

Abstract
Chromatin immunoprecipitation (ChIP) is a powerful method to determine protein binding to chromatin DNA. Fiber-rich skeletal muscle, however, has been a challenge for ChIP due to technical difficulty in isolation of high-quality nuclei with minimal contamination of myofibrils. Previous protocols have attempted to purify nuclei before cross-linking, which incurs the risk of altered DNA-protein interaction during the prolonged nuclei preparation process. In the current protocol, we first cross-linked the skeletal muscle tissue collected from mice, and the tissues were minced and sonicated. Since we found that ultracentrifugation was not able to separate nuclei from myofibrils using cross-linked muscle tissue, we devised a sequential filtration procedure to obtain high-quality nuclei devoid of significant myofibril contamination. We subsequently prepared chromatin by using an ultrasonicator, and ChIP assays with anti-BMAL1 antibody revealed robust circadian binding pattern of BMAL1 to target gene promoters. This filtration protocol constitutes an easily applicable method to isolate high-quality nuclei from cross-linked skeletal muscle tissue, allowing consistent sample processing for circadian and other time-sensitive studies. In combination with next-generation sequencing (NGS), our method can be deployed for various mechanistic and genomic studies focusing on skeletal muscle function.
Keywords: Biochemistry, Issue 125, Skeletal muscle, cross-linking, nuclei isolation, chromatin immunoprecipitation, sequential filtration, circadian time
Introduction
Skeletal muscle plays important roles in physiology and behavior. The multi-nucleated muscle fiber consists of myofibrils where actin and myosin form functional units called sarcomeres to generate contractile force. Skeletal muscle is also the largest metabolic organ in the body, accounting for >80% postprandial glucose intake and regulating insulin response and metabolic homeostasis1,2. Muscle physiology and metabolism are closely regulated by the circadian clock, an intrinsic biological timer3,4,5,6. For example, the skeletal muscle-specific deletion of Bmal1, one of the core circadian clock components, resulted in insulin resistance and diminished glucose uptake in skeletal muscle, and the animals were found to develop type 2 diabetes7. In addition, skeletal muscle is also increasingly being appreciated as an endocrine organ8, secreting myokines to regulate systemic metabolism and physiology. Mechanistic studies are required to fully understand these regulatory functions in skeletal muscle.
ChIP is a powerful approach to delineate promoter recruitment of DNA binding proteins. ChIP was initially developed to identify nucleosome organization on chromatin DNA9,10. A variety of methods have since been reported to cross-link proteins and chromatin DNA using formaldehyde, dimethyl sulfate or ultraviolet irradiation (UV)11,12. Formaldehyde cross-linking is the most commonly used, preserving chromatin structure and DNA-protein interactions9,13,14. Cross-linked chromatin is shredded by sonication and immunoprecipitated with antibody against the particular DNA binding protein of interest15,16. In recent years, ChIP-sequencing (ChIP-seq), a method combining ChIP with NGS, has been developed to interrogate genome-wide transcription factor binding17, and in some cases to monitor dynamic changes over a time course18,19,20. For example, circadian ChIP-seq studies have revealed a highly orchestrated sequence of genomic binding of circadian clock components and histone markers, which drives temporally precise gene expression throughout the ~ 24 h circadian cycle18.
Most available ChIP protocols are designed for soft tissues (e.g. liver, brain, etc.), and very few have been published for hard tissues including skeletal muscle. It is technically challenging to homogenize fiber-rich skeletal muscle and isolate high-quality nuclei21, especially for ChIP experiments which require cross-linking. In a recent muscle ChIP study22, satellite cells were separated from myofibers, and nuclei were prepared from both cell types through a prolonged procedure involving tissue digestion. The entire process took approximately three hours to complete before formaldehyde cross-linking was performed on isolated nuclei. While this procedure avoided cross-linking muscle fiber, which makes muscle tissue even more refractory to efficient homogenization, and was able to produce high-quality nuclei, the significant time-lag from tissue collection to nuclei cross-linking incurs the risk of altered DNA-protein interaction. In contrast, most studies performed cross-linking immediately after experimental treatment or tissue collection in order to preserve the real-time DNA-protein binding12. A second drawback of nuclei isolation before cross-linking is that it precludes time-sensitive applications such as circadian sample collection which typically occurs at 3 - 4 h intervals. Without cross-linking the nuclei, the isolation needs to proceed immediately after dissection, whereas cross-linked samples can be processed together after the entire time course is completed, thus ensuring greater experimental consistency.
