, C(CH3)3]; 13C NMR (CDCl3) δ 155.84, 149.78, 146.4, 132.88, 120.71, 114.61, 112.65, 81.32, 68.7, 68.54, 55.91, 41.81, 35.77, 28.39; ESI-HRMS calculated for C16H24FNO4 313.1689, found 314.1678 [M + H]+, 336.1504 [M + Na]+.
2-(4-(2-Fluoroethoxy)-3-methoxyphenyl)ethanamine (8)
tert-Butyl 4-(2-fluoroethoxy)-3-methoxyphenethylcarbamate (6, 200 mg, 0.638 mmol) was dissolved in 4 mL of DCM and 4 mL of TFA and stirred at room temperature for 3 h. The solvent was evaporated in vacuo, and the crude product was purified using flash column chromatography (15–20% MeOH/DCM) to obtain the desired product (125 mg, 0.586 mmol, 92.0%) as white crystals. 1H NMR (DMSO-d6) δ 6.93–6.76 [3H, m, CHAR], 4.71 [2H, dt, J = 48 and 3.8 Hz, CH2F], 4.17 [2H, dt, J = 30 and 3.9 Hz, OCH2], 3.77 [3H, s, CH3], 3.03 [2H, t, J = 6.7 Hz, H2NCH2], 2.77 [2H, t, J = 6.7 Hz, H2NCH2CH2]; 13C NMR (CDCl3) δ 149.82, 146.84, 129.87, 120.63, 114.61, 112.47, 82.47, 81.11, 55.69, 40.62, 32.90; ESI-HRMS calculated for C11H16FNO2 213.1165, found 214.1238 [M + H]+.
2-(4-(2-((tert-Butoxycarbonyl)amino)ethyl)-2-methoxyphenoxy)ethyl 4-Methylbenzenesulfonate (5)
tert-Butyl 4-hydroxy-3-methoxyphenethylcarbamate (4, 613 mg, 2.29 mmol) was dissolved in 15 mL of DMF, and ethane-1,2-diyl bis(4-methylbenzenesulfonate) (849 mg, 2.29 mmol) and cesium carbonate (149 mg, 4.59 mmol) were added. The reaction mixture was stirred at room temperature for 3 h, quenched with water, extracted with EtOAc, washed with brine, and dried over Na2SO4. The solvent was removed in vacuo, and crude product was purified using flash column chromatography (15–35% EtOAc/hex) to obtain the desired product as a white solid (440 mg, 0.945 mmol, 41% yield). 1H NMR (CDCl3) δ 7.82 [2H, d, J = 8.2 Hz, OTs], 7.33 [2H, d, J = 8.2 Hz, OTs], 6.76–6.68 [3H, m, CHAR], 4.35 [2H, t, J = 5.0 Hz, (OCH2CH2OTs)], 4.19 [2H, t, J = 5.0 Hz, (OCH2CH2OTs)], 3.81 [3H, s, OCH3], 3.33 [2H, q, J = 6.6 Hz, NHCH2], 2.72 [2H, t, J = 6.8 Hz, NHCH2CH2], 2.44 [3H, s, SC6H4CH3], 1.43 [9H, s, C(CH3)3]; 13C NMR (CDCl3) δ 155.83, 149.82, 145.97, 144.85, 133.26, 132.78, 129.81, 128.01, 120.75, 115.21, 112.71, 68.1, 67.12, 60.39, 55.86, 41.81, 35.77, 28.39, 21.66; ESI-HRMS calculated for C23H31NO7S 465.1821, found 488.1719 [M + Na]+.
2-(4-(2-Aminoethyl)-2-methoxyphenoxy)ethyl 4-methylbenzenesulfonate (7)
2-(4-(2-((tert-Butoxycarbonyl)amino)ethyl)-2-methoxyphenoxy)ethyl 4-methylbenzenesulfonate (440 mg, 0.945 mmol) was dissolved in 5 mL of DCM and 5 mL of TFA and stirred for 30 min at room temperature. The solvent was evaporated, and the crude product was purified on flash column chromatography (5–10% MeOH/DCM) to obtain the purified product 2-(4-(2-aminoethyl)-2-methoxyphenoxy)ethyl 4-methylbenzenesulfonate (350 mg, 0.958 mmol, quantitative yield) as a white powder. 1H NMR (MeOD) δ 7.81 [2H, d, J = 8.4 Hz, OTs], 7.42 [2H, d, J = 8.4 Hz, OTs], 6.88–6.74 [3H, m, CHAR], 4.32 [2H, m, (OCH2CH2OTs)], 4.15 [2H, m, (OCH2CH2OTs)], 3.82 [3H, s, OCH3], 3.15 [2H, q, J = 6.6 Hz, NHCH2], 2.88 [2H, t, J = 7.1 Hz, NHCH2CH2], 2.45 [3H, s, SC6H4CH3]; 13C NMR (MeOD) δ 151.78, 148.41, 146.64, 134.46, 132.07, 131.19, 129.21, 122.25, 116.79, 114.32, 70.33, 68.66, 56.66, 42.12, 34.32, 21.74; ESI-HRMS calculated for C18H23NO5S 365.1297, found 366.1435 [M + H]+.
