

Abstract
The pathogenesis of acute aortic dissection (AAD) complicated acute lung injury (ALI) is not currently well defined. At present, no effective animal model has been established for AAD complicated ALI, which hinders research and development of an appropriate treatment regimen for the concurrent conditions. The aim of the present study was to evaluate the therapeutic effects of bindarit (Bnd), an indazolic derivative, on the production of monocyte chemoattractant protein (MCP)-1 in angiotensin II (AngII)-induced complicated ALI in rats. An AAD complicated ALI rat model was established using aminopropionitrile (BAPN) and AngII. The pathological features of AAD complicated ALI were assessed via biochemical and histopathological evaluations. AngII-stimulated human pulmonary microvascular endothelial cells (hPMVECs) were used to assess the effects of Bnd on MCP-1 expression. Western blot analysis was performed to analyze the expression of proteins that may be associated with the process. AAD complicated ALI was established following BAPN and AngII interference, and a massive accumulation of macrophages was observed in the lung tissues of the study rats. Bnd was able to significantly attenuate the incidence of AAD complicated ALI (P<0.05), and significantly inhibit the accumulation of macrophages (P<0.05). The overexpression of MCP-1 induced by AngII in hPMVECs was significantly inhibited by Bnd (P<0.05), which may be associated with downregulation of the classical nuclear factor-κB pathway. Bnd was able to attenuate the incidence of AAD complicated ALI, and inhibit the accumulation of macrophages in vivo. These findings provide a basis for future applications of Bnd as part of a therapeutic treatment schedule for aortic dissection complicated lung injury.
Keywords: acute aortic dissection, lung injury, nuclear factor-κB pathway, bindarit
Introduction
Acute aortic dissection (AAD), which is the most frequent and severe manifestation of acute aortic syndrome, is one of the primary causes of cardiovascular disease-related mortality worldwide (1–3). In clinical practice, a large number of AAD patients (~35%) are diagnosed with concurrent acute lung injury (ALI) presenting with hypoxemia prior to surgery (2). Concurrent ALI prior to surgery is a risk factor for postoperative acute respiratory distress syndrome, and patients with concurrent AAD and ALI have been demonstrated to have a poor prognosis (4).
The pathogenesis of AAD complicated ALI is not currently well understood. At present, animal models of AAD are typically established based on surgical methods or the β-aminopropionitrile (BAPN) treatment (5). Such models contribute to the investigation on the pathogenesis of AAD; however, no animal model has yet been established for AAD complicated ALI, which hinders the research and development of an appropriate treatment regimen for the concurrent conditions. There is therefore a need to establish an animal model of AAD complicated ALI in order to elucidate the exact mechanism of and appropriate treatment regimen for these concurrent diseases. Bindarit (Bnd) is able to downregulate the gene expression of monocyte chemoattractant protein-1 (MCP-1) and the expression of the MCPs cluster members such as MCP2/CCL8 and MCP-3/CCL7 (6). These processes promote prolonged and sustained inflammation through monocyte-macrophage recruitment, which serves crucial roles in the homeostatic positioning and trafficking of immune cells (6). In the present study, Bnd was demonstrated to reduce the incidence of AAD complicated ALI. In vitro experiments revealed the effects of Bnd on AAD complicated ALI may be associated with the inhibition of MCP1 mRNA and protein expression, as well as modulating the NF-κB pathway.
Materials and methods
In vivo experiments
Development of AAD complicated lung injury model in rats
For the animal model, 3-week-old male Sprague-Dawley rats provided by the Animal Center of Wuhan University (Wuhan, China) were randomly divided into three groups: i) Control group (n=7), provided with a standard laboratory diet for 4 weeks; ii) BAPN group (n=7), provided with a standard laboratory diet and administered with 0.25% BAPN (M27603; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) dissolved in drinking water (0.1 g/kg per day) for 4 weeks; and iii) BAPN+AngII group (n=7), provided with a standard laboratory diet and administered with 0.25% BAPN dissolved in drinking water (0.1 g/kg per day) for 4 weeks, together with subcutaneous implantation of AngII (1 µg/kg per minute; Sigma-Aldrich; Merck KGaA) via osmotic mini pumps (ALZET Osmotic Pumps; DURECT Corp, Cupertino, CA, USA) as previously described (7). Rats were sacrificed 24 h post-treatment following administration of 1% phenobarbital as an anesthetic. All animal handling was performed in accordance with the Wuhan Directive for Animal Research and the current Guidelines for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH publication no. 85-23, revised 1996). The animal study was approved by the Ethical Committee of the Renmin Hospital of Wuhan University.
Roles of macrophages in the pathogenesis of AAD complicated ALI
Following the establishment of the animal model, AAD was confirmed by conventional pathological examination. Rats exhibiting any of the following were deemed to have ALI: PaO2/FiO2 ≤300 mmHg, presence of edema in the alveolar septum, or infiltration of inflammatory cells in the alveolar interstitium. To confirm the role of macrophages in AAD complicated ALI, Bnd (cat. no. S3032; Selleck Chemicals, Houston, TX, USA), which is an inhibitor of MCP-1 reported to be associated with the recruitment of macrophages (8), was administered. A total of 5 groups were used in the present study: Control group (n=7), BAPN group (n=7), BAPN+AngII group (n=7), BAPN+AngII+Bnd group (n=7), and Bnd group (n=7). The dose of Bnd was 200 mg/kg per day for 2 days prior to AngII administration until sacrifice (8). Following sacrifice, the accumulation of macrophages in the lung tissues was assessed in triplicate.
Histopathological examination
Lung tissues were harvested following sacrifice and samples were fixed and embedded. The fixing step was performed at room temperature as follows: Dehydrated alcohol (Sinopharm Chemical Reagent Co., Ltd., Shanghai, China) combined with Xylene (Sinopharm Chemical Reagent Co., Ltd.; vol/vol=1:1) for 10 min, followed by Xylene I (Sinopharm Chemical Reagent Co., Ltd.) for 10 min and xylene II (Sinopharm Chemical Reagent Co., Ltd.) for 7 min. The samples were embedded in paraffin at 60°C for 30 min using neutral resin (Sinopharm Chemical Reagent Co., Ltd.). Hematoxylin and eosin (H&E) staining, and immunohistostaining were performed as previously described (9). Tissues were observed using a CKX41SF light microscope (Olympus Corporation, Tokyo, Japan) and images were captured to assess the histopathological changes in lung tissues and the expression of cluster of differentiation (CD) 68.
Determination of methane dicarboxylic aldehyde (MDA) and superoxide dismutase (SOD)
Lung tissues were homogenated and placed in a water bath at 95°C for 40 min. Tissues were cooled to room temperature and the mixture was subsequently centrifuged at 4,865 × g for 10 min at 4°C. The supernatant was subsequently collected and the absorbance of MDA was determined at 532 nm. The absorbance of SOD was determined at 550 nm.
Evaluation of pulmonary wet/dry weight ratio (W/D)
Immediately following sacrifice, lung tissues were harvested and the surface was dried using a sterilized filter paper. Tissues were weighed to determine the wet weight and subsequently stored in an oven at 100°C for 24 h until the weight was constant. The W/D ratio was calculated.
In vitro experiments
To investigate the potential mechanism of in vivo results, in vitro experiments were performed based on cultured human pulmonary microvascular endothelial cells (hPMVECs).
Experimental design
hPMVECs of passages 2–8 were purchased from Wuhan Biofavor Biotech Services Co., Ltd. (Wuhan, China; cat. no. CP-H001). Cells were cultured in RPMI-1640 medium (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Selleck Chemicals) 1% penicillin/streptomycin and 0.5% fungizone (Invitrogen; Thermo Fisher Scientific, Inc.) and maintained at 37°C in a humidified atmosphere containing 5% CO2 for 2 days. Cell growth was arrested by replacing the medium with FBS-free RPMI-1640 for 24 h. Cells were subsequently incubated at 25°C in low-serum RPMI-1640 supplemented with 2% FBS, AngII (1 µM; Sigma-Aldrich; Merck KGaA) (10), and Bnd with or without AngII for 12 h. The MCP-1 inhibitor Bnd (7 mg/ml, dissolved in 0.5% CMC) was added for 30 min prior to the addition of AngII (11).
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated from hPMVECs using TRIzol reagent (Sigma-Aldrich; Merck KGaA) according to the manufacturer's protocol. cDNA synthesis was performed using the SuperScript commercial kit (Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol. PCR was performed using an ABI 7500 system (Applied Biosystems; Thermo Fisher Scientific, Inc. The specific primers were designed from the coding regions of human MCP-1 as follows: Forward, 5′-AACTGAAGCTCGCACTCTCG-3′ and reverse, 5′-TCAGCACAGATCTCCTTGGC-3′. GAPDH was used as a control, and the primer sequences were as follows: Forward, 5′-ACCACAGTCCATGCCATCAC-3′ and reverse, 5′-TCCHCCACCCTGTTGCTGTA-3′. A total reaction volume of 25 µl was used for PCR, containing 10x buffer, 2 µl DNA temperate, 20 pmol/l GADPH, 50 pmol/l each primer, 0.5 µl Taq DNA polymerase (5 u/µl). The PCR conditions were 94°C for 5 min followed by 34 cycles at 94°C for 30 sec, 55°C for 30 sec and 72°C for 60 sec. The PCR product was sequenced and the amplified productions of human MCP-1 and GAPDH were 258 and 452 bp, respectively. The experiment was performed at least in triplicate. Quantification was performed using the 2−∆∆Cq method (12).
Western blotting
Cells were harvested using Commercial Cell Lysis buffer (Beijing BioDev-Tech, Beijing, China) at 72 h and centrifuged at 16,000 × g for 5 min at 4°C to extract the protein according to the manufacturer's protocol. The cells were washed using PBS buffer at 4°C three times for 5 min. Protein concentrations were determined via bicinchoninic acid assay using a commercial kit (BCAP-1-W; Suzhou Coming Chengye Medical Technology Co., Ltd., Suzhou, China) for a minimum of three times (13). Proteins (40 µg per lane) were separated by 10% SDS-PAGE. The expressions of MCP-1, IkBα, phosphorylated (P)-IkBα, transcription factor p65 (p65) and P-p65 were detected using a standard western blotting protocol. The transferred polyvinylidene fluoride membrane was blocked with 10% skimmed milk for 1 h at room temperature. The membrane was subsequently incubated with primary antibodies for MCP-1 (1:1,000; BA1843-2; Wuhan Boster Biotechnology, Ltd., Wuhan, China), IκBα (1:2,000; 4812, Cell Signaling Technology, Inc., Danvers, MA, USA), P-IκBα (Ser32/36; 1:1,000; 5209, Cell Signaling Technology, Inc.), p65/Rel-A (1:1,000; 4764; Cell Signaling Technology, Inc.), P-p65/Rel-A (Ser 536; 1:1,000; 4025; Cell Signaling Technology, Inc.), and β-actin (1:700; sc-81178, Santa Cruz Biotechnology, Inc., Dallas, TX, USA) overnight at 4°C, respectively. Membranes were subsequently incubated with the horseradish peroxidase-conjugated secondary antibody (1:5,000; ZB-2301, OriGene Technologies, Inc., Beijing, China) for 1 h at room temperature and the immunoblotting signals were visualized using a Western Luminescent Detection kit (Vigorous Biotechnology, Beijing, China).
Statistical analysis
All data are expressed as the mean ± standard error of the mean. All experiments were performed with at least 6 independent hPMVEC cultures. Statistical analyses were performed using one-way analysis of variance using SPSS 17.0 software (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Establishment of AAD complicated lung injury model
A significantly higher incidence of lung injury was observed in the BAPN+AngII group compared with the BAPN group (P<0.05, Fig. 1). H&E staining revealed massive infiltration of inflammatory cells in the lung tissues and injury to the pulmonary alveoli in the BAPN+AngII group. Red blood cells, inflammatory cells and exudate were also observed in the alveolar space (Fig. 2). The lung MDA content and W/D ratio in the BAPN+AngII group were significantly elevated compared with the control (both P<0.01) and there was a significant decrease in SOD activity (P<0.01; Table I). These findings suggest that lung injury was successfully induced in the BAPN+AngII group.
Figure 1.
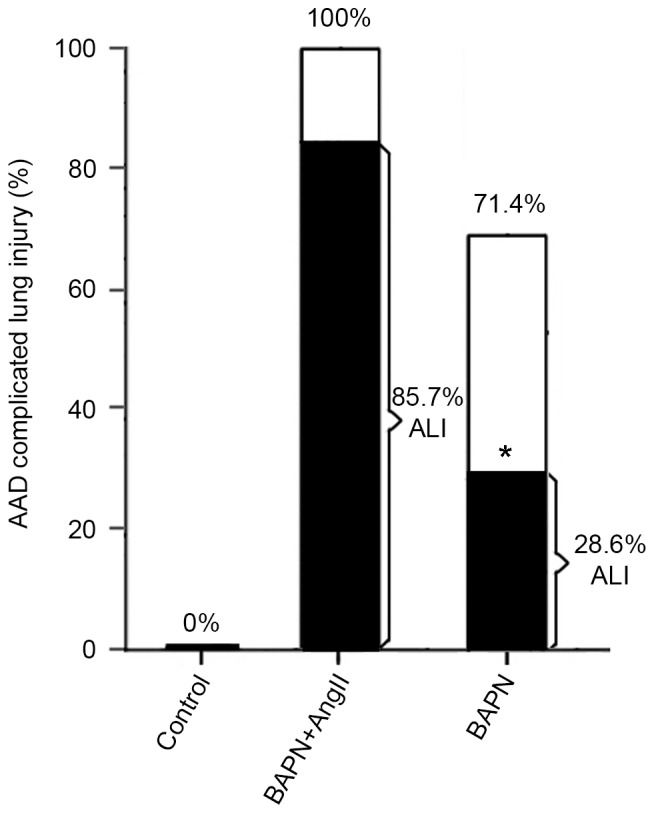
The incidence of AAD complicated ALI in each group. *P<0.05 vs. the BAPN+AngII group. AAD, acute aortic dissection; ALI, acute lung injury; BAPN, β-aminopropionitrile; AngII, angiotensin II.