Other protocols for nuclei isolation from uncrosslinked skeletal muscle have also been reported. Two studies described the use of gradient ultracentrifugation to separate nuclei from remaining myofibrils and cell debris23,24. While sucrose or colloidal gradient ultracentrifugation is effective with uncrosslinked muscle tissues, our experiments revealed that after crosslinking, gradient ultracentrifugation failed to separate nuclei from cell debris on the gradient.
We therefore developed a procedure to isolate high-quality nuclei using cross-linked mouse skeletal muscle tissues. Rather than gradient ultracentrifugation, we devised a serial filtration method to effectively separate nuclei from debris. Following ultrasonication, the chromatin samples were successfully applied for ChIP studies which showed a circadian pattern of BMAL1 protein binding to target promoters. Our method can be broadly applicable to various mechanistic studies of muscle tissues.
Protocol
Animal care was performed under Institutional Animal Care and Use Committee (IACUC) guidelines, and the procedures were conducted according to an animal protocol approved by the University of Texas Health Science Center at Houston.
1. Nuclei Isolation from Cross-linked Skeletal Muscle
- Weigh and mince hind limb skeletal muscle, isolated from approximately 20-week old C57BL/6 male mice, in ice-cold phosphate buffered saline (PBS) as described in detail previously22. NOTE: We usually obtain 1.0 - 1.5 g of skeletal muscle from one mouse.
- Place minced skeletal muscle tissue in a 50-mL conical tube containing 10 mL PBS on ice.
Centrifuge at 300 x g at 4 °C for 5 min.
Carefully remove PBS supernatant by aspiration.
Estimate pellet volume in each 50-mL tube by using reference tubes with 1, 2, 3 and 4 mL of water.
Add 7 volumes of ice-cold 1% formaldehyde in PBS and homogenize the samples on ice using a probe tissue homogenizer (see Table of Materials). NOTE: Optional: The homogenization procedure can be conducted in a cold room.
Cross-link the sample by incubating at room temperature for 10 min.
Quench the cross-linking reaction by the addition of 1 M glycine to a final concentration of 0.125 M and incubate for 5 min at room temperature.
Centrifuge the samples at 3,000 x g for 5 min at 4 °C. Remove the supernatant via aspiration.
Rinse with 10 mL of ice-cold base buffer (10 mM HEPES-KOH at pH 7.3, 10 mM EDTA, 10 mM KCl, 5 mM MgCl2, 0.5 mM DTT) containing freshly added protease inhibitors (0.2 mM phenylmethylsulfonyl fluoride (PMSF), and 10 µg/mL leupeptin). Centrifuge at 3,000 g for 5 min at 4 °C and carefully remove the supernatant by aspiration.
Resuspend the pellet in 6 mL of lysis buffer (10 mM HEPES-KOH at pH 7.3, 10 mM EDTA, 10 mM KCl, 5 mM MgCl2, 0.5 mM DTT, 1% Triton X-100) containing freshly added protease inhibitors (0.2 mM PMSF, and 10 µg/mL leupeptin).
Transfer the sample to a prechilled 15 mL Dounce homogenizer and incubate for 10 min on ice.Dounce homogenize each sample on ice with 15 slow strokes using loose pestle followed by 15 strokes with tight pestle to release the nuclei.
- Filter the homogenate (6 mL) through cell strainer (pore size: 100 µm) and rinse with 4 mL of lysis buffer. Centrifuge the samples at 1,000 x g for 10 min at 4 °C. Resuspend into 5 mL of ice-cold base buffer and dounce 10 times with tight pestle to release nuclei.
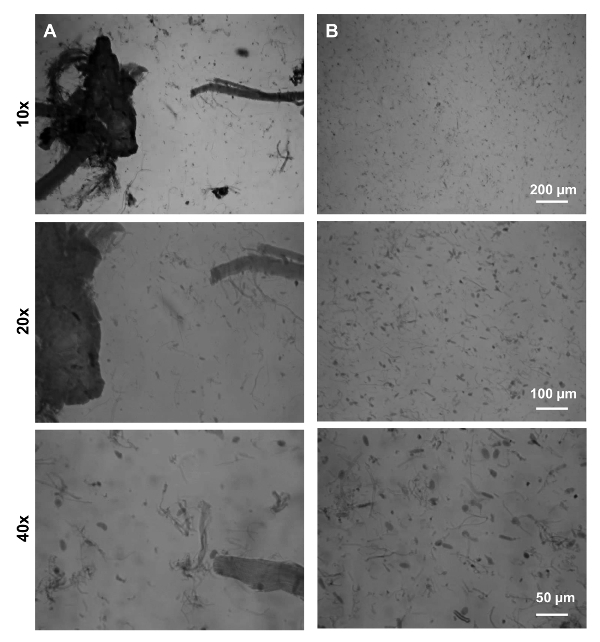
- Remove an aliquot of 20 µL for DNA concentration measurement with a spectrophotometer (OD260/OD280) and microscopic observation with trypan blue if necessary (see below) (Figure 1A).
Filter the suspension through a cell strainer (Pore size: 70 µm), rinse the tube and filter again with 2 mL base buffer as above.
Repeat the filtration as in step 1.13 with cell strainers of gradually reduced pore size (40 µm, 30 µm, 20 µm and 10 µm). NOTE: In total, strainers of 6 pore sizes were used in steps 1.12-1.14.
Centrifuge at 1,000 g at 4 °C for 10 min. Remove supernatant and resuspend the pellet in 500 µL base buffer.
Measure DNA concentration as above. NOTE: Approximately 200 µg of nucleic DNA was obtained from combining both hind limbs from one animal. Optionally, save 20 µL for microscopic observation with trypan blue staining (stock concentration 0.4%, 1:5 dilution); nuclei will stain blue (Figure 1B).
Centrifuge at 1,000 g at 4 °C for 10 min, and discard the supernatant. Store pellet at -80 °C.
2. Sonication
Thaw samples in 500 µL of SDS lysis buffer and resuspend with gentle pipetting.
Transfer the nuclei DNA suspension to a glass vial on ice.
Run the sonication with focused bursts of ultrasonic acoustic energy from a dish-shaped transducer. See Table of Materials for equipment details. Use the following setting: cycles per burst: 200; intensity: 5; duty cycle: 20; temperature 4 - 6 °C; 30 s on/off. NOTE: An example for evaluating sonication efficacy is shown in Figure 2.
Transfer the sonicated chromatin to a 1.5 mL tube. Centrifuge at 12,000 x g for 15 min and transfer the supernatant to a new tube. Transfer 50 µL of sonicated samples (~ 7 - 10 µg/muscle sample) into a new 1.5 mL tube for reverse crosslinking overnight at 65 °C. Freeze the remaining samples at -80 °C.

3. Evaluation of Sonication and Quantitation
Add 1.0 µL of RNase A (500 U/mL RNase A: 20,000 U/mL RNase T1) to the saved 50 µL sonicated samples and incubate for 30 min at 37 °C.
Add 8 µL of protease K (10 mg/mL) and incubate 30 min at 55 °C.
- Harvest the DNA by using a DNA extraction kit.
- Add 5 volumes of binding buffer, apply it to spin column, and centrifuge at 4,000 x g, 10 min.
- Apply the pass-through onto the same column and centrifuge again.
- Wash with washing buffer two times at 13,000 x g, 1 min.
- Centrifuge again at 13,000 x g, 1 min to dry the column.
- Add 50 µL of H2O and centrifuge at 13,000 x g, 1 min to elute DNA.
- Apply the pass-through onto the same column and centrifuge again.
Quantify the amount of DNA with a spectrophotometer (OD260/OD280). Run samples on 0.8% gels to evaluate the size and amount (1 µg) of the sonicated products (Figure 2). NOTE: The average chromatin yield is approximately ~ 20%.
4. ChIP
- Thaw the previously saved chromatin at -80 °C and aliquot the chromatin to ~ 100 - 120 µg per tube. Dilute 1:10 with radioimmunoprecipitation assay (RIPA) buffer (20mM Tris-HCl (pH 7.4), 150mM NaCl, 1mM EDTA, 1mM NaF, 1mM Na3VO4, 1mM PMSF, 1x Protease inhibitor cocktail (PIC), 0.1% Na-deoxycholate (w/v), 1% Triton X-100).
- If there are multiple samples in each group, combine equal amounts of chromatin DNA to the final amount of ~ 100 - 120 µg/sample. Save 10% as input.
Add 40 µL (50% v/v slurry in RIPA buffer) of BSA-pre-blocked (~ 2 h, 4 °C) Protein A/G agarose (non-Chicken antibody) or IgY beads (Chicken antibody) and incubate on a rotator for 3 h at 4 °C.
Centrifuge at 1,000 x g, 10 min and carefully transfer the supernatant to new tubes.
Add antibodies at 1 µg antibody per ~ 25 - 100 µg chromatin DNA.
Rotate gently at 4 °C overnight at approximately 15 rpm.
Add 10 µL of BSA-pre-blocked (~2 h, 4 °C) Protein A/G agarose (non-Chicken antibody) or IgY beads (Chicken antibody) mix and rotate in the cold room for 2 h.
- Wash beads as follows. Wash with 1 mL of RIPA buffer for 3 min twice, wash with 1 mL of high salt buffer (20mM Tris-HCl (pH 7.4), 500mM NaCl, 2mM EDTA, 1mM PMSF, 1% Triton X-100) for 3 min twice, wash with 1 mL of LiCl buffer (20mM Tris-HCl (pH 7.4), 250mM LiCl, 2mM EDTA, 1mM PMSF, 1% Na-deoxycholate, 0.5% NP40) for 3 min twice.
- Wash with 1 mL of Tris-EDTA (TE) buffer (10mM Tris-HCl (pH 7.4), 1mM EDTA) for 3 min once. Then elute DNA with elution buffer (20 mM Tris-HCl, 5 mM EDTA and 0.5% SDS).
Centrifuge at 1,000 g, 1 min and carefully remove the supernatant.
Resuspend the bead with 50 µL of elution buffer.
Incubate for 10 min at 65 °C.
Centrifuge at 12,000 g for 5 min.
Transfer the supernatant to a new tube, add another 50 µL and centrifuge once again at 12,000 g for 5 min. The final elution volume will be 100 µL.
- Reverse crosslinking and DNA elution.
- Incubate the eluted chromatin overnight at 65 °C.
- Add 1.0 µL of RNase A and incubate for 30 min at 37 °C.
- Add 8 µL of protease K (10 mg/mL) and incubate 30 min at 55 °C.
- Harvest the DNA by using a PCR cleanup kit with elution volume at 50 µL according to the manufacturer's protocol.
Analyze the profiles by Real-time qPCR as previously described25,26. NOTE: The primer sequences are listed in Figure 3 Legend. Data are presented as mean ± SEM (Figure 3).
Representative Results
Here we performed formaldehyde cross-linking immediately after tissue collection to preserve real-time DNA-protein interaction. However, we found that sucrose or colloidal gradient, commonly used for nuclei isolation23,24, was not effective in separating nuclei from myofibrils (data not shown). The reason may be that cross-linking conferred similar gravity for nuclei and myofibrils. Therefore, we developed a serial filtration process to effectively remove large myofibrils and other debris from the nuclei fraction. After 100 µm filtration, there were still large tissue debris and myofibrils (Figure 1A). In comparison, at the end of the sequential filtration, the majority of large tissue debris, intact cells and large myofibrils were successfully removed (Figure 1B).
Those nuclei were used for dish-shaped ultrasonication. Nuclei stored at -80 °C were suspended in SDS lysis buffer and immediately subjected to sonication. Although incubation of nuclei in SDS lysis buffer on ice or room temperature may improve sonication for some tissue such as liver27 or mouse embryonic fibroblast (MEF)28, in our experience it interfered with sonication of skeletal muscle nuclei and resulted in broad smearing, indicating ineffective sonication. Using the current protocol with immediate sonication after suspension in SDS lysis buffer we observed efficient sonication with chromatin gradually shredded in a cycle-dependent manner and eventually producing ~ 500 bp DNA fragments (Figure 2). Pre-incubation in the lysis buffer detectably compromised the sonication efficiency.
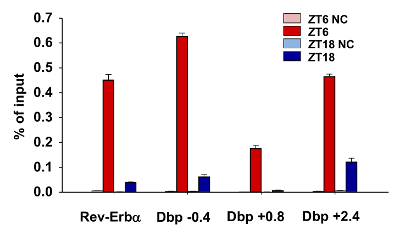
To confirm the quality of chromatin preparation for ChIP, we examined DNA binding of the circadian transcription factor BMAL1, which showed binding peak and trough at Zeitgeber time (ZT)6 and ZT18, respectively18,29. Skeletal muscle samples from C57B/6J mice were collected at ZT6 and ZT18, and chromatin samples were prepared as above. Briefly, after sonication, shredded chromatin samples were pre-cleared with IgY beads that have been pre-blocked with BSA followed by incubation with anti-BMAL1 antibody at 4 °C overnight. After elution and purification of chromatin, we performed RT-qPCR to detect BMAL1 binding on E-Box elements of two target genes, Nr1d1 and Dbp30. We detected robust binding of BMAL1 to the E-box elements at ZT6 and minimal binding at ZT18 (Nr1d1: 0.452 ± 0.022 vs. 0.039 ± 0.002, Dbp -0.4: 0.627 ± 0.013 vs. 0.062 ± 0.009, Dbp +0.8: 0.176 ± 0.013 vs. 0.008 ± 0.001, Dbp +2.4: 0.466 ± 0.010 vs. 0.122 ± 0.014; all values are mean ± SEM) (Figure 3), validating the protocol for time-sensitive transcription factor binding analysis in skeletal muscle.
Figure 1: Sequential Filtration Effectively Removed Tissue Debris.(A) Representative images showing samples after 100 µm filtration. Large tissue and fiber debris are observed. (B) Representative images showing samples after serial filtration. Large fiber debris were cleared. Only isolated nuclei and small myofibril fragments are observed. Pictures were taken by using a light microscope at 10X, 20X and 40X magnifications. Scale bars are shown on the right hand side panels. Please click here to view a larger version of this figure.
Figure 2: Progressive Chromatin Shredding Through 10 Cycles of Sonication. Ten cycles of sonication with digested chromatin DNA to ~ 500 bp, as revealed in a 0.8 % agarose gel, run at 150 V for 60 min. The right panel indicates a lower sonication efficiency after pre-incubation in ice-cold SDS lysis buffer for 1 h. Please click here to view a larger version of this figure.
Figure 3: Representative qPCR Results for BMAL1 ChIP with Mouse Skeletal Muscle Samples Collected at ZT6 and ZT18. Data are presented as mean ± SEM. Dbp -0.4, +0.8 and +2.4 indicate locations of the E-Box elements on the Dbp gene. NC: negative control with IgY. The temporal pattern of BMAL1 binding is consistent with previous results showing BMAL1 binding peak at around ZT618. The forward and reverse primers are as follows. Rev-erba: 5'-GTAGACTACAAATCCCAACAATCCTG, and 5'-TGGAGCAGGTACCATGTGATTC; Dbp -0.4: 5'-ACACCCGCATCCGATAGC, and 5'-CCACTTCGGGCCAATGAG; Dbp +0.8: 5'- ATGCTCACACGGTGCAGACA, and 5'- CTGCTCAGGCACATTCCTCAT; Dbp +2.4: 5'- TGGGACGCCTGGGTACAC, and 5'- GGGAATGTGCAGCACTGGTT. Please click here to view a larger version of this figure.
Discussion
Here we describe a robust method where cross-linked skeletal muscle tissues were used to isolate high-quality nuclei. Sequential filtration was carried out to effectively separate nuclei from debris, and ultrasonic acoustic energy from dish-shaped transducer sheared the chromatin for ChIP analysis. The results showed circadian time-specific binding of BMAL1 to target promoters.
ChIP can be employed to capture real-time protein occupancy on genomic DNA when cross-linking takes place. To take advantage of this potential, we aimed to develop a method to allow cross-linking of skeletal muscle at the time of tissue dissection and to streamline the nuclei isolation without gradient ultracentrifugation. Due to the difficulty of homogenizing fiber-rich skeletal muscle compared with soft tissues such as liver, we minced muscle tissue in ice-cold PBS and then homogenized the sample in a formaldehyde buffer. After quenching, tissue suspension was centrifuged and rinsed with ice-cold base buffer to rinse out any remaining formaldehyde. Nuclei were released by Dounce homogenization, and the homogenates were sequentially filtered to gradually remove cell debris and myofibrils. We devised the series of filtration to minimize filter clogging which could adversely impact yield. Only very short myofibrils remained when the sequential filtration was completed.
The sonication and ChIP procedures were adapted from a previous report12 with modifications including sonication timing and SDS buffer amount. The dish-shaped sonicator allows exposure to centralized ultrasonic wave for samples in glass vials in a cold water bath. Compared with probe sonicators, this sonicator controls the sample temperature to avoid overheating, and also prevents sample cross-contamination. If probe sonicators are used, optimal sonication conditions need to be determined empirically. We also reduced the amount of SDS buffer since the yield of muscle chromatin is lower than that in liver12. Several protocols28,31,32 include incubation on ice or at room temperature prior to sonication. However, in our experience, pre-incubation on ice did not improve the sonication efficiency. In fact, in some instances the sonication was compromised. It is possible that residual myofibrils entangled the chromatin DNA during incubation and attenuated sonication efficacy. With immediate sonication after nuclei suspension in SDS lysis buffer, we succeeded in obtaining progressive chromatin fragmentation with increasing sonication cycles (Figure 2).
We validated the quality of chromatin with ChIP qPCR. As shown in Figure 3, the BMAL1 promoter occupancy was robust at ZT6 and minimal at ZT18, consistent with previously shown BMAL1 circadian promoter binding18. This functional assay confirmed the quality of nuclei and chromatin. In recent years, rapid development in NGS opened up a new horizon for ChIP application where ChIP-seq can quantitatively interrogate genomic binding with high sensitivity17. Particularly for ChIP-seq, high-quality nuclei and chromatin are required to consistently capture protein-DNA interaction. The procedure described herein may constitute a valuable resource for ChIP-seq studies using skeletal muscle. Of note, ChIP-seq library preparation requires additional measures to improve signal resolution, such as PAGE-based size selection18.