(R)-2-(4-(2-((tert-Butoxycarbonyl)(4-cyano-4-(3,4-dimethoxyphenyl)-5-methylhexyl)amino)ethyl)-2-methoxyphenoxy)ethyl 4-Methylbenzenesulfonate (13)
Na2SO4 and (R)-2-(3,4-dimethoxyphenyl)-2-isopropyl-5-oxopentanenitrile (12, 151 mg, 0.547 mmol) in 2.5 mL of DCE were added to a solution of 2-(4-(2-aminoethyl)-2-methoxyphenoxy)ethyl 4-methylbenzenesulfonate (7, 300 mg, 0.821 mmol) in 2.5 mL of DCE. The reaction mixture was stirred at room temperature overnight under argon. Sodium triacetoxyhydroborate (174 mg, 0.821 mmol) was added to the mixture, and the mixture was stirred for 1.5 h at room temperature. The reaction was quenched with 1 M NaHCO3, extracted with EtOAc (15 mL), washed with water (2×) and brine, and the organic layers were dried over Na2SO4 and used as such in the next step. Di-tert-butyl dicarbonate (239 mg, 1.095 mmol) and triethylamine (152 μL, 1.09 mmol) were added to the diluted reaction mixture, and the mixture was stirred at room temperature for 1.5 h. The reaction mixture was diluted with EtOAc, washed with water and brine, and dried over Na2SO4, and solvent was removed in vacuo. The crude mixture was purified by flash column chromatography (30–50% EtOAc/hexane) to obtain the purified product as colorless oil (174 mg, 0.240 mmol, 43.9% yield). 1H NMR (CDCl3) δ 7.82 (2H, d, J = 8.0 Hz, OTs), 7.33 (2H, d, J = 8.0 Hz, OTs), 6.89–6.61 (6H, m, CHAR), 4.34 [2H, t, J = 4.9 Hz, (OCH2CH2OTs)], 4.18 [2H, t, J = 5.0 Hz, (OCH2CH2OTs)], 3.88 (3H, s, OCH3), 3.86 (3H, s, OCH3), 3.79 (3H, s, OCH3), 3.25–2.99 (4H, m, CH2NCH2), 2.67 (2H, t, J = 6.5 Hz, NCH2CH2Ar), 2.44 (3H, s, TsCH3), 2.05 (2H, m, CH2CH2CH2), 2.04–1.54 (3H, m, CH(CH3)2 and CCH2), 1.42 (9H, m, Boc),, 1.17 and 0.78 (3H each, d, J = 6.7 Hz, CH(CH3)2); 13C NMR (CDCl3) δ (ppm) 149.69, 148.93, 148.2, 145.85, 145.78, 144.82, 133.42, 132.71, 130.34, 129.78, 127.95, 121.31, 120.73, 118.63, 115.18, 112.75, 110.95, 109.24, 79.35, 68.09, 67.07, 55.89, 55.81, 55.79, 49.06, 48.66, 47.27, 37.84, 35.13, 34.77, 28.34, 24.58, 21.61, 18.89, 18.50; ESI-HRMS calculated for C39H52N2O9S 724.3394, found 725.3477 [M + H]+ and 747.3318 [M + Na]+.
(R)-2-(3,4-Dimethoxyphenyl)-5-((4-(2-fluoroethoxy)-3-methoxyphenethyl)amino)-2-isopropylpentanenitrile (2)
To a solution of (R)-2-(3,4-dimethoxyphenyl)-2-isopropyl-5-oxopentanenitrile (12, 108 mg, 0.392 mmol) in 5 mL of dry MeOH were added Na2SO4 (167 mg, 1.18 mmol) and 2-(4-(2-fluoroethoxy)-3-methoxyphenyl)ethanamine (8, 125 mg, 0.588 mmol) under argon and stirred overnight at room temperature. Sodium triacetoxyhydroborate (125 mg, 0.588 mmol) was added, and the mixture was stirred for 2 h at room temperature under argon. The reaction was quenched with saturated NaHCO3 and diluted with ether, and the crude mixture was filtered. The filtrate was washed with brine and dried over Na2SO4 and the solvent was removed in vacuo. The crude product was purified by flash column chromatography (1–10% MeOH/DCM) to obtain the desired product as an oil (20 mg, 0.042 mmol, 10% yield). 1H NMR (CDCl3) δ 6.93–6.66 [6H, m, CHAR], 4.74 [2H, m, CH2CH2F], 4.22 [2H, m, CH2CH2F], 3.87, 3.85, and 3.82 [9H, 3xs, OCH3], 2.80 [4H, m, CH2NHCH2CH2], 2.67 [2H, m, NHCH2CH2], 2.06 [1H, m, CH(CH3)2], 2.16 and 1.88 [1H each, dt, J = 4.6 and 13 Hz, CCH2], 1.61 and 1.23 [1H each, m, CH2CH2CH2], 1.15 and 0.76 [3H each, d, J = 7 Hz, CH(CH3)2]; 13C NMR (CDCl3) δ (ppm) 149.74, 148.96, 148.23, 146.41, 132.52, 130.13, 121.2, 120.52, 118.59, 114.51, 112.53, 110.97, 109.31, 82.61, 81.25, 68.61, 68.44, 55.94, 55.88, 55.79, 49.90, 48.35, 37.85, 35.21, 34.42, 24.58, 18.86, 18.53; ESI-HRMS calculated for C27H37FN2O4 472.2737, found [M + H]+ 473.2745.
(R)-5-((3,4-Dimethoxyphenethyl)(2-fluoroethyl)amino)-2-(3,4-dimethoxyphenyl)-2-isopropylpentanenitrile (1)
To a solution of (R)-5-((3,4-dimethoxyphenethyl)amino)-2-(3,4-dimethoxyphenyl)-2-isopropylpentanenitrile (74 mg, 0.17 mmol) in 2 mL of DMF, 1-bromo-2-fluoroethane (500 μL, 6.72 mmol) and potassium carbonate (255 mg, 1.85 mmol) were added, and the mixture was stirred at 65 °C for 4 h. The reaction mixture was extracted with EtOAc, washed with water and brine, and dried over Na2SO4, and the solvent was removed in vacuo. The crude product was purified by flash column chromatography (0–1% MeOH/DCM) to obtain the desired product as an oil (40 mg, 0.082 mmol, 49% yield). 1H NMR (CDCl3) δ 6.91–6.66 [6H, m, CHAR], 4.46 [2H, m, CH2CH2F], 3.89, 3.88, 3.86, and 3.85 [12H, 4xs, OCH3], 2.81–2.62 [6H, m, CH2N(CH2)CH2CH2], 2.54 [2H, m, NCH2CH2], 2.05 [1H, m, CH(CH3)2], 2.12 and 1.85 [1H each, dt, J = 4.3 and 13 Hz, CCH2], 1.52 and 1.15 [1H each, m, CH2CH2CH2], 1.18 and 0.79 [3H each, d, J = 6.7 Hz, CH(CH3)2]; 13C NMR (CDCl3) δ (ppm) 148.96, 148.78, 148.23, 147.28, 132.8, 130.6, 121.47, 120.5, 118.64, 112.04, 111.17, 111.02, 109.57, 83.25, 81.92, 56.36, 55.95, 55.88, 55.84, 55.80, 53.92, 53.70, 53.3, 37.93, 35.41, 32.98, 23.35, 18.91, 18.57; ESI-HRMS calculated for C28H39FN2O4 486.2894, found 487.2975 [M + H]+.
Radiochemistry
(R)-5-((3,4-Dimethoxyphenethyl)(2-[18F]fluoroethyl)amino)-2-(3,4-dimethoxyphenyl)-2-isopropylpentanenitrile ([18F]1)
[18F]F– was produced by the 18O(p,n)18F nuclear reaction using an IBA (Louvain-la-Neuve, Belgium) Cyclone 18/9 cyclotron. Radioactivity levels were measured using a Veenstra (Joure, The Netherlands) VDC-405 dose calibrator. Radiochemistry was carried out in homemade, remotely controlled synthesis units.29 After irradiation, [18F]fluoride was trapped on a PS-HCO3 column and eluted with 1 mL of MeCN/H2O (9:1, v/v) containing 13 mg (35 μmol) of Kryptofix 2.2.2 and 2 mg (14 μmol) of K2CO3 into a screw cap reaction vial. The [18F]K222/KF/K2CO3 complex was dried at 90 °C under a helium flow of 50 mL·min–1 and reduced pressure for 6 min. MeCN (0.5 mL) was added, and the complex was dried for 3 min resulting in a white tarnish at the bottom of the vial. 2-Bromoethyltosylate (10 mg, 36 μmol) was dissolved in 0.5 mL of DMF and added to the vial containing the dried complex, and this reaction mixture was heated to 90 °C. After 10 min, the formed volatile intermediate 1-bromo-2-[18F]fluoroethane was distilled at 100 °C through a preheated silver triflate column at 200 °C resulting in [18F]fluoroethyltriflate, which was bubbled to the second reaction vial containing a reaction mixture with 1.5 mg (3.4 μmol) of (R)-desmethyl-norverapamil, 1.5 mg (11 μmol) of K2CO3, and a stirring bar in 0.5 mL of ACN at 0 °C (Scheme 4). After distillation, the reaction was stirred for 15 min at 120 °C, quenched with 1 mL of water, and purified by semipreparative HPLC (method E). The product eluted at 8 min and was collected for 1.5 min, and the fraction was diluted with 40 mL of water. The mixture was passed through a Sep-Pak Plus tC18 cartridge and subsequently rinsed with 20 mL water. The product was eluted from the Sep-Pak Plus tC18 cartridge with 1 mL of ethanol (96%). The final product containing [18F]1 was diluted with a solution of 7.11 mM NaH2PO4 in 0.9% NaCl (w/v in water), pH 5.2, resulting in a final solution with 5% ethanol. The purity was >95%, and the specific activity was 143 ± 88 GBq/μmol, which was derived from a calibration curve of reference compound 1 using HPLC (methods A and B). In the end, 400–1600 MBq was isolated, and the overall radiochemical yield was 2.7% ± 1.2% DC, starting from 20 to 50 GBq [18F]F– (n = 7).
(R)-2-(3,4-Dimethoxyphenyl)-5-((4-(2-[18F]fluoroethoxy)-3-methoxyphenethyl)amino)-2-isopropylpentanenitrile ([18F]2)
The [18F]K222/KF/K2CO3 complex was dried as described above, 1.0 mg of precursor 13 in 0.5 mL of MeCN was added to the reaction vial, and the reaction mixture was heated at 90 °C for 15 min. The reaction mixture was cooled down to room temperature, and 0.2 mL of TFA was added. After 10 min, the reaction was quenched with 0.9 mL of 2.5 M NaOH and purified by semipreparative HPLC (method F). The product eluted at 15 min and was collected for 1.5 min, and the fraction was diluted with 40 mL of water. The mixture was passed through the Sep-Pak Plus tC18 cartridge and subsequently rinsed with 20 mL water. The product [18F]2 was eluted with 1 mL of ethanol (96%) and diluted with a solution of 7.11 mM NaH2PO4 in 0.9% NaCl (w/v in water), pH 5.2, resulting in a final solution with 5% ethanol with a purity of >95%. The specific activity was 151 ± 74 GBq/μmol, which was derived from a calibration curve of reference compound 2 using HPLC (method C). At the end of the synthesis, 300–8000 MBq was isolated, and the overall radiochemical yield was 17.2% ± 9.9% DC, starting from 15–50 GBq [18F]F– (n = 7).
(R)-[11C]Verapamil
Radiolabeling of (R)-[11C]verapamil was performed as described previously.30 The product was purified by semipreparative HPLC (method E) with a retention time of 10 min. The collected HPLC fraction was diluted with 40 mL of water and the mixture was passed through a Sep-Pak Plus tC18 cartridge and subsequently rinsed with 20 mL of water. The product was eluted with 1 mL of ethanol (96%) and diluted with a solution of 7.11 mM NaH2PO4 in 0.9% NaCl (w/v in water), pH 5.2, resulting in a final solution with 5% ethanol, with a purity of >95% and a specific activity of 118 GBq/μmol (n = 1) as determined on HPLC (method B). At the end of the synthesis, 5.2 GBq was isolated, and overall radiochemical yield was 28.2% DC, starting from 60 GBq [11C]CO2 (n = 1).
Log D7.4
The distribution of [18F]1 and [18F]2 between equal volumes of 0.2 M phosphate buffer (pH 7.4) and 1-octanol was measured in triplicate at room temperature. One milliliter of a 1–5 MBq/mL solution of [18F]1 or [18F]2 in 0.2 M phosphate buffer (pH 7.4) was vigorously mixed with 1 mL of 1-octanol for 1 min at room temperature using a vortex. After 30 min, five samples of 100 μL were taken from both layers, avoiding cross-contamination. To determine recovery, 5 samples of 100 mL were taken from the 1–5 MBq/mL solution. All samples were counted for radioactivity. The log Doct,7.4 value was calculated according to log Doct,7.4 = 10log(Aoct/Abuffer), where Aoct and Abuffer represent average radioactivity of 5 1-octanol and 5 buffer samples, respectively.31
Animals
Healthy male Wistar rats were obtained from Harlan Netherlands B.V. (Horst, the Netherlands) and male wild-type mice, Mdr1a/b(−/−) mice, and Bcrp1(−/−) mice developed from the FVB line were purchased from Taconic (Hudson, USA). All animals were housed in groups of four to six per cage until treatment and were kept at room temperature of 20–24 °C with a relative humidity of 50–70% and under a 12 h light/dark cycle. Animals had unrestricted access to food (Teklad Global 16% Protein Rodent Diet, Harlan, Madison, WI, USA) and tap water. All animal experiments were performed in compliance with Dutch laws on animal experimentation and after approval by the local animal ethics committee.
Biodistribution
Healthy Wistar rats (218–244 g) were injected with 50 ± 2 MBq of either [18F]1 or [18F]2 in the tail vein under isoflurane anesthesia (2% in O2 at 1 L·min–1). Rats were conscious for the allowed time between injection and sacrificing, except for the animals of the 5 min time point, which were left unconscious for the whole time. Animals were sacrificed under isoflurane anesthesia at time points 5, 15, or 60 min (n = 3). Blood was collected via a heart puncture from each rat. Heart, lungs, liver, kidneys, bone, cerebral cortex, cerebellum, and the rest of the brain were collected, weighed, and counted for radioactivity in a Wallac Universal Gamma Counter 1282 (PerkinElmer, Waltham, MA, USA). Biodistribution data were expressed as percentage injected dose per gram tissue (%ID/g).
Metabolite Analysis
Healthy Wistar rats (230–291 g) were injected with 28 ± 4 or 43 ± 24 MBq of [18F]1 or [18F]2, respectively, in the tail vein under isoflurane anesthesia. After injection, rats were conscious for the allowed time and sacrificed under isoflurane anesthesia at time point 5, 15, or 60 min (n = 3). Blood samples were collected via heart puncture, and the brain was removed from the skull and cut in half. Blood was collected in a heparin tube and centrifuged for 5 min at 4000 rpm (Hettich universal 16, Depex B.V., the Netherlands). Plasma was separated from blood cells, 1 mL was loaded onto an Sep-Pak tC18 cartridge (Waters, Etten-Leur, the Netherlands), and the cartridge was washed with 20 mL of water. This eluate was defined as the polar radiolabeled metabolite fraction. Next, the Sep-Pak cartridge was eluted with 1.5 mL of methanol. This eluate was defined as the nonpolar fraction and contains the parent tracer. This eluate was analyzed using HPLC (method G). The recovery from the Sep-pak procedure was >85%, and remaining activity was not taken into account. One half of the brain was counted for activity, and the other half was homogenized with an IKA T18 ULTRA-TURRAX Basic Homogenizer (IKA, Germany) in cold H2O/MeCN (1:1, v/v) under ice cooling and subsequently centrifuged at 4000 rpm for 5 min. Separated supernatants were analyzed using HPLC.
PET Imaging and Data Analysis
For all experiments, animals were anesthetized via a nose mask initially using 4% isoflurane in oxygen at a rate of 1 L/min. One hour prior to each study, a jugular vein was cannulated for administration of the radiotracer. Rats were positioned in pairs, and mice were scanned in groups of 4–6 using a double LSO/LYSO layer High Resolution Research Tomograph (HRRT; Siemens/CTI, Knoxville, TN, USA).32 During scanning, anesthesia was maintained using 2% isoflurane in oxygen. For each scanning session, first a transmission scan was acquired using a 740 MBq two-dimensional (2D) fan-collimated 137Cs (662 keV) moving point source.33 This scan was used to correct subsequent emission scans for attenuation and scatter. As the HRRT was decommissioned, the final study (Mdr1a/b(−/−) vs WT mice of [18F]2) had to be performed on nanoPET/CT and nanoPET/MR scanners (Mediso Ltd., Budapest, Hungary)34 with identical PET components. In this study, the CT scan was used for attenuation correction and the MR scan for co-registration purposes. For the emission scans, Wistar rats (341–450 g) were injected with 9.9 ± 3.2 MBq of [18F]1 (n = 4) or 9.9 ± 0.6 MBq of [18F]2 (n = 6) and scanned for 1 h. Next, a second emission scan (30 min) following injection of 9.0 ± 1.7 MBq of [18F]NaF was performed to delineate bone. The following day, the same rats received an intravenous bolus injection of 15 mg/kg (3.5 mg/mL) tariquidar, dissolved in a vehicle consisting of 5% glucose in saline. Thirty to forty minutes after tariquidar administration, rats were injected with 9.8 ± 1.4 MBq of [18F]1 or 8.7 ± 0.5 MBq of [18F]2. Again, this was followed by an [18F]NaF (7.9 ± 3.5 MBq) scan. [18F]1 and [18F]2 scans were acquired in list mode and rebinned into the following frame sequence: 10 × 60, 4 × 300, and 3 × 600 s. The [18F]NaF scans were processed as a single static scan. Mice (28.3–39.7 g) were injected with 3.6 ± 0.6 MBq of [18F]1 or 3.8 ± 0.4 MBq of [18F]2 and scanned for 60 min. Emission scans were acquired in list mode and rebinned into the following frame sequence: 7 × 10, 1 × 20, 3 × 30, 2 × 60, 2 × 150, 4 × 300, and 3 × 600 s for all HRRT scans. For the nanoPET scans, the following frame sequence was used: 4 × 5, 4 × 10, 2 × 30, 3 × 60, 2 × 300, 3 × 600, and 1 × 900 s. Following corrections for decay, dead time, scatter, and randoms, HRRT emission scans were reconstructed using an iterative 3D-ordered subset weighted least-squares algorithm (3D-OSWLS).33 The point source resolution varied across the field of view from approximately 2.3 to 3.2 mm full width at half-maximum in the transaxial direction and from 2.5 to 3.4 mm in the axial direction. Reconstruction of the nanoPET emission scans was performed using an iterative 3D Poisson ordered-subset expectation-maximization algorithm (Tera-Tomo; Mediso Ltd.34) with 4 iterations and 6 subsets, resulting in an isoptropic 0.4 mm voxel dimension. PET images were analyzed using the freely available AMIDE software (version 0.9.2).35 An MR based rat brain atlas was used to define a whole brain region of interest in rats. For each rat, this validated MR atlas was aligned visually with the [18F]NaF image using a procedure described previously to obtain a corresponding whole brain region of interest (ROI).36 Next, these ROIs were projected onto the dynamic [18F]1 and [18F]2 image sequence, generating whole brain time–activity curves (TACs). For mice, ellipsoidal shaped ROIs were drawn manually over the brain. Again, these ROIs were projected onto the dynamic image sequences, generating whole brain TACs. All TACs were expressed as standardized uptake values (SUV), that is, mean ROI radioactivity concentration normalized to injected dose and body weight. In addition, an ellipsoid was drawn over the complete animal (excluding the cannula) to obtain the image derived percentage injected dose per cc (%ID/cc). Finally, ellipsoid shaped ROIs manually drawn over the left ventricle were used to obtain a blood curve. The area under the curve (AUC) of the blood and brain curves were determined by Graphpad PRISM (v 5.02, Graphpad Software Inc.) and brain-to-blood AUC ratios were derived from these values.
Author Contributions
R.M.R. was involved with all aspects of the studies and performed (radio)synthesis of compounds. E.J.M.K. and R.C.S. assisted with the metabolite and PET studies. R.M.R., G.L., P.H.E., A.A.L., and A.D.W. designed the studies, performed analysis of the data, and wrote the manuscript.
This research was financially supported by STW (OTP-project 11741). Fluorine-18 was kindly provided by BV Cyclotron (Amsterdam).
The authors declare no competing financial interest.
References
- Zlokovic B. V. (2008) The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57, 178–201. 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- van Assema D. M.; van Berckel B. N. (2016) Blood-Brain Barrier ABC-transporter P-glycoprotein in Alzheimer’s Disease: Still a Suspect?. Curr. Pharm. Des. 22, 5808. 10.2174/1381612822666160804094544. [DOI] [PubMed] [Google Scholar]
- Potschka H. (2012) Role of CNS efflux drug transporters in antiepileptic drug delivery: overcoming CNS efflux drug transport. Adv. Drug Delivery Rev. 64, 943–952. 10.1016/j.addr.2011.12.007. [DOI] [PubMed] [Google Scholar]
- Raaphorst R. M.; Windhorst A. D.; Elsinga P. H.; Colabufo N. A.; Lammertsma A. A.; Luurtsema G. (2015) Radiopharmaceuticals for assessing ABC transporters at the blood-brain barrier. Clin. Pharmacol. Ther. 97, 362–371. 10.1002/cpt.73. [DOI] [PubMed] [Google Scholar]
- Luurtsema G.; Molthoff C. F. M.; Windhorst A. D.; Smit J. W.; Keizer H.; Boellaard R.; Lammertsma A. A.; Franssen E. J. F. (2003) R)- and (S)-[11C]verapamil as PET-tracers for measuring P-glycoprotein function: in vitro and in vivo evaluation. Nucl. Med. Biol. 30, 747–751. 10.1016/S0969-8051(03)00078-7. [DOI] [PubMed] [Google Scholar]
- Deo A. K.; Borson S.; Link J. M.; Domino K.; Eary J. F.; Ke B.; Richards T. L.; Mankoff D. A.; Minoshima S.; O’Sullivan F.; Eyal S.; Hsiao P.; Maravilla K.; Unadkat J. D. (2014) Activity of P-Glycoprotein, a beta-Amyloid Transporter at the Blood-Brain Barrier, Is Compromised in Patients with Mild Alzheimer Disease. J. Nucl. Med. 55, 1106–1111. 10.2967/jnumed.113.130161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeek J.; Syvanen S.; Schuit R. C.; Eriksson J.; de Lange E. C.; Windhorst A. D.; Luurtsema G.; Lammertsma A. A. (2012) Synthesis and preclinical evaluation of [11C]D617, a metabolite of (R)-[11C]verapamil. Nucl. Med. Biol. 39, 530–539. 10.1016/j.nucmedbio.2011.10.017. [DOI] [PubMed] [Google Scholar]
- Slobbe P.; Poot A. J.; Windhorst A. D.; van Dongen G. A. (2012) PET imaging with small-molecule tyrosine kinase inhibitors: TKI-PET. Drug Discovery Today 17, 1175–1187. 10.1016/j.drudis.2012.06.016. [DOI] [PubMed] [Google Scholar]
- Vlaming M. L.; Lappchen T.; Jansen H. T.; Kivits S.; van Driel A.; van de Steeg E.; van der Hoorn J. W.; Sio C. F.; Steinbach O. C.; DeGroot J. (2015) PET-CT imaging with [18F]-gefitinib to measure Abcb1a/1b (P-gp) and Abcg2 (Bcrp1) mediated drug-drug interactions at the murine blood-brain barrier. Nucl. Med. Biol. 42, 833–841. 10.1016/j.nucmedbio.2015.07.004. [DOI] [PubMed] [Google Scholar]
- Sander K.; Galante E.; Gendron T.; Yiannaki E.; Patel N.; Kalber T. L.; Badar A.; Robson M.; Johnson S. P.; Bauer F.; Mairinger S.; Stanek J.; Wanek T.; Kuntner C.; Kottke T.; Weizel L.; Dickens D.; Erlandsson K.; Hutton B. F.; Lythgoe M. F.; Stark H.; Langer O.; Koepp M.; Arstad E. (2015) Development of Fluorine-18 Labeled Metabolically Activated Tracers for Imaging of Drug Efflux Transporters with Positron Emission Tomography. J. Med. Chem. 58, 6058–6080. 10.1021/acs.jmedchem.5b00652. [DOI] [PubMed] [Google Scholar]
- Savolainen H.; Cantore M.; Colabufo N. A.; Elsinga P. H.; Windhorst A. D.; Luurtsema G. (2015) Synthesis and Preclinical Evaluation of Three Novel Fluorine-18 Labeled Radiopharmaceuticals for P-Glycoprotein PET Imaging at the Blood-Brain Barrier. Mol. Pharmaceutics 12, 2265–2275. 10.1021/mp5008103. [DOI] [PubMed] [Google Scholar]
- Savolainen H.; Windhorst A. D.; Elsinga P. H.; Cantore M.; Colabufo N. A.; Willemsen A. T.; Luurtsema G. (2017) Evaluation of [18F]MC225 as a PET radiotracer for measuring P-glycoprotein function at the blood-brain barrier in rats: Kinetics, metabolism, and selectivity. J. Cereb. Blood Flow Metab. 37, 1286–1298. 10.1177/0271678X16654493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorner B.; Kuntner C.; Bankstahl J. P.; Wanek T.; Bankstahl M.; Stanek J.; Mullauer J.; Bauer F.; Mairinger S.; Loscher W.; Miller D. W.; Chiba P.; Muller M.; Erker T.; Langer O. (2011) Radiosynthesis and in vivo evaluation of 1-[18F]fluoroelacridar as a positron emission tomography tracer for P-glycoprotein and breast cancer resistance protein. Bioorg. Med. Chem. 19, 2190–2198. 10.1016/j.bmc.2011.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura K.; Yamasaki T.; Konno F.; Yui J.; Hatori A.; Yanamoto K.; Wakizaka H.; Ogawa M.; Yoshida Y.; Nengaki N.; Fukumura T.; Zhang M. R. (2011) Synthesis and in vivo evaluation of [18F]fluoroethyl GF120918 and XR9576 as positron emission tomography probes for assessing the function of drug efflux transporters. Bioorg. Med. Chem. 19, 861–870. 10.1016/j.bmc.2010.12.004. [DOI] [PubMed] [Google Scholar]
- Weast R. C. (1974) Handbook of Chemistry and Physics, Vol. 55, CRC Press, Inc.. [Google Scholar]
- von Richter O.; Eichelbaum M.; Schonberger F.; Hofmann U. (2000) Rapid and highly sensitive method for the determination of verapamil, [2H7]verapamil and metabolites in biological fluids by liquid chromatography–mass spectrometry. J. Chromatogr., Biomed. Appl. 738, 137–147. 10.1016/S0378-4347(99)00508-3. [DOI] [PubMed] [Google Scholar]
- Pauli-Magnus C.; Von Richter O.; Burk O.; Ziegler A.; Mettang T.; Eichelbaum M.; Fromm M. F. (2000) Characterization of the Major Metabolites of Verapamil as Substrates and Inhibitors of P-glycoprotein. J. Pharmacol. Exp. Ther. 293, 376–382. [PubMed] [Google Scholar]
- Luurtsema G.; Molthoff C. F.; Schuit R. C.; Windhorst A. D.; Lammertsma A. A.; Franssen E. J. (2005) Evaluation of (R)-[11C]verapamil as PET tracer of P-glycoprotein function in the blood-brain barrier: kinetics and metabolism in the rat. Nucl. Med. Biol. 32, 87–93. 10.1016/j.nucmedbio.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Echizen H.; Brecht T.; Niedergesäss S.; Vogelgesang B.; Eichelbaum M. (1985) The effect of dextro-, levo-, and racemic verapamil on atrioventricular conduction in humans. Am. Heart J. 109, 210–217. 10.1016/0002-8703(85)90585-X. [DOI] [PubMed] [Google Scholar]
- Gilmore J.; Prowse W.; Steggles D.; Urquhart M.; Olkowski J. (1996) Convenient synthesis of (2R)- and (2S)-2-(1-methylethyl)-5-oxo-2-phenylpentanenitrile, intermediates in the preparation of phenylalkylamine calcium channel blockers. J. Chem. Soc., Perkin Trans. 1 1, 2845–2850. 10.1039/p19960002845. [DOI] [Google Scholar]
- Elsinga P. H.; Franssen E. J. F.; Hendrikse N. H.; Fluks L.; Weemaes A. A.; van der Graaf W. T. A.; de Vries E. G. E.; Visser G. M.; Vaalburg W. (1996) Carbon-11-Labeled Daunorubicin and Verapamil for Probing P-Glycoprotein in Tumors with PET. J. Nucl. Med. 37, 1571–1575. [PubMed] [Google Scholar]
- Lubberink M.; Luurtsema G.; van Berckel B. N.; Boellaard R.; Toornvliet R.; Windhorst A. D.; Franssen E. J.; Lammertsma A. A. (2007) Evaluation of tracer kinetic models for quantification of P-glycoprotein function using (R)-[11C]verapamil and PET. J. Cereb. Blood Flow Metab. 27, 424–433. 10.1038/sj.jcbfm.9600349. [DOI] [PubMed] [Google Scholar]
- Bankstahl J. P.; Kuntner C.; Abrahim A.; Karch R.; Stanek J.; Wanek T.; Wadsak W.; Kletter K.; Muller M.; Loscher W.; Langer O. (2008) Tariquidar-induced P-glycoprotein inhibition at the rat blood-brain barrier studied with (R)-11C-verapamil and PET. J. Nucl. Med. 49, 1328–1335. 10.2967/jnumed.108.051235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan P.; Telu S.; Shukla S.; Ambudkar S. V.; Pike V. W.; Halldin C.; Gottesman M. M.; Innis R. B.; Hall M. D. (2011) The ″specific″ P-glycoprotein inhibitor Tariquidar is also a substrate and an inhibitor for breast cancer resistance protein (BCRP/ABCG2). ACS Chem. Neurosci. 2, 82–89. 10.1021/cn100078a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntner C.; Bankstahl J. P.; Bankstahl M.; Stanek J.; Wanek T.; Stundner G.; Karch R.; Brauner R.; Meier M.; Ding X.; Muller M.; Loscher W.; Langer O. (2010) Dose-response assessment of tariquidar and elacridar and regional quantification of P-glycoprotein inhibition at the rat blood-brain barrier using (R)-[(11)C]verapamil PET. Eur. J. Nucl. Med. Mol. Imaging 37, 942–953. 10.1007/s00259-009-1332-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syvanen S.; Lindhe O.; Palner M.; Kornum B. R.; Rahman O.; Langstrom B.; Knudsen G. M.; Hammarlund-Udenaes M. (2009) Species differences in blood-brain barrier transport of three positron emission tomography radioligands with emphasis on P-glycoprotein transport. Drug Metab. Dispos. 37, 635–643. 10.1124/dmd.108.024745. [DOI] [PubMed] [Google Scholar]
- Wanek T.; Romermann K.; Mairinger S.; Stanek J.; Sauberer M.; Filip T.; Traxl A.; Kuntner C.; Pahnke J.; Bauer F.; Erker T.; Loscher W.; Muller M.; Langer O. (2015) Factors Governing P-Glycoprotein-Mediated Drug-Drug Interactions at the Blood-Brain Barrier Measured with Positron Emission Tomography. Mol. Pharmaceutics 12, 3214–3225. 10.1021/acs.molpharmaceut.5b00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan P.; Brimacombe K. R.; Kreisl W. C.; Liow J. S.; Zoghbi S. S.; Telu S.; Zhang Y.; Pike V. W.; Halldin C.; Gottesman M. M.; Innis R. B.; Hall M. D. (2011) Lysosomal trapping of a radiolabeled substrate of P-glycoprotein as a mechanism for signal amplification in PET. Proc. Natl. Acad. Sci. U. S. A. 108, 2593–2598. 10.1073/pnas.1014641108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windhorst A. D.; Linden T. T.; de Nooij A.; Keus J. F.; Buijs F. L.; Schollema P. E.; van Rooij L. F.; Herscheid J. D. M. (2001) A complete, multipurpose, low cost, fully automated and GMP compliant radiosynthesis system. J. Labelled Compd. Radiopharm. 44, S1052–S1054. 10.1002/jlcr.25804401375. [DOI] [Google Scholar]
- Luurtsema G.; Windhorst A. D.; Mooijer M. P. J.; Herscheid J. D. M.; Lammertsma A. A.; Franssen E. J. F. (2002) Fully automated high yield synthesis of (R)- and (S)-[11C]verapamil for measuring P-glycoprotein function with positron emission tomography. J. Labelled Compd. Radiopharm. 45, 1199–1207. 10.1002/jlcr.632. [DOI] [Google Scholar]
- Klein P. J.; Chomet M.; Metaxas A.; Christiaans J. A.; Kooijman E.; Schuit R. C.; Lammertsma A. A.; van Berckel B. N.; Windhorst A. D. (2016) Synthesis, radiolabeling and evaluation of novel amine guanidine derivatives as potential positron emission tomography tracers for the ion channel of the N-methyl-d-aspartate receptor. Eur. J. Med. Chem. 118, 143–160. 10.1016/j.ejmech.2016.04.022. [DOI] [PubMed] [Google Scholar]
- de Jong H. W.; van Velden F. H.; Kloet R. W.; Buijs F. L.; Boellaard R.; Lammertsma A. A. (2007) Performance evaluation of the ECAT HRRT: an LSO-LYSO double layer high resolution, high sensitivity scanner. Phys. Med. Biol. 52, 1505–1526. 10.1088/0031-9155/52/5/019. [DOI] [PubMed] [Google Scholar]
- van Velden F. H.; Kloet R. W.; van Berckel B. N.; Molthoff C. F.; de Jong H. W.; Lammertsma A. A.; Boellaard R. (2008) Impact of attenuation correction strategies on the quantification of High Resolution Research Tomograph PET studies. Phys. Med. Biol. 53, 99–118. 10.1088/0031-9155/53/1/007. [DOI] [PubMed] [Google Scholar]
- Szanda I.; Mackewn J.; Patay G.; Major P.; Sunassee K.; Mullen G. E.; Nemeth G.; Haemisch Y.; Blower P. J.; Marsden P. K. (2011) National Electrical Manufacturers Association NU-4 performance evaluation of the PET component of the NanoPET/CT preclinical PET/CT scanner. J. Nucl. Med. 52, 1741–1747. 10.2967/jnumed.111.088260. [DOI] [PubMed] [Google Scholar]
- Loening A. M.; Gambhir S. S. (2003) AMIDE: A Free Software Tool for Multimodality Medical Image Analysis. Mol. Imaging 2, 131–137. 10.1162/153535003322556877. [DOI] [PubMed] [Google Scholar]
- Buiter H. J.; van Velden F. H.; Leysen J. E.; Fisher A.; Windhorst A. D.; Lammertsma A. A.; Huisman M. C. (2012) Reproducible Analysis of Rat Brain PET Studies Using an Additional [(18)F]NaF Scan and an MR-Based ROI Template. Int. J. Mol. Imaging 2016, 580717. 10.1155/2012/580717. [DOI] [PMC free article] [PubMed] [Google Scholar]