

Abstract
Since the discovery of the multiple endocrine neoplasia type 1 (MEN1) gene in 1997, elucidation of the molecular function of its protein product, menin, has been a challenge. Biochemical, proteomics, genetics and genomics approaches have identified various potential roles, which converge on gene expression regulation. The most consistent findings show that menin connects transcription factors and chromatin modifying enzymes, in particular the histone H3K4 methyltransferase complexes MLL1 and MLL2. Chromatin immunoprecipitation combined with next generation sequencing has enabled studying genome-wide dynamics of chromatin binding by menin. We propose that menin regulates cell type-specific transcriptional programs by linking chromatin regulatory complexes to specific transcription factors. In this fashion, the MEN1 gene is a tumor suppressor gene in the endocrine tissues that are affected in MEN1. Recent studies have hinted at possibilities to pharmacologically restore the epigenetic changes caused by loss of menin function as therapeutic strategies for MEN1, for example by inhibition of histone demethylases. The current lack of appropriate cellular model systems for MEN1-associated tumors is a limitation for compound testing, which needs to be addressed in the near future. In this review, we look back at the past twenty years of research on menin and the mechanism of disease of MEN1. In addition, we discuss how the current understanding of the molecular function of menin offers future directions to develop novel treatments for MEN1-associated endocrine tumors.
Keywords: multiple endocrine neoplasia type 1 (MEN1), menin, transcriptional regulation, histone H3K4 trimethylation
Introduction
In the 1997 paper reporting the positional cloning of the multiple endocrine neoplasia type 1 (MEN1) gene, the authors proposed the name menin for its protein product. Chandrasekharappa and colleagues noted that analysis of the 610 amino acid sequence provided few clues to menin’s function and that there were no apparent molecular similarities to previously known proteins (Chandrasekharappa, et al. 1997). The MEN1 gene is expressed very widely and menin was found to reside primarily in the cell nucleus (Guru, et al. 1998).
MEN1 patients are predisposed to develop parathyroid adenomas, neuroendocrine tumors (NETs) of the duodenum and the pancreas and pituitary adenomas, which often manifest at a young age. In addition, adrenal adenomas, neuroendocrine tumors of the stomach, thymus or lungs, lipomas, leiomyomas and meningiomas can be observed (Thakker, et al. 2012). Recently, an increased risk to develop breast cancer was added to the MEN1 spectrum (Dreijerink, et al. 2014). Metastatic pancreatic neuroendocrine tumours (pNETs) are the most frequent MEN1-related cause of death in MEN1 patients (Conemans, et al. 2017). MEN1 patients and MEN1 gene mutation carriers are monitored at regular intervals in order to detect and treat manifestations in a timely manner.
The first genetics studies showed that MEN1 germ line mutations found in MEN1 families were mostly scattered through the coding exons and had clearly inactivating effects on menin (Chandrasekharappa et al. 1997; Lemmens, et al. 1997). The identification of inactivating mutations, together with the previously reported loss of heterozygosity (LOH) at the MEN1 locus at chromosome 11q13 in MEN1-related insulinoma demonstrated that the MEN1 gene acts as a classic tumor suppressor gene in MEN1 (Larsson, et al. 1988). This was confirmed in mouse models: homozygous deletion of the Men1 gene is embryonic lethal at the mid-gestation stage. However, mice carrying heterozygous Men1 germ line mutations develop phenotypes that show close resemblance to MEN1 manifestations in humans as well as loss of the wild type allele in the tumor tissues [reviewed in (Agarwal 2014)]. In addition to inherited tumors in MEN1 patients, the MEN1 gene is also commonly mutated in sporadic parathyroid adenomas and pNETs (Heppner, et al. 1997; Jiao, et al. 2011; Scarpa, et al. 2017).
The majority of MEN1 germ line mutations are nonsense and frameshift mutations causing premature stop codons (Lemos and Thakker 2008). Expression of truncated menin proteins is probably suppressed by nonsense-mediated mRNA decay (Zetoune, et al. 2008). In addition, the majority of missense MEN1 mutations lead to unstable proteins, which are prone to ubiquitin-dependent proteasomal degradation (Yaguchi, et al. 2004). Proteasomal inhibition or silencing of expression of the responsible carboxy-terminus of Hsc70 interacting protein (CHIP) E3 ubiquitin ligase was demonstrated to increase expression of menin mutant proteins (Canaff, et al. 2012). The solution of the crystal structure of menin has enabled three-dimensional mapping of MEN1 mutations (Huang, et al. 2012). Although there have been reports about selected missense mutations and intron mutations occurring in familial isolated hyperparathyroidism or in families with relatively mild phenotypes, there is no clear genotype- phenotype correlation in MEN1 [reviewed by (Lips, et al. 2012)]. The penetrance of MEN1 manifestations is highly variable, even within MEN1 families carrying the same gene mutation.
In this paper, we review the literature addressing the molecular function of menin and the pathogenesis of MEN1. We highlight a number of mechanisms that have been validated in a translational fashion in MEN1 model systems. Recent studies have indicated potential therapeutic interventions for MEN1. Strategies aimed to restore the function of menin could result in mechanism-based therapies for MEN1 and MEN1-associated tumors, in particular pNETs.
Menin is involved in transcriptional regulation
After the discovery of the MEN1 gene, many research groups set out to identify menin-interacting proteins in order to understand the pathogenesis of MEN1 [reviewed in (Matkar, et al. 2013)]. The vast majority of menin-interacting partners include proteins involved in transcriptional regulation and chromatin modification. Transcription of genes into messenger RNA is carried out by the RNA polymerase II (pol II) enzyme together with the basal transcription machinery, which direct initiation of mRNA synthesis at transcription start sites (TSS) of gene promoters (Figure 1). Gene-specific transcription factors co-regulate transcription by binding to specific DNA-sequences that may either be located close to gene promoters but also at distant regulatory elements termed enhancers. Although enhancers may be far away from TSS from a linear perspective, these sites can be looped to TSS to enable interactions of DNA-bound proteins and thus affect transcription initiation. Gene-specific transcription factors are often expressed in certain tissues, which results in tissue-specific enhancer function. Prominent enhancer clusters are sometimes referred to as super-enhancers (Whyte, et al. 2013). Chromatin accessibility and modification constitute additional layers of transcriptional regulation. DNA is wrapped around histone proteins into chromatin. The functional unit of chromatin is called a nucleosome and consists of a histone octamer (each containing two sets of the core histones H2A, H2B, H3 and H4) and ~150 base pairs of DNA. Post-translational modifications of histones such as acetylation and methylