

Abstract
Histone deacetylase inhibitors are emerging therapies for many diseases including cancers and neurological disorders; however, these drugs are teratogens to the developing skeleton. Hdac3 is essential for proper endochondral ossification as its deletion in chondrocytes increases cytokine signaling and the expression of matrix remodeling enzymes. Here we explored the mechanism by which Hdac3 controls Mmp13 expression in chondrocytes. In Hdac3-depleted chondrocytes, Erk1/2 as well as its downstream substrate, Runx2, were hyperphosphorylated as a result of decreased expression and activity of the Erk1/2 specific phosphatase, Dusp6. Erk1/2 kinase inhibitors and Dusp6 adenoviruses reduced Mmp13 expression and partially rescued matrix production in Hdac3-deficient chondrocytes. Postnatal chondrocyte-specific deletion of Hdac3 with an inducible Col2a1-Cre caused premature production of pErk1/2 and Mmp13 in the growth plate. Thus, Hdac3 controls the temporal and spatial expression of tissue-remodeling genes in chondrocytes to ensure proper endochondral ossification during development.
Keywords: Hdac3, cartilage, Dusp6, Erk1/2, MAPK, Runx2
Introduction
Long bones form through the process of endochondral ossification when mesenchymal progenitor cells differentiate into chondrocytes, which form a cartilage intermediate that serves as a template for bone formation. As committed chondrocytes mature, they proliferate and eventually enter into hypertrophy, stimulate vasculogenesis, and recruit osteoclasts and osteoblasts that remodel and ossify bone (1–4). Most hypertrophic chondrocytes will undergo apoptosis, though some of these chondrocytes are able to escape cell death and contribute to the osteogenic pool in trabecular bone (5, 6). Various signaling and transcriptional pathways are necessary to orchestrate this complex developmental process. The Erk/MAPK signaling pathway affects ossification during skeletal development by phosphorylating transcription factors such as AP-1, Runx2, and Elk1 to promote transcription of target genes, such as matrix metalloproteinase (Mmp)-13 in osteoblasts (7–15). Disruptions in Erk/MAPK signaling during skeletogenesis cause several skeletal disorders, namely achondroplasia and hypochondroplasia (16, 17), suggesting an important role for Erk/MAPK signaling in chondrocytes. While the exact mechanism of Erk/MAPK in chondrocytes remains elusive (17–24), it is evident is that the activity of Erk1/2 requires temporal and spatial regulation to ensure optimal bone growth.
Histone deacetylases (Hdacs) are enzymes that epigenetically control gene transcription by removing acetyl groups from lysine side chains of histone tails, leading to chromatin compaction and repression of gene expression (25–27). Hdacs also modify other proteins such as transcription factors (i.e., Runx2, p53, Stat3, etc.) and influence their stability and activity to further modulate gene expression (28–33). Thus, Hdacs are important during developmental and cellular differentiation when temporal gene expression is necessary to ensure proper growth and development. Hdac inhibitors have been widely used in the clinic to treat several diseases including many cancers, neurological disorders and arthritis (34–36). However, because these inhibitors are target multiple Hdacs, adverse side effects are possible, particularly in the skeleton. Hdac inhibitors are teratogenic to fetuses and long-term exposure increases fracture risk in epileptic patients (37–46). Therefore, understanding the roles of individual Hdacs in the skeleton is important.
Several Hdacs participate in endochondral bone formation (34). Hdac3 is strongly expressed in osteoblasts and chondrocytes and acts as a corepressor of Runx2, Zfp521 and other transcription factors (47, 48). While germline deletion of Hdac3 is embryonic lethal (49) conditional knockout mouse models have demonstrated the importance of Hdac3 in maintaining proper bone mass during aging, as well as long bone development through actions in osteoblasts and chondrocytes (50–52). We recently showed that conditional deletion of Hdac3 in chondrocytes (with inducible type II collagen alpha 1, (Col2ERT)-Cre) alters cytokine signaling and matrix metalloproteinase expression suggesting an important role in regulating extracellular matrix remodeling (53).
Here we describe a mechanism whereby Hdac3 controlsMmp13 expression and chondrocyte maturation. Hdac3 deficiency reduced matrix production and increased expression of Mmp13 in primary mouse chondrocytes. Both Erk1/2 and its downstream substrate, Runx2, were hyperphosphorylated in Hdac3 deficient chondrocytes due to reduced expression of the Erk1/2 specific phosphatase, Dusp6. Inhibition of the Erk1/2 kinase, MEK, reduced Mmp13 expression and partially restored matrix production in Hdac3-CKO chondrocytes. Dusp6 overexpression also partially rescued matrix production and reduced elevated Mmp13 expression in Hdac3-deficient chondrocytes. Finally, pErk1/2 and Mmp13 were misexpressed in the growth plates of Hdac3-deficient mice. These data demonstrate that Hdac3 controls the temporal and spatial regulation of Erk1/2 phosphorylation and subsequent Mmp13 expression to ensure proper chondrocyte maturation during endochondral ossification.
Materials and Methods
Isolation and culture of immature murine chondrocytes (IMCs)
IMCs were isolated and cultured as previously described (54). Briefly, the femoral heads and tibial plateaus were harvested from 1-week-old Hdac3fl/fl pups. Articular cartilage was digested in 3 mg/ml collagenase for 1 hour and then overnight in 0.5 mg/ml collagenase. The resulting IMC suspensions were brought to a concentration of 2 × 107 cells/ml and plated in micromasses with 10 μl drops, each containing 2 × 105 cells in DMEM. Three micromasses were plated together in 35 mm plates. After 1 hour, micromasses were covered with 3 ml DMEM and 5% FBS.
Adenoviral transductions and Erk1/2 inhibitor treatments of IMCs
On day 3 of culture, micromasses were transduced with adenoviruses expressing Cre recombinase (Ad-Cre, Vector Biolabs) or green fluorescent protein (Ad-GFP, Vector Biolabs) at 1000 MOI. For experiments without inhibitors, cells were collected 48 hr following transduction for RNA and protein extraction and matrix staining with Alcian blue. The MEK (Mapk/Erk Kinase) inhibitor, U0126 (1 μM, Millipore 662005), or vehicle (0.01% v/v DMSO) was added to IMC micromass cultures 48 hr after adenoviral transduction. RNA and protein extracts and plates for Alcian blue staining were collected 24 hr later. For Dusp6 overexpression experiments, micromasses were transduced simultaneously with Ad-Cre or Ad-GFP (1000 MOI) and either Ad-GFP or Ad-Dusp6 (Vector Biolabs) (1000 MOI). RNA, protein extracts, and plates for Alcian blue staining were collected 48 hr later.
Alcian blue staining
IMCs were fixed with 10% neutral buffered formalin and stained with 0.5% Alcian blue, 3% acetic acid for 2 hr, then destained with 70% ethanol and rinsed twice with water.
RNA isolation and qPCR
Total RNA was isolated from IMC micromass cultures with TRIzol reagent (Invitrogen) and phenol/chloroform. RNA (3 ug) was reverse transcribed to cDNA with the SuperScript III First-Strand Synthesis cDNA kit (Invitrogen), which was then used in real-time semi-quantitative PCR (qPCR) reactions containing gene-specific primers (Table 1). Transcript levels were normalized to reference gene Gapdh. Transcript levels were quantified using the 2−ΔΔCt method.
Table 1.
Primer Sequences and Gene Abbreviations
| Gene | Gene Abbreviation | Forward Primer | Reverse Primer |
|---|---|---|---|
| DUSP1 | Dual Specificity Phosphatase 1 | TTCTCCAAGGAGGATATGAAGCG | CTGCATCCGGATTCTGCACT |
| DUSP11 | Dual Specificity Phosphatase 11 | CCGGCAGCTGTTGAACAATTC | TTATCGGTGTCCACTGCACC |
| DUSP2 | Dual Specificity Phosphatase 2 | TTCTTGCGAGGCGGTTTCA | GTTGCTATTTTCGGCCCCAG |
| DUSP3 | Dual Specificity Phosphatase 3 | TCTGTGGCTCAGGACATCAC | GGCCCTTTCAAAGTAAGCACTG |
| DUSP4 | Dual Specificity Phosphatase 4 | AGTACAAGTGCATCCCCGTC | ACAGTCCTTTACTGCGTCGAT |
| DUSP5 | Dual Specificity Phosphatase 5 | TGCACCACCCACCTACACTA | CCTTCTTCCCTGACACAGTCAATA |
| DUSP6 | Dual Specificity Phosphatase 6 | TTACTGAAGCCACCTTCCAGG | ACGAGAATAGCAGCGACTGG |
| DUSP7 | Dual Specificity Phosphatase 7 | CTTGGAGCGGGCTTCATCAAT | TTTGAGCACGGTGGTGAGTT |
| DUSP8 | Dual Specificity Phosphatase 8 | AGCCTAAGGGCTTGGTAGGTA | GCCCAGTAAGAATGGGGGTAG |
| DUSP9 | Dual Specificity Phosphatase 9 | TGCGACAAGGCCTCATCAAT | CACCCCCAACCTTCCTAACC |
| Gapdh | Glyceraldehyde-3-Phosphate Dehydrogenase | GCCTCACCCCATTTGATGTT | GGGAAGCCCATCACCATCTT |
| Map2k1 | Mitogen-Activated Protein Kinase Kinase 1 | TCGGCTTCTTCTTGGGCATC | GTGTGCTGAGGCGAGAGTTC |
| Map2k2 | Mitogen-Activated Protein Kinase Kinase 2 | ATGACCTACACTGATGGGGC | GGCCAACTCGTTTGTAGGGA |
| Map2k3 | Mitogen-Activated Protein Kinase Kinase 3 | GCCACAGTGTACGAAGTCCA | TTCCTGTCGGTCTCTGCAAC |
| Map2k4 | Mitogen-Activated Protein Kinase Kinase 4 | GTGCTTTGCGCTTACCCTG | GCTCGGCTCTTCACTTCCAA |
| Map2k5 | Mitogen-Activated Protein Kinase Kinase 5 | TGACTGGTCCTCAGTGGTCA | CTGAGGAGCTGATGGGTCAC |
| Map2k6 | Mitogen-Activated Protein Kinase Kinase 6 | CGCTTCTTGCCTTTCGACTG | CCTGCAGCTTGCATCTTTGT |
| Map2k7 | Mitogen-Activated Protein Kinase Kinase 7 | CAGACTCCCACTGAAGAAGGG | GTCGCGTCCTGGTTTAAGGA |
| Map3k1 | Mitogen-Activated Protein Kinase Kinase Kinase 1 | ATGAGCAGGTTGGCACCTTT | AGGATCTGTGGCTCACCTCT |
| Map3k2 | Mitogen-Activated Protein Kinase Kinase Kinase 2 | CTTTATTGCCCGGCCGCTG | TGTCCCATGTGAAGCAGGTT |
| Map3k3 | Mitogen-Activated Protein Kinase Kinase Kinase 3 | GTTTGGGTGACCTGTGTCCT | TCGCCACCATGGATGAACA |
| Map3k4 | Mitogen-Activated Protein Kinase Kinase Kinase 4 | CAACATGCACTCCTGCCTCG | CTTGGTGCACGGATGAGAGA |
| Map4k1 | Mitogen-Activated Protein Kinase Kinase Kinase Kinase 1 | CAGAGCAGACGGAAGGTGAG | GATCGACTCGAGGCCTTCTG |
| Mmp13 | Matrix Metalloproteinase 13 | TGAACCGCAGCGCTCAGTCTC | GCACCCTCAGCAGGTTGAGGC |
| PP2A | Protein Phosphatase 2A | CGAGAGCGCATCACCATACT | AGAAGATCTGCCCATCCACC |
| Ppp4r1 | Serine/threonin-protein phosphatase 4 regulatory subunit 1 | ATCCACATCAAGTCCGTCCG | CACCGGCTCTCTACTTGAGC |
| Ptpla | Protein tyrosine phosphatase-like, member A | AAAGCACCACGCTCTCTTCA | TGCTTTGCTTGAGGTAGTCCAT |
| Ptpn5 | Protein tyrosine phosphatase, non-receptor type 9 | ACGCAGCAGTTTTACATGGC | TATAACGGGCGCTTTGGTGA |
| Ptpn7 | Protein tyrosine phosphatase non-receptor type 7 | AGTCTGCTACAGTGTTGGGC | CTACATCCGGGGCTATGACG |
| Ptpn9 | Protein tyrosine phosphatase, non-receptor type 9 | AGAAACTGCTTGGTAGCCTGC | GTGAAGCAAACAGGAGGCATT |
| Ptprd | Protein tyrosine phosphatase, receptor type, D | CATTTCTTCACTAGGGCAGCCG | CTCTCCCTGTCTGCAGTTGA |
| Ptprr | Protein tyrosine phosphatase, receptor type, R | AAACAACCTGCAGCATGAAGA | GTATCTCCCCATCCCTTGCG |
| Ptprv | Protein tyrosine phosphatase, receptor type, V | CCCATGAAAGGTGTCCAGCA | TCTGCTTGGACCTACCCACT |
Western blotting
IMC micromasses were lysed in SDS sample buffer (0.1% glycerol, 0.01% SDS, 0.1 M Tris, pH 6.8) on ice. Total protein concentrations were determined using the Bio-Rad DC assay (Bio-Rad). Proteins (20 μg) were separated by SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blotted using antibodies for Hdac3 (Abcam, ab7030, 1:10,000 dilution), p-Thr202/Tyr204 Erk1/2 (Cell Signaling, #9101S, 1:2000 dilution), total Erk (Cell Signaling, #9102, 1:5000 dilution), p-Ser217/221 MEK (Cell Signaling, #9121, 1:5000 dilution), α-tubulin (Developmental Studies Hybridoma Bank, E7, 1:5000 dilution), β-actin (Sigma Aldrich, A5316, 1:10,000), Mmp13 (Abcam, ab39012, 1:2000), p-Ser319 Runx2 (gift from Dr. Renny Franceschi (University of Michigan), 1:1000), p-MAPK Family Antibody Sampler kit (p-p38 MAPK (Thr180/Tyr182), p-SAPK/JNK (Thr183/Tyr185), p-p44/42 MAPK (Erk1/2) (Thr202/Tyr204),) (Cell Signaling, #9910S, 1:1000), Dusp6 (Abcam, ab182605, 1:5000), Dusp7 (Abcam, ab100921, 1:5000) and corresponding secondary antibodies (Santa Cruz Biotechnology). Chemiluminescent detection was performed with Pierce femto reagent (Pierce).
Zymography
Conditioned media (CM) were collected from IMC micromass cultures 48 hr following Ad-Cre or Ad-GFP transduction. CM were spun for 2 min at 6000 rpm to remove cellular debris. Supernatant was collected and equal volumes of each sample were incubated with zymography sample buffer (62.5 mM Tris-HCl, pH 6.8, 25% glycerol, 4% SDS, 0.01% Bromophenol blue, Bio-Rad) for 10 min at room temperature. Proteins were resolved on 8% SDS-PAGE gel containing 0.3 mg/ml type I collagen from rat tail (BD Biosciences, 354236). Following electrophoresis, gels were washed in 2.5% Triton X-100 for 40 min at room temperature with constant agitation. Gels were incubated in 50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 10 mM CaCl, 0.05% NaN3 at 37°C for 20–24 hr with constant agitation. Gels were washed in distilled water 3 times and stained with 0.5% Coomassie Brilliant Blue G-250 (Bio-Rad) (25% methanol; 10% glacial acetic acid) for 40 min at room temperature and destained with 4% methanol and 8% glacial acetic acid. Gels were imaged under the Coomassie filter on the gel documentation system (Fotodyne) and dried on a gel drying system (Bio-Rad).
Generation of Hdac3 deficient animals
Hdac3fl/fl mice were crossed to tamoxifen-inducible type II collagen alpha 1Cre (Col2a1ERT-Cre) mice as previously reported (53, 55). To generate Hdac3-CKOCol2ERT mice, 5-day-old pups were administered a single intraperitoneal injection of tamoxifen (1 mg/kg, Sigma Aldrich, St. Louis, MO) or corn oil (53, 55). All animals were housed in an accredited facility under a 12-hour light/dark cycle and provided water and food (PicoLab® RodentDiet20, LabDiet) ad libitum. All animal research was conducted according to guidelines provided by the National Institute of Health and the Institute of Laboratory Animal Resources, National Research Council. The Mayo Clinic Institutional Animal Care and Use Committee approved all animal studies.
Immunohistochemistry
Tibiae from 9-day-old Hdac3-CKOCol2ERT mice were fixed in formalin for 48 hr and decalcified in 15% EDTA for 7 days. Decalcified bones were embedded in paraffin and sectioned to a thickness of 5 μm. Immunohistochemistry was performed with antibodies (diluted in 1% bovine serum albumin in Tris-buffered saline) directed to p-p44/42 MAPK (Erk 1/2) (Cell Signaling, #9101S, 1:50) and Mmp13 (Abcam, ab39012, 1:50) or a non-specific IgG control. Chromogens were detected with a polyvalent secondary HRP kit (Abcam ab93697) and 3,3-diaminobenzidine (DAB) (Sigma Aldrich, D3939). Sections were counterstained with 0.5% Alcian blue for 1 min.
Statistical analysis
Data collected represent the mean ± standard error of mean (SEM). P values were determined with the Student’s t test.
Results
Hdac3 deficiency increases Mmp13 expression and enzymatic activity in vitro
We previously demonstrated Hdac3 is expressed during skeletal development, as early as E14.5 and continues to be robustly expressed by growth plate chondrocytes into adulthood (34, 52, 53). Tissue-specific deletion of Hdac3 in chondrocytes with tamoxifen-inducible type II collagen alpha 1 Cre (Col2a1ERT-Cre) caused dramatic delays in endochondral ossification (34, 53). Genome-wide transcriptional profiling of Hdac3-depleted chondrocytes showed that markers of chondrocyte maturation, extracellular matrix, and bone development were suppressed with Hdac3-deficiency, while cartilage catabolic genes, like matrix metalloproteinases (e.g., Mmp13) were highly induced (53). To understand these Hdac3-dependent changes in matrix degrading enzymes, immature murine chondrocytes (IMCs) were isolated from 1-week-old Hdac3fl/fl mice, plated in micromasses, and transduced with Ad-GFP or Ad-Cre to generate control and Hdac3-deficient IMCs, respectively. Hdac3 mRNA transcripts were 40–50% decreased following adenviral transduction (Fig 1A). Consistent with previous studies (52, 53), Hdac3-deficient IMC micromasses exhibited reduced Alcian blue staining of proteoglycans within the cartilage matrix (Fig 1B). Real-time semi-quantitative PCR (QPCR) on RNA isolated from these Hdac3-deficient IMCs showed elevated Mmp13 mRNA transcripts (Fig 1C). Mmp13 enzymatic activity was also increased in conditioned medium from Hdac3-deficient IMC cultures as indicated by type I collagen zymography (Fig 1D). While pro-Mmp2 levels were elevated, active Mmp2 levels were not different between Ad-GFP and Ad-Cre cultures.
Figure 1. Hdac3 deficiency increases Mmp13 activity in chondrocytes.
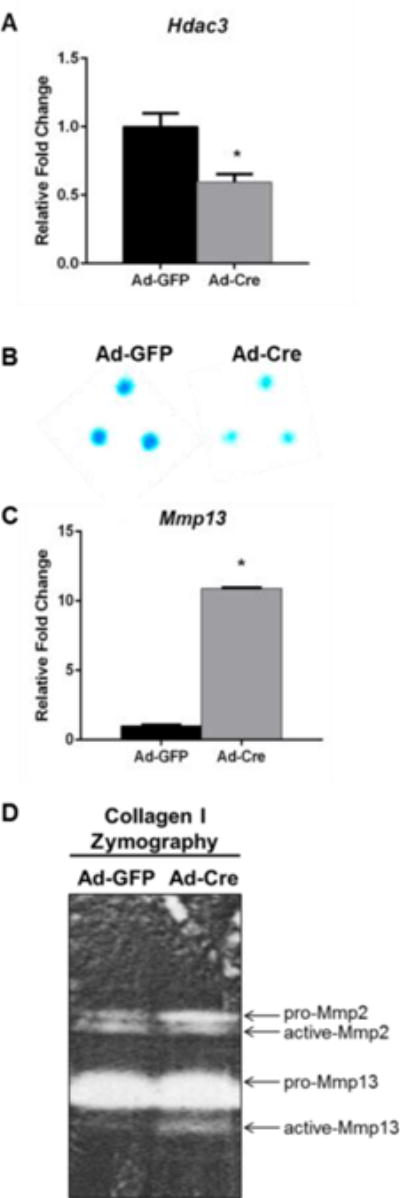
Primary chondrocytes from 1-week-old Hdac3fl/fl mice were plated in micromasses and transduced with Ad-GFP or Ad-Cre 3 days later. (A) QPCR was performed for Hdac3 mRNA transcripts. (B) Micromasses were stained with Alcian blue 48 hr after transduction. (C) QPCR was performed for Mmp13 mRNA transcripts. (D) Type I collagen zymography was performed with the conditioned media from transduced micromass cultures. * p ≤ 0.05
Hdac3 deficiency increases Erk phosphorylation
Mmp13 expression is regulated by Erk1/2-dependent phosphorylation of Runx2 and other transcription factors in osteoblasts (8–15). To determine if similar mechanisms are responsible for increased Mmp13 activity in Hdac3 deficient chondrocytes, western blotting analysis was performed for MAPK/Erk signaling pathways. Hdac3 deficiency increased phosphorylation of Erk1/2, but did not alter phosphorylation of p38, SAPK/JNK, or MAPK/Erk kinase (MEK). However, phosphorylation of the Erk1/2 substrate, Ser319 of Runx2 was increased (Fig 2). These results suggest that Hdac3 suppresses phosphorylation and activation of Erk1/2 and Runx2 in chondrocytes.
Figure 2. Hdac3 deletion in chondrocytes increases phosphorylation of Erk1/2 and Runx2.

Primary chondrocytes from Hdac3fl/fl mice were plated in micromasses and transduced with Ad-GFP or Ad-Cre on day 3 of culture. (A) Western blotting for the indicated proteins was performed 48 hr later. (B) Quantification of relative band intensity normalized to actin. * p ≤ 0.05, # p ≤ 0.1
Erk1/2 inhibition in Hdac3-depleted chondrocytes decreases Mmp13 expression and restores matrix production
To determine if enhanced Erk1/2 activity contributed to Mmp13 expression in Hdac3 deficient IMCs, U0126, a MEK inhibitor, was added to micromass cultures. Inhibition of the MAPK/Erk kinase did not affect Hdac3 or Erk1/2 expression in Hdac3 deficient IMCs, but reduced pErk1/2 and Mmp13 levels (Fig 3A). Matrix production and proteoglycan content returned to normal in Hdac3 deficient IMCs following U0126 treatment as indicated by Alcian blue staining of micromasses (Fig 3B). Mmp13 transcript levels were significantly reduced in U0126 treated Hdac3 deficient chondrocytes (Fig 3C). These results support a mechanism whereby Hdac3 indirectly controls Erk1/2 phosphorylation and subsequent Runx2 activation to control Mmp13 gene expression.
Figure 3. MEK inhibition reduces Mmp13 expression and restores matrix production in Hdac3-deficient chondrocytes.

Primary chondrocytes from Hdac3fl/fl mice were plated in micromasses, transduced with Ad-GFP or Ad-Cre on day 3 of culture and treated with U-126 for 24 hours. (A) Western blotting was performed for the indicated proteins 48 hrs following addition of U0126. (B) Micromasses were stained with Alcian blue. (C) QPCR was performed for Mmp13 mRNA transcripts. * p ≤ 0.05
Hdac3 controls dual-specificity phosphatase (Dusp)6 expression in chondrocytes
To understand the mechanisms responsible for increased Erk1/2 phosphorylation and signaling in Hdac3-deficient chondrocytes, a QPCR-based candidate screen was performed on a panel of well-known Erk1/2 phosphatases and kinases. Only expression of the dual-specificity phosphatases (Dusp)6 and Dusp7 were significantly decreased in Hdac3 depleted chondrocytes by more than two-fold (Fig 4A). There were no significant changes in any known Erk1/2 kinases (Fig 4A). Decreased Dusp6 protein expression was confirmed by western blotting, but Dusp7 protein levels did not change (Fig 4B). Dusp6 is a cytosolic class II Dusp that specifically dephosphorylates Erk1/2 but not other MAPKs (56). Dusp6 reduction is thus consistent with increased phosphorylation of Erk1/2, but not p38 or JNK/SAPK in Hdac3 CKO chondrocytes (Fig 2).
Figure 4. Dusp6 controls Erk1/2 phosphorylation and chondrogenesis of Hdac3-deficient IMCs.

(A–B) Primary chondrocytes from Hdac3fl/fl mice were plated in micromasses and transduced with Ad-GFP or Ad-Cre on day 3. (A) QPCR candidate screen was performed for known Erk1/2 kinase and phosphatase mRNA transcripts 48 hrs following transduction. (B) Western blotting was performed for Dusp6 and Dusp7 48 hrs following transduction. (C–E) Primary chondrocytes from Hdac3fl/fl P7 pups were isolated and plated in micromasses. Cultures were simultaneously transduced with Ad-GFP or Ad-Cre and either Ad-GFP or Ad-Dusp6 on day 3. (C) Western blotting was performed for the indicated proteins 48 hrs post-transduction. Note that the Dusp6 antibody recognizes an epitope near the C-terminus and may be blocked by the V5-tag. (D) Mmp13 mRNA transcripts were measured by QPCR. (E) Micromasses were fixed and stained with Alcian blue. * p ≤ 0.05
Adenoviral reintroduction of V5-Dusp6 in Hdac3-CKO chondrocytes reduced Erk1/2 phosphorylation (Fig 4C) and subsequent Mmp13 gene expression (Fig 4D). Matrix production was also partially restored in Hdac3-CKO chondrocytes with Dusp6 overexpression (Fig 4E). These results suggest Dusp6 acts as a mediator between Hdac3 and Erk1/2 activity in chondrocytes.
Growth plate chondrocytes in Hdac3 deficient mice aberrantly express pErk1/2 and Mmp13
To validate the in vitro studies of Hdac3-CKO chondrocytes, pErk1/2 expression was evaluated by immunohistochemistry in the tibiae of 9-day-old Hdac3-CKOCol2ERT and control mice (Fig 5A). Phosphorylated Erk1/2 was restricted to pre-hypertrophic and hypertrophic chondrocytes in control mice, but was dramatically increased and prematurely expressed in proliferating chondrocytes and present at greater levels in pre-hypertrophic and hypertrophic chondrocytes in Hdac3-CKOCol2ERT mice (Fig 5A). Likewise, Mmp13 protein expression was restricted to hypertrophic chondrocytes in control animals, but was prematurely expressed in proliferating and prehypertrophic chondrocytes in Hdac3-CKOCol2ERT mice (Fig 5B) (53). Altogether, these results demonstrate the importance of Hdac3 in controlling the temporal and spatial activity of Erk1/2 and Mmp13 in growth plate chondrocytes (Fig 5C).
Figure 5. Chondrocyte-specific deletion of Hdac3 increases Erk1/2 phosphorylation and Mmp13 expression in murine growth plates.

(A–B) Hdac3fl/fl mice were crossed to mice expressing the Col2ERT-Cre transgene. Five-day-old pups were injected with tamoxifen or vehicle. Immunohistochemistry was performed on tibiae of P9 animals for Mmp13 (A) or p-Erk1/2 (B). Scale bars = 100 μm. (C) Working model. Hdac3 indirectly modulatesDusp6 activity, which inhibits Erk1/2 activity, Runx2 phosphorylation and Mmp13 expression. Hdac3 may also suppress Mmp13 expression by affecting histone acetylation and Runx2 activity.
Discussion
In this study, we explored molecular mechanisms by which Hdac3 controls Mmp13 expression during chondrocyte maturation. Ablation of Hdac3 in chondrocytes increased the temporal and spatial activation of Erk1/2 by decreasing expression of the dual specific phosphatase Dusp6. Downstream effects of increased Erk activity included increased Runx2 phosphorylation and Mmp13 activation. When Hdac3 was absent, pErk and Mmp13 were aberrantly expressed in developing growth plates. Hdac3 also interacts with Runx2 and the Mmp13 promoter (10, 11). It is likely that Hdac3 deletion is directly affecting Runx2 activity and histone acetylation of the Mmp13 gene, in addition to controlling Erk1/2 activity through Dusp6.
Hdac3 has many roles during chondrocyte maturation (34, 52, 53) and these studies demonstrate that Hdac3 controls Erk1/2 phosphorylation in growth plate chondrocytes. The role of Erk1/2 in endochondral bone formation remains elusive and complex with both positive and negative activity reported in chondrocytes. In some studies, Erk1/2 signaling suppresses hypertrophic chondrocyte maturation, resulting in dwarfism and delayed primary and secondary ossification center formation (21, 53). This phenotype is similar to that of the Hdac3-CKOCol2ERT mice (21, 53). Conversely, other groups reported that phosphorylated Erk1/2 is restricted to hypertrophic chondrocytes and its activity is necessary for chondrocyte hypertrophy (22). The specific roles of Erk1/2 signaling in chondrocytes needs further investigation, but and these results further support the idea that Erk1/2 phosphorylation is crucial during the final stages of chondrocyte maturation.
To understand how loss of Hdac3 could for suppress the phosphorylation of a cytosolic kinase, a candidate screen approach was used and the Erk1/2 specific phosphatase, Dusp6, was identified as a mediator between Hdac3 and Erk1/2 activation. Dusp6 mRNA and protein expression were decreased in Hdac3-deficient chondrocytes. Overexpressing Dusp6 in Hdac3-CKO chondrocytes restored Erk1/2 phosphorylation and partially reduced Mmp13 expression and matrix production. Interestingly, loss of Dusp6 in mice causes skeletal dwarfism, disrupted growth plates and delayed ossification (57), similar to the phenotype observed in Hdac3-CKOCol2ERT animals (53). This supports a mechanism by which Hdac3 mediates the timely activation or deactivation of Erk1/2 signaling to ensure optimal bone growth. However, further investigation is needed to determine the intermediate mechanisms or secondary events by which Hdac3 controls Dusp6 levels in chondrocytes.
In summary, Hdac3 mediates temporal and spatial Erk1/2 activation and subsequent expression of downstream Erk1/2 target genes such as Mmp13 to control chondrocyte hypertrophy, terminal chondrocyte hypertrophy and the coupling to ossification during endochondral bone formation. Loss of Hdac3 activity in chondrocytes during development can disrupt chondrocyte maturation and lead to long term detrimental effects on overall skeletal growth.
Acknowledgments
The authors would like to thank Mr. Xiaodong Li, Ms. Oksana Pichurin, Dr. David Razidlo, and Ms. Bridget Stensgard for technical assistance. We thank Dr. Renny Franceschi for kindly providing the pRunx2 antibody.
Funding
This work was supported by research and training grants from the National Institutes of Health (R01 AR68103, R01 DE20194, T32 AR56950, F31 AR067646, K01 AR65397).
Footnotes
Declaration of Interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the article.
References
- 1.Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003 May 15;423(6937):332–6. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- 2.Day TF, Guo X, Garrett-Beal L, Yang Y. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Developmental cell. 2005 May;8(5):739–50. doi: 10.1016/j.devcel.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 3.Hall BK, Miyake T. All for one and one for all: condensations and the initiation of skeletal development. BioEssays : news and reviews in molecular, cellular and developmental biology. 2000 Feb;22(2):138–47. doi: 10.1002/(SICI)1521-1878(200002)22:2<138::AID-BIES5>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 4.Barna M, Niswander L. Visualization of cartilage formation: insight into cellular properties of skeletal progenitors and chondrodysplasia syndromes. Developmental cell. 2007 Jun;12(6):931–41. doi: 10.1016/j.devcel.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 5.Yang L, Tsang KY, Tang HC, Chan D, Cheah KS. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc Natl Acad Sci U S A. 2014 Aug 19;111(33):12097–102. doi: 10.1073/pnas.1302703111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou X, von der Mark K, Henry S, Norton W, Adams H, de Crombrugghe B. Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS Genet. 2014 Dec;10(12):e1004820. doi: 10.1371/journal.pgen.1004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luo Y, Jian W, Stavreva D, Fu X, Hager G, Bungert J, et al. Trans-regulation of histone deacetylase activities through acetylation. The Journal of biological chemistry. 2009 Dec 11;284(50):34901–10. doi: 10.1074/jbc.M109.038356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. The Journal of biological chemistry. 1995 Jul 14;270(28):16483–6. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- 9.Cruzalegui FH, Cano E, Treisman R. ERK activation induces phosphorylation of Elk-1 at multiple S/T-P motifs to high stoichiometry. Oncogene. 1999 Dec 23;18(56):7948–57. doi: 10.1038/sj.onc.1203362. [DOI] [PubMed] [Google Scholar]
- 10.Vincenti MP, Brinckerhoff CE. Transcriptional regulation of collagenase (MMP-1, MMP-13) genes in arthritis: integration of complex signaling pathways for the recruitment of gene-specific transcription factors. Arthritis research. 2002;4(3):157–64. doi: 10.1186/ar401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Selvamurugan N, Shimizu E, Lee M, Liu T, Li H, Partridge NC. Identification and characterization of Runx2 phosphorylation sites involved in matrix metalloproteinase-13 promoter activation. FEBS letters. 2009 Apr 2;583(7):1141–6. doi: 10.1016/j.febslet.2009.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Franceschi RT, Ge C, Xiao G, Roca H, Jiang D. Transcriptional regulation of osteoblasts. Ann N Y Acad Sci. 2007 Nov;1116:196–207. doi: 10.1196/annals.1402.081. [DOI] [PubMed] [Google Scholar]
- 13.Ge C, Yang Q, Zhao G, Yu H, Kirkwood KL, Franceschi RT. Interactions between extracellular signal-regulated kinase 1/2 and p38 MAP kinase pathways in the control of RUNX2 phosphorylation and transcriptional activity. J Bone Miner Res. 2012 Mar;27(3):538–51. doi: 10.1002/jbmr.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao G, Jiang D, Thomas P, Benson MD, Guan K, Karsenty G, et al. MAPK pathways activate and phosphorylate the osteoblast-specific transcription factor, Cbfa1. J Biol Chem. 2000 Feb 11;275(6):4453–9. doi: 10.1074/jbc.275.6.4453. [DOI] [PubMed] [Google Scholar]
- 15.Ge C, Xiao G, Jiang D, Yang Q, Hatch NE, Roca H, et al. Identification and functional characterization of ERK/MAPK phosphorylation sites in the Runx2 transcription factor. The Journal of biological chemistry. 2009 Nov 20;284(47):32533–43. doi: 10.1074/jbc.M109.040980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsushita T, Murakami S. The ERK MAPK Pathway in Bone and Cartilage Formation: InTech. 2012 [cited 2012. Available from: http://www.intechopen.com/books/protein-kinases/the-erk-mapk-pathway-in-bone-and-cartilage-formation.
- 17.Bobick BE, Kulyk WM. Regulation of cartilage formation and maturation by mitogen-activated protein kinase signaling. Birth defects research Part C, Embryo today : reviews. 2008 Jun;84(2):131–54. doi: 10.1002/bdrc.20126. [DOI] [PubMed] [Google Scholar]
- 18.Bobick BE, Kulyk WM. MEK-ERK signaling plays diverse roles in the regulation of facial chondrogenesis. Experimental cell research. 2006 Apr 15;312(7):1079–92. doi: 10.1016/j.yexcr.2005.12.028. [DOI] [PubMed] [Google Scholar]
- 19.Bobick BE, Kulyk WM. The MEK-ERK signaling pathway is a negative regulator of cartilage-specific gene expression in embryonic limb mesenchyme. The Journal of biological chemistry. 2004 Feb 6;279(6):4588–95. doi: 10.1074/jbc.M309805200. [DOI] [PubMed] [Google Scholar]
- 20.Schindeler A, Little DG. Ras-MAPK signaling in osteogenic differentiation: friend or foe? Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2006 Sep;21(9):1331–8. doi: 10.1359/jbmr.060603. [DOI] [PubMed] [Google Scholar]
- 21.Murakami S, Balmes G, McKinney S, Zhang Z, Givol D, de Crombrugghe B. Constitutive activation of MEK1 in chondrocytes causes Stat1-independent achondroplasia-like dwarfism and rescues the Fgfr3-deficient mouse phenotype. Genes & development. 2004 Feb 1;18(3):290–305. doi: 10.1101/gad.1179104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Provot S, Nachtrab G, Paruch J, Chen AP, Silva A, Kronenberg HM. A-raf and B-raf are dispensable for normal endochondral bone development, and parathyroid hormone-related peptide suppresses extracellular signal-regulated kinase activation in hypertrophic chondrocytes. Molecular and cellular biology. 2008 Jan;28(1):344–57. doi: 10.1128/MCB.00617-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsushita T, Chan YY, Kawanami A, Balmes G, Landreth GE, Murakami S. Extracellular signal-regulated kinase 1 (ERK1) and ERK2 play essential roles in osteoblast differentiation and in supporting osteoclastogenesis. Molecular and cellular biology. 2009 Nov;29(21):5843–57. doi: 10.1128/MCB.01549-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sebastian A, Matsushita T, Kawanami A, Mackem S, Landreth GE, Murakami S. Genetic inactivation of ERK1 and ERK2 in chondrocytes promotes bone growth and enlarges the spinal canal. Journal of orthopaedic research : official publication of the Orthopaedic Research Society. 2011 Mar;29(3):375–9. doi: 10.1002/jor.21262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997 Sep 25;389(6649):349–52. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- 26.Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- 27.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009 Jan;10(1):32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005 Dec 19;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 29.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009 Aug 14;325(5942):834–40. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 30.Jeon EJ, Lee KY, Choi NS, Lee MH, Kim HN, Jin YH, et al. Bone morphogenetic protein-2 stimulates Runx2 acetylation. J Biol Chem. 2006 Jun 16;281(24):16502–11. doi: 10.1074/jbc.M512494200. [DOI] [PubMed] [Google Scholar]
- 31.Juan LJ, Shia WJ, Chen MH, Yang WM, Seto E, Lin YS, et al. Histone deacetylases specifically down-regulate p53-dependent gene activation. J Biol Chem. 2000 Jul 7;275(27):20436–43. doi: 10.1074/jbc.M000202200. [DOI] [PubMed] [Google Scholar]
- 32.Luo J, Su F, Chen D, Shiloh A, Gu W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature. 2000 Nov 16;408(6810):377–81. doi: 10.1038/35042612. [DOI] [PubMed] [Google Scholar]
- 33.Yuan ZL, Guan YJ, Chatterjee D, Chin YE. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science. 2005 Jan 14;307(5707):269–73. doi: 10.1126/science.1105166. [DOI] [PubMed] [Google Scholar]
- 34.Bradley EW, Carpio LR, van Wijnen AJ, McGee-Lawrence ME, Westendorf JJ. Histone Deacetylases in Bone Development and Skeletal Disorders. Physiol Rev. 2015 Oct;95(4):1359–81. doi: 10.1152/physrev.00004.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lakshmaiah KC, Jacob LA, Aparna S, Lokanatha D, Saldanha SC. Epigenetic therapy of cancer with histone deacetylase inhibitors. J Cancer Res Ther. 2014 Jul-Sep;10(3):469–78. doi: 10.4103/0973-1482.137937. [DOI] [PubMed] [Google Scholar]
- 36.Dinarello CA, Fossati G, Mascagni P. Histone deacetylase inhibitors for treating a spectrum of diseases not related to cancer. Mol Med. 2011 May-Jun;17(5–6):333–52. doi: 10.2119/molmed.2011.00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Di Renzo F, Broccia ML, Giavini E, Menegola E. Relationship between embryonic histonic hyperacetylation and axial skeletal defects in mouse exposed to the three HDAC inhibitors apicidin, MS-275, and sodium butyrate. Toxicol Sci. 2007 Aug;98(2):582–8. doi: 10.1093/toxsci/kfm115. [DOI] [PubMed] [Google Scholar]
- 38.Di Renzo F, Cappelletti G, Broccia ML, Giavini E, Menegola E. Boric acid inhibits embryonic histone deacetylases: a suggested mechanism to explain boric acid-related teratogenicity. Toxicol Appl Pharmacol. 2007 Apr 15;220(2):178–85. doi: 10.1016/j.taap.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 39.Menegola E, Di Renzo F, Broccia ML, Prudenziati M, Minucci S, Massa V, et al. Inhibition of histone deacetylase activity on specific embryonic tissues as a new mechanism for teratogenicity. Birth Defects Res B Dev Reprod Toxicol. 2005 Oct;74(5):392–8. doi: 10.1002/bdrb.20053. [DOI] [PubMed] [Google Scholar]
- 40.Eikel D, Lampen A, Nau H. Teratogenic effects mediated by inhibition of histone deacetylases: evidence from quantitative structure activity relationships of 20 valproic acid derivatives. Chem Res Toxicol. 2006 Feb;19(2):272–8. doi: 10.1021/tx0502241. [DOI] [PubMed] [Google Scholar]
- 41.Paradis FH, Hales BF. The Effects of Class-Specific Histone Deacetylase Inhibitors on the Development of Limbs During Organogenesis. Toxicol Sci. 2015 Nov;148(1):220–8. doi: 10.1093/toxsci/kfv174. [DOI] [PubMed] [Google Scholar]
- 42.McGee-Lawrence ME, McCleary-Wheeler AL, Secreto FJ, Razidlo DF, Zhang M, Stensgard BA, et al. Suberoylanilide hydroxamic acid (SAHA; vorinostat) causes bone loss by inhibiting immature osteoblasts. Bone. 2011 May 1;48(5):1117–26. doi: 10.1016/j.bone.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pratap J, Akech J, Wixted JJ, Szabo G, Hussain S, McGee-Lawrence ME, et al. The histone deacetylase inhibitor, vorinostat, reduces tumor growth at the metastatic bone site and associated osteolysis, but promotes normal bone loss. Mol Cancer Ther. 2010 Dec;9(12):3210–20. doi: 10.1158/1535-7163.MCT-10-0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sato Y, Kondo I, Ishida S, Motooka H, Takayama K, Tomita Y, et al. Decreased bone mass and increased bone turnover with valproate therapy in adults with epilepsy. Neurology. 2001 Aug 14;57(3):445–9. doi: 10.1212/wnl.57.3.445. [DOI] [PubMed] [Google Scholar]
- 45.Vestergaard P, Rejnmark L, Mosekilde L. Fracture risk associated with use of antiepileptic drugs. Epilepsia. 2004 Nov;45(11):1330–7. doi: 10.1111/j.0013-9580.2004.18804.x. [DOI] [PubMed] [Google Scholar]
- 46.Guo CY, Ronen GM, Atkinson SA. Long-term valproate and lamotrigine treatment may be a marker for reduced growth and bone mass in children with epilepsy. Epilepsia. 2001 Sep;42(9):1141–7. doi: 10.1046/j.1528-1157.2001.416800.x. [DOI] [PubMed] [Google Scholar]
- 47.Schroeder TM, Kahler RA, Li X, Westendorf JJ. Histone deacetylase 3 interacts with runx2 to repress the osteocalcin promoter and regulate osteoblast differentiation. J Biol Chem. 2004 Oct 1;279(40):41998–2007. doi: 10.1074/jbc.M403702200. [DOI] [PubMed] [Google Scholar]
- 48.Hesse E, Saito H, Kiviranta R, Correa D, Yamana K, Neff L, et al. Zfp521 controls bone mass by HDAC3-dependent attenuation of Runx2 activity. J Cell Biol. 2010 Dec 27;191(7):1271–83. doi: 10.1083/jcb.201009107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bhaskara S, Chyla BJ, Amann JM, Knutson SK, Cortez D, Sun ZW, et al. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol Cell. 2008 Apr 11;30(1):61–72. doi: 10.1016/j.molcel.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McGee-Lawrence ME, Bradley EW, Dudakovic A, Carlson SW, Ryan ZC, Kumar R, et al. Histone deacetylase 3 is required for maintenance of bone mass during aging. Bone. 2013 Jan;52(1):296–307. doi: 10.1016/j.bone.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Razidlo DF, Whitney TJ, Casper ME, McGee-Lawrence ME, Stensgard BA, Li X, et al. Histone deacetylase 3 depletion in osteo/chondroprogenitor cells decreases bone density and increases marrow fat. PloS one. 2010;5(7):e11492. doi: 10.1371/journal.pone.0011492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bradley EW, Carpio LR, Westendorf JJ. Histone deacetylase 3 suppression increases PH domain and leucine-rich repeat phosphatase (Phlpp)1 expression in chondrocytes to suppress Akt signaling and matrix secretion. J Biol Chem. 2013 Apr 5;288(14):9572–82. doi: 10.1074/jbc.M112.423723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carpio LR, Bradley EW, McGee-Lawrence ME, Weivoda MM, Poston DD, Dudakovic A, et al. Histone deacetylase 3 supports endochondral bone formation by controlling cytokine signaling and matrix remodeling. Science signaling. 2016;9(440):ra79. doi: 10.1126/scisignal.aaf3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gosset M, Berenbaum F, Thirion S, Jacques C. Primary culture and phenotyping of murine chondrocytes. Nat Protoc. 2008;3(8):1253–60. doi: 10.1038/nprot.2008.95. [DOI] [PubMed] [Google Scholar]
- 55.Nakamura E, Nguyen MT, Mackem S. Kinetics of tamoxifen-regulated Cre activity in mice using a cartilage-specific CreER(T) to assay temporal activity windows along the proximodistal limb skeleton. Dev Dyn. 2006 Sep;235(9):2603–12. doi: 10.1002/dvdy.20892. [DOI] [PubMed] [Google Scholar]
- 56.Bermudez O, Pages G, Gimond C. The dual-specificity MAP kinase phosphatases: critical roles in development and cancer. Am J Physiol Cell Physiol. 2010 Aug;299(2):C189–202. doi: 10.1152/ajpcell.00347.2009. [DOI] [PubMed] [Google Scholar]
- 57.Li C, Scott DA, Hatch E, Tian X, Mansour SL. Dusp6 (Mkp3) is a negative feedback regulator of FGF-stimulated ERK signaling during mouse development. Development. 2007 Jan;134(1):167–76. doi: 10.1242/dev.02701. [DOI] [PMC free article] [PubMed] [Google Scholar]