Figure 2.

Pathological features of lung tissue from rats in the (A) control group, (B) BAPN+AngII group and (C) BAPN group. Massive edema, inflammation and cell infiltration were observed in the BAPN+AngII group. Magnification, ×200. BAPN, β-aminopropionitrile; AngII, angiotension II.
Table I.
MDA, SOD and W/D in each group.
| Group | MDA (nmol/mgprot) | SOD (U/mgprot) | W/D |
|---|---|---|---|
| Normal control | 1.34±0.31 | 48.19±4.24 | 3.25±0.37 |
| BAPN | 1.77±0.26 | 41.74±2.83 | 3.48±0.54 |
| BAPN+AngII | 4.84±0.49a | 20.70±3.17a | 4.57±0.66a |
P<0.01 vs. control. MDA, methane dicarboxylic aldehyde; SOD, superoxide dismutase; W/D, wet/dry ratio; BAPN, aminopropionitrile; AngII, angiotension II.
Bnd induces a decrease of AAD complicated ALI
Following infusion of AngII, a marked accumulation of macrophages was observed in the lung tissues of rats with lung injury, which confirmed the recruitment of macrophages in the pathogenesis of AAD complicated ALI. However, the accumulation of monocytes and macrophages was markedly inhibited following the administration of Bnd. To investigate the roles of MCP-1 in the pathogenesis of AAD complicated ALI, Bnd was administered to the BAPN+AngII group. There was no marked decrease in the incidence of AAD following the administration of Bnd; however, CD68 staining revealed that the accumulation of macrophages was markedly attenuated (Fig. 3). Furthermore, the incidence of lung injury was significantly decreased by 42.8% (85.7% vs. 42.9%, P<0.05; Fig. 4). This suggests that Bnd was able to attenuate AAD complicated ALI in rats via modulating the recruitment of macrophages.
Figure 3.

Immunohistochemical analysis of cluster of differentiation 68 in the control, BAPN+AngII, BAPN+AngII+Bnd and Bnd groups. Magnification, ×400. BAPN, β-aminopropionitrile; AngII, angiotension II; Bnd, bindarit; H&E, hematoxylin and eosin.
Figure 4.
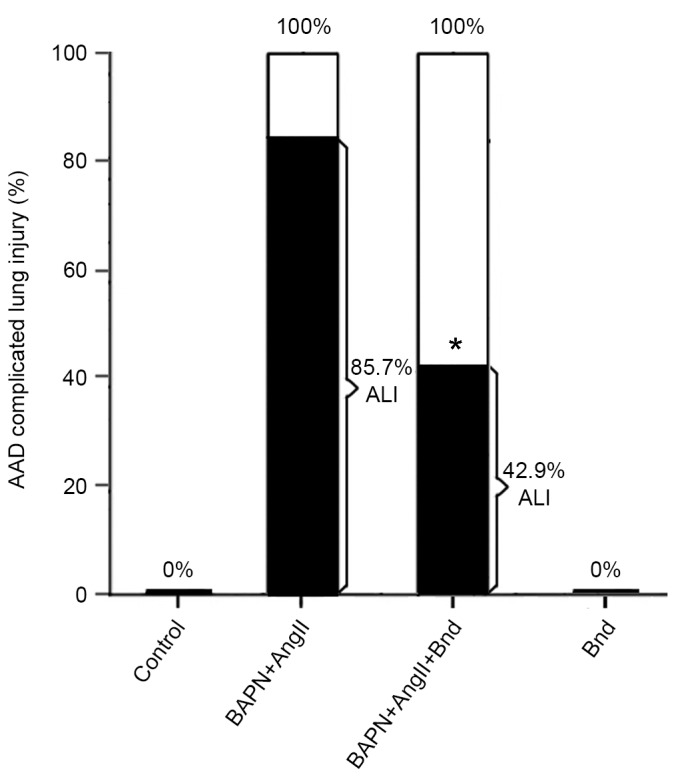
Incidence of AAD complicated ALI in different groups. The incidence of AAD complicated ALI was significantly reduced by Bnd. *P<0.05 vs. the BAPN+AngII group. AAD, acute aortic dissection; ALI, acute lung injury; Bnd, bindarit; BAPN, β-aminopropionitrile; AngII, angiotension II.
Bnd induces a decrease of MDA and increase of SOD in lung tissues
The MDA and W/D ratio were significantly decreased in the BAPN+AngII+Bnd group compared with the BAPN+AngII group (P<0.01; Table II). Furthermore, there was a significant increase in SOD levels in the BAPN+AngII+Bnd group compared with the BAPN+AngII group (P<0.01; Table II). These results suggest that massive endogenous SOD was consumed during the oxidative stress, which resulted in a decrease in the antioxidant capacity and formation of lipid peroxides (e.g. MDA).
Table II.
MDA, SOD and W/D in each group.
| Group | MDA (nmol/mgprot) | SOD (U/mgprot) | W/D |
|---|---|---|---|
| Normal control | 1.34±0.31 | 48.19±4.24 | 3.25±0.37 |
| BAPN+AngII | 4.84±0.49a | 20.70±3.17a | 4.57±0.66a |
| BAPN+AngII+Bnd | 3.12±0.40b | 38.77±5.93b | 4.16±0.69b |
| Bnd | 1.31±0.39 | 49.53±5.88 | 3.18±0.34 |
P<0.01 vs. control
P<0.01 vs. BAPN+AngII. MDA, methane dicarboxylic aldehyde; SOD, superoxide dismutase; W/D, wet/dry ratio; BAPN, aminopropionitrile; AngII, angiotension II, Bnd, bindarit.
Bnd inhibits AngII-induced MCP-1 overexpression in hPMVECs
To investigate the potential mechanism of Bnd in the attenuation of AAD complicated ALI, the expression of MCP-1 mRNA and protein in hPMVECs was determined following stimulation of AngII. The results indicated AngII was able to induce MCP-1 mRNA and protein overexpression; however, administration of Bnd was able to significantly inhibit this effect (P<0.01; Fig. 5A and B).
Figure 5.

The effect of Bnd on the AngII-induced expression of MCP-1 (A) mRNA and (B) protein. *P<0.01 vs. AngII; **P<0.001 vs. AngII. Bnd, bindarit; AngII, angiotensin II; MCP-1, monocyte chemotactic protein 1.
Nuclear factor (NF)-κB pathway is associated with the MCP-1 expression mediated by Bnd
To investigate how Bnd is associated with the downregulation of MCP-1 expression in hPMVECs subject to AngII interference, the expressions of P-IkBα, IkBα, P-p65 and p65 were determined. Western blot analysis revealed that the expression of P-IkBα was significantly increased in the AngII group compared with the control group (P<0.05, Fig. 6). P-IkBα expression was significantly downregulated in the AngII+Bnd group compared with the AngII group (P<0.05; Fig. 6). Furthermore, the expression of P-p65 was significantly decreased in the AngII+Bnd group compared with the AngII group (P<0.05; Fig. 6). These results suggest that Bnd is able to inhibit the synthesis of MCP-1 via modulating the NF-κB pathway.
Figure 6.
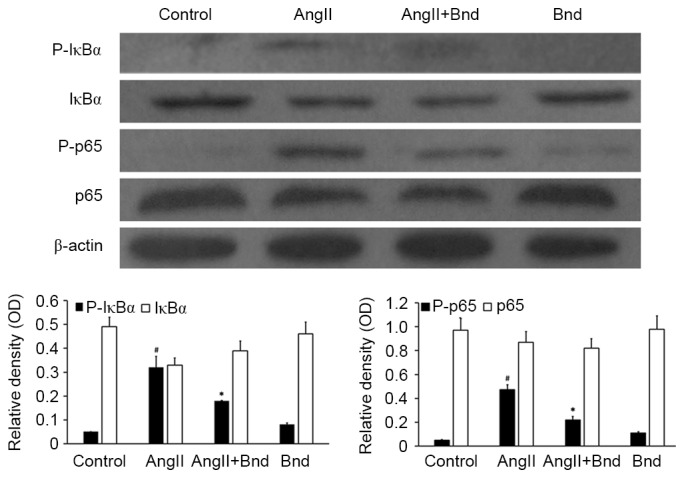
The effect of Bnd on the AngII-induced activation of the NF-κB pathway. Western blot analysis of IkBa and p65, and their respective phosphorylated isoforms was performed with β-actin as a loading control. #P<0.05 vs. control; *P<0.05 vs. AngII. Bnd, bindarit; AngII, angiotensin II; NF, nuclear factor; P, phosphorylated; p65, transcription factor p65.
Discussion
In the present study, a rat model of AAD complicated ALI was established based on a combination of AngII and BAPN treatment. In lung tissues harvested from the AAD complicated ALI rat model, a large number of macrophages were accumulated in the pulmonary mesenchymal tissues. This observation suggests that the AngII-induced macrophage infiltration may be associated with the onset of AAD complicated ALI. Furthermore, the results of the present study revealed a marked elevation in MDA and decrease in SOD in the lung tissues, which may reflect the extent of lung injury. It was speculated that changes in MDA and SOD may be associated with a higher consumption of SOD, which in turn results in the accumulation of MDA due to a decrease in the in vivo antioxidant capacity.
The number of macrophages was markedly lower in the BAPN+AngII+Bnd group compared with the BAPN+AngII group and the incidence of lung injury was also decreased. This suggests that Bnd was not able to attenuate the incidence of AAD, whereas it was able to attenuate the incidence of ALI via inhibiting the accumulation of macrophages in lung tissues. The anti-inflammatory activity of Bnd has previously been ascribed to its ability to impair monocyte/macrophage recruitment in inflamed tissues (14). In the present study, the effect of Bnd on the regulation of MCP-1 in AngII-induced hPMVECs was investigated to better define the molecular mechanisms involved. The results of the present study demonstrated that AngII was able to upregulate MCP-1 expression in hPMVECs, and that this was completely reversed by the administration of Bnd.
MCP-1, which is associated with the accumulation and migration of monocytes and macrophages, serves an important role in the pathogenesis and development of ALI. Several factors have been reported to be responsible for the expression of MCP-1 in endothelial cells, including the NF-κB signaling pathway, the tyrosine kinase-activator protein (AP)-1 pathway and the PKC signaling pathway (15,16). It is well known that several factors are able to contribute to the upregulation of MCP-1 by modulating different signal pathways (17,18). For example, AngII may stimulate the expression of MCP-1 by activating NF-κB and AP-1 and also the expression of c-fos and c-jun (18). Such a process is mediated by tyrosine protein kinase and mitogen activated protein kinase (18). Additionally, several factors including hydrogen peroxidase, apocynin (NADH/NADPH blocker), diphenylene iodonium and ectogeneous nitric oxide may inhibit the AngII-induced expression of MCP-1induced by AngII (19). As an inner κB sequence was identified in the promoter of MCP-1, the expression of MCP-1 is largely dependent on the NF-κB signaling pathway (20). Therefore, the AngII-induced MCP-1 expression in hPMVECs may be associated with the NF-κB signaling pathway.
Nuclear factor-kappa B (NF-κB), a ubiquitous transcription factor, typically presents as a heterodimer of 50 kDa (p50) and 65 kDa (p65) subunits (21). It is typically present in the cytoplasm in an inactive form through its association with IkBα, the prototype of a family of NF-κB inhibitory proteins (IκB) (21). Under in vitro stimulations, IκB was phosphorylated by the IκB kinase (IKK), which is recognized by ubiquitin (22). The complex was subsequently degraded by the 26S proteasome, based on which the nucleic localization sequences of the NF-κB were exposed (23). Upon entering the nucleus, NF-κB was able to bind with the κB sequence of the corresponding promoters, which triggered the transcription of certain genes (24–26). Extensive previous studies have revealed that activated NF-κB is a complex of p65 and p50 (27,28). This process is mediated by IKKβ and IκBα, particularly the phosphorylation of Ser32 and Ser36 in IκBα (27,28). In the present study, it was demonstrated that the expression of P-IκBα and P-p65 were elevated in hPMVECs following AngII interference. This indicates that AngII may be able to activate the NF-κB signaling pathway. However, this effect was completely reversed by administration of Bnd. On this basis, it is reasonable to conclude that Bnd may inhibit the expression of MCP-1 via inhibiting NF-κB activity.
In conclusion, AngII-mediated recruitment of macrophages serves an important role in the development of AAD complicated ALI. This process is associated with the expression of MCP-1, which was confirmed by the inhibition of macrophage recruitment following Bnd treatment. An in vitro study demonstrated that AngII was able to induce overexpression of MCP-1 in hPMVECs via activating the NF-κB signaling pathway. Furthermore, Bnd may inhibit the expression of MCP-1 in hPMVECs via inhibiting the activity of NF-κB.
References
- 1.Kamberi LS, Gorani DR, Karabulut AM, Beqiri AI, Mustafai AI. Follow-up of acute aortic dissection. Med Arh. 2011;65:89–91. [PubMed] [Google Scholar]
- 2.Bonser RS, Ranasinghe AM, Loubani M, Evans JD, Thalji NM, Bachet JE, Carrel TP, Czerny M, Di Bartolomeo R, Grabenwöger M, et al. Evidence, lack of evidence, controversy, and debate in the provision and performance of the surgery of acute type A aortic dissection. J Am Coll Cardiol. 2011;58:2455–2474. doi: 10.1016/j.jacc.2011.06.067. [DOI] [PubMed] [Google Scholar]
- 3.LeMaire SA, Russell L. Epidemiology of thoracic aortic dissection. Nat Rev Cardiol. 2011;8:103–113. doi: 10.1038/nrcardio.2010.187. [DOI] [PubMed] [Google Scholar]
- 4.Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med. 2000;342:1301–1308. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- 5.Nagashima H, Uto K, Sakomura Y, Aoka Y, Sakuta A, Aomi S, Hagiwara N, Kawana M, Kasanuki H. An angiotensin-converting enzyme inhibitor, not an angiotensin II type-1 receptor blocker, prevents beta-aminopropionitrile monofumarate-induced aortic dissection in rats. J Vasc Surg. 2002;36:818–823. doi: 10.1067/mva.2002.126091. [DOI] [PubMed] [Google Scholar]
- 6.Luster AD, Rothenberg ME. Role of the monocyte chemoattractant protein and eotaxin subfamily of chemokines in allergic inflammation. J Leukoc Biol. 1997;62:620–633. doi: 10.1002/jlb.62.5.620. [DOI] [PubMed] [Google Scholar]
- 7.Kurihara T, Shimizu-Hirota R, Shimoda M, Adachi T, Shimizu H, Weiss SJ, Itoh H, Hori S, Aikawa N, Okada Y. Neutrophil-derived matrix metalloproteinase 9 triggers acute aortic dissection. Circulation. 2012;126:3070–3080. doi: 10.1161/CIRCULATIONAHA.112.097097. [DOI] [PubMed] [Google Scholar]
- 8.Guglielmotti A, D'Onofrio E, Coletta I, Aquilini L, Milanese C, Pinza M. Amelioration of rat adjuvant arthritis by therapeutic treatment with bindarit, an inhibitor of MCP-1 and TNF-alpha production. Inflamm Res. 2002;51:252–258. doi: 10.1007/PL00000301. [DOI] [PubMed] [Google Scholar]
- 9.Barbash IM, Chouraqui P, Baron J, Feinberg MS, Etzion S, Tessone A, Miller L, Guetta E, Zipori D, Kedes LH, et al. Systemic delivery of bone marrow-derived mesenchymal stem cells to the infarcted myocardium: Feasibility, cell migration, and body distribution. Circulation. 2003;108:863–868. doi: 10.1161/01.CIR.0000084828.50310.6A. [DOI] [PubMed] [Google Scholar]
- 10.Lee YH, Mungunsukh O, Tutino RL, Marquez AP, Day RM. Angiotensin-II-induced apoptosis requires regulation of nucleolin and Bcl-xL by SHP-2 in primary lung endothelial cells. J cell Sci. 2010;123:1634–1643. doi: 10.1242/jcs.063545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mirolo M, Fabbri M, Sironi M, Vecchi A, Guglielmotti A, Mangano G, Biondi G, Locati M, Mantovani A. Impact of the anti-inflammatory agent bindarit on the chemokinome: Selective inhibition of the monocyte chemotactic proteins. Eur Cytokine Netw. 2008;19:119–122. doi: 10.1684/ecn.2008.0133. [DOI] [PubMed] [Google Scholar]
- 12.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-tie quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 13.Morton RE, Evans TA. Modification of the bicinchoninic acid protein assay to eliminate lipid interference in determining lipoprotein protein content. Anal Biochem. 1992;204:332–334. doi: 10.1016/0003-2697(92)90248-6. [DOI] [PubMed] [Google Scholar]
- 14.Sironi M, Guglielmotti A, Polentarutti N, Fioretti F, Milanese C, Romano M, Vigini C, Coletta I, Sozzani S, Bernasconi S, et al. A small synthetic molecule capable of preferentially inhibiting the production of the CC chemokine monocyte chemotactic protein-1. Eur Cytokine Netw. 1999;10:437–442. [PubMed] [Google Scholar]
- 15.Burysek L, Syrovets T, Simmet T. The serine protease plasmin triggers expression of MCP-1 and CD40 in human primary monocytes via activation of p38 MAPK and janus kinase (JAK)/STAT signaling pathways. J Biol Chem. 2002;277:33509–33517. doi: 10.1074/jbc.M201941200. [DOI] [PubMed] [Google Scholar]
- 16.Matsuo H, Tamura M, Kabashima N, Serino R, Tokunaga M, Shibata T, Matsumoto M, Aijima M, Oikawa S, Anai H, Nakashima Y. Prednisolone inhibits hyperosmolarity-induced expression of MCP-1 via NF-kappaB in peritoneal mesothelial cells. Kidney Int. 2006;69:736–746. doi: 10.1038/sj.ki.5000131. [DOI] [PubMed] [Google Scholar]
- 17.Funakoshi Y, Ichiki T, Shimokawa H, Egashira K, Takeda K, Kaibuchi K, Takeya M, Yoshimura T, Takeshita A. Rho-kinase mediates angiotensin II-induced monocyte chemoattractant protein-1 expression in rat vascular smooth muscle cells. Hypertension. 2001;38:100–104. doi: 10.1161/01.HYP.38.1.100. [DOI] [PubMed] [Google Scholar]
- 18.Leach HG, Chrobak I, Han R, Trojanowska M. Endothelial cells recruit macrophages and contribute to a fibrotic milieu in bleomycin lung injury. Am J Respir Cell Mol Biol. 2013;49:1093–1101. doi: 10.1165/rcmb.2013-0152OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tanifuji C, Suzuki Y, Geot WM, Horikoshi S, Sugaya T, Ruiz-Ortega M, Egido J, Tomino Y. Reactive oxygen species-mediated signaling pathways in angiotensin II-induced MCP-1 expression of proximal tubular cells. Antioxid Redox Signal. 2005;7:1261–1268. doi: 10.1089/ars.2005.7.1261. [DOI] [PubMed] [Google Scholar]
- 20.Goebeler M, Gillitzer R, Kilian K, Utzel K, Bröcker EB, Rapp UR, Ludwig S. Multiple signaling pathways regulate NF-kappaB-dependent transcription of the monocyte chemoattractant protein-1 gene in primary endothelial cells. Blood. 2001;97:46–55. doi: 10.1182/blood.V97.1.46. [DOI] [PubMed] [Google Scholar]
- 21.Wen Y, Yang S, Liu R, Perez E, Yi KD, Koulen P, Simpkins JW. Estrogen attenuates nuclear factor-kappa B activation induced by transient cerebral ischemia. Brain Res. 2004;1008:147–154. doi: 10.1016/j.brainres.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 22.Haase R, Kirschning CJ, Sing A, Schröttner P, Fukase K, Kusumoto S, Wagner H, Heesemann J, Ruckdeschel K. A dominant role of Toll-like receptor 4 in the signaling of apoptosis in bacteria-faced macrophages. J Immunol. 2003;171:4294–4303. doi: 10.4049/jimmunol.171.8.4294. [DOI] [PubMed] [Google Scholar]
- 23.Li W, Zhang X, Olumi AF. MG-132 sensitizes TRAIL-resistant prostate cancer cells by activating c-Fos/c-Jun heterodimers and repressing c-FLIP(L) Cancer Res. 2007;67:2247–2255. doi: 10.1158/0008-5472.CAN-06-3793. [DOI] [PubMed] [Google Scholar]
- 24.Baeuerle PA, Baltimore D. NF-kappa B: Ten years after. Cell. 1996;87:13–20. doi: 10.1016/S0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 25.Van Huffel S, Delaei F, Heyninck K, De Valck D, Beyaert R. Identification of a novel A20-binding inhibitor of nuclear factor-kappa B activation termed ABIN-2. J Biol Chem. 2001;276:30216–30223. doi: 10.1074/jbc.M100048200. [DOI] [PubMed] [Google Scholar]
- 26.Shambharkar PB, Blonska M, Pappu BP, Li H, You Y, Sakurai H, Darnay BG, Hara H, Penninger J, Lin X. Phosphorylation and ubiquitination of the IkappaB kinase complex by two distinct signaling pathways. EMBO J. 2007;26:1794–1805. doi: 10.1038/sj.emboj.7601622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Minhajuddin M, Fazal F, Bijli KM, Amin MR, Rahman A. Inhibition of mammalian target of rapamycin potentiates thrombin-induced intercellular adhesion molecule-1 expression by accelerating and stabilizing NF-kappa B activation in endothelial cells. J Immunol. 2005;174:5823–5829. doi: 10.4049/jimmunol.174.9.5823. [DOI] [PubMed] [Google Scholar]
- 28.Ten RM, McKinstry MJ, Trushin SA, Asin S, Paya CV. The Signal Transduction Pathway of CD23 (Fc epsilon RIIb) targets I kappa B kinase. J Immunol. 1999;163:3851–3857. [PubMed] [Google Scholar]