In conclusion, we developed a filtration-based protocol to prepare high-quality nuclei from cross-linked skeletal muscle. We remove the need for ultracentrifugation, making it easily applicable. In addition to ChIP for genes of interest, the nuclei and chromatin prepared as described can be broadly applicable to ChIP-seq studies.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Karyn Esser, Nobuya Koike and Noheon Park for helpful advice. This work was in part supported by NIH/NIGMS (R01GM114424) to S.-H.Y., and the Robert A. Welch Foundation (AU-1731) and NIH/NIA (R01AG045828) to Z.C.
References
- Bouzakri K, et al. siRNA-based gene silencing reveals specialized roles of IRS-1/Akt2 and IRS-2/Akt1 in glucose and lipid metabolism in human skeletal muscle. Cell Metab. 2006;4(1):89–96. doi: 10.1016/j.cmet.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Boucher J, Kleinridders A, Kahn CR. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol. 2014;6(1) doi: 10.1101/cshperspect.a009191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green CB, Takahashi JS, Bass J. The meter of metabolism. Cell. 2008;134(5):728–742. doi: 10.1016/j.cell.2008.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu AC, Lewis WG, Kay SA. Mammalian circadian signaling networks and therapeutic targets. Nat Chem Biol. 2007;3(10):630–639. doi: 10.1038/nchembio.2007.37. [DOI] [PubMed] [Google Scholar]
- Harfmann BD, Schroder EA, Esser KA. Circadian rhythms, the molecular clock, and skeletal muscle. J Biol Rhythms. 2015;30(2):84–94. doi: 10.1177/0748730414561638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nohara K, Yoo SH, Chen ZJ. Manipulating the circadian and sleep cycles to protect against metabolic disease. Front Endocrinol (Lausanne) 2015;6:35. doi: 10.3389/fendo.2015.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harfmann BD, et al. Muscle-specific loss of Bmal1 leads to disrupted tissue glucose metabolism and systemic glucose homeostasis. Skelet Muscle. 2016;6:12. doi: 10.1186/s13395-016-0082-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen BK, Febbraio MA. Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Rev Endocrinol. 2012;8(8):457–465. doi: 10.1038/nrendo.2012.49. [DOI] [PubMed] [Google Scholar]
- Solomon MJ, Varshavsky A. Formaldehyde-mediated DNA-protein crosslinking: a probe for in vivo chromatin structures. Proc Natl Acad Sci U S A. 1985;82(19):6470–6474. doi: 10.1073/pnas.82.19.6470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson V. Studies on histone organization in the nucleosome using formaldehyde as a reversible cross-linking agent. Cell. 1978;15(3):945–954. doi: 10.1016/0092-8674(78)90278-7. [DOI] [PubMed] [Google Scholar]
- Gilmour DS, Lis JT. Detecting protein-DNA interactions in vivo: distribution of RNA polymerase on specific bacterial genes. Proc Natl Acad Sci U S A. 1984;81(14):4275–4279. doi: 10.1073/pnas.81.14.4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi JS, et al. ChIP-seq and RNA-seq methods to study circadian control of transcription in mammals. Methods Enzymol. 2015;551:285–321. doi: 10.1016/bs.mie.2014.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo MH, Allis CD. In vivo cross-linking and immunoprecipitation for studying dynamic Protein:DNA associations in a chromatin environment. Methods. 1999;19(3):425–433. doi: 10.1006/meth.1999.0879. [DOI] [PubMed] [Google Scholar]
- Chen Z, Manley JL. Core promoter elements and TAFs contribute to the diversity of transcriptional activation in vertebrates. Mol Cell Biol. 2003;23(20):7350–7362. doi: 10.1128/MCB.23.20.7350-7362.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menet JS, Abruzzi KC, Desrochers J, Rodriguez J, Rosbash M. Dynamic PER repression mechanisms in the Drosophila circadian clock: from on-DNA to off-DNA. Genes Dev. 2010;24(4):358–367. doi: 10.1101/gad.1883910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T, Matsumoto T, Zhao Z, Lee CC. p53 regulates Period2 expression and the circadian clock. Nat Commun. 2013;4:2444. doi: 10.1038/ncomms3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furey TS. ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nat Rev Genet. 2012;13(12):840–852. doi: 10.1038/nrg3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike N, et al. Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science. 2012;338(6105):349–354. doi: 10.1126/science.1226339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Yu W, Hardin PE. ChIPping away at the Drosophila clock. Methods Enzymol. 2015;551:323–347. doi: 10.1016/bs.mie.2014.10.019. [DOI] [PubMed] [Google Scholar]
- Takahashi JS. Transcriptional architecture of the mammalian circadian clock. Nat Rev Genet. 2016. [DOI] [PMC free article] [PubMed]
- Edelman JC, Edelman PM, Kniggee KM, Schwartz IL. Isolation of skeletal muscle nuclei. J Cell Biol. 1965;27(2):365–377. doi: 10.1083/jcb.27.2.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkawa Y, Mallappa C, Vallaster CS, Imbalzano AN. Isolation of nuclei from skeletal muscle satellite cells and myofibers for use in chromatin immunoprecipitation assays. Methods Mol Biol. 2012;798:517–530. doi: 10.1007/978-1-61779-343-1_31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkie GS, Schirmer EC. Purification of nuclei and preparation of nuclear envelopes from skeletal muscle. Methods Mol Biol. 2008;463:23–41. doi: 10.1007/978-1-59745-406-3_2. [DOI] [PubMed] [Google Scholar]
- Hahn CG, Covault J. Isolation of transcriptionally active nuclei from striated muscle using Percoll density gradients. Anal Biochem. 1990;190(2):193–197. doi: 10.1016/0003-2697(90)90180-h. [DOI] [PubMed] [Google Scholar]
- Jeong K, et al. Dual attenuation of proteasomal and autophagic BMAL1 degradation in Clock Delta19/+ mice contributes to improved glucose homeostasis. Scientific reports. 2015;5:12801. doi: 10.1038/srep12801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nohara K, et al. Ammonia-lowering activities and carbamoyl phosphate synthetase 1 (Cps1) induction mechanism of a natural flavonoid. Nutrition & metabolism. 2015;12:23. doi: 10.1186/s12986-015-0020-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Rubins NE, Ahima RS, Greenbaum LE, Kaestner KH. Foxa2 integrates the transcriptional response of the hepatocyte to fasting. Cell Metab. 2005;2(2):141–148. doi: 10.1016/j.cmet.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Gade P, Kalvakolanu DV. Chromatin immunoprecipitation assay as a tool for analyzing transcription factor activity. Methods Mol Biol. 2012;809:85–104. doi: 10.1007/978-1-61779-376-9_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondratov RV, et al. BMAL1-dependent circadian oscillation of nuclear CLOCK: posttranslational events induced by dimerization of transcriptional activators of the mammalian clock system. Genes Dev. 2003;17(15):1921–1932. doi: 10.1101/gad.1099503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripperger JA, Schibler U. Rhythmic CLOCK-BMAL1 binding to multiple E-box motifs drives circadian Dbp transcription and chromatin transitions. Nat Genet. 2006;38(3):369–374. doi: 10.1038/ng1738. [DOI] [PubMed] [Google Scholar]
- Laplante M, et al. DEPTOR cell-autonomously promotes adipogenesis, and its expression is associated with obesity. Cell Metab. 2012;16(2):202–212. doi: 10.1016/j.cmet.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blechman J, Amir-Zilberstein L, Gutnick A, Ben-Dor S, Levkowitz G. The metabolic regulator PGC-1alpha directly controls the expression of the hypothalamic neuropeptide oxytocin. J Neurosci. 2011;31(42):14835–14840. doi: 10.1523/JNEUROSCI.1798-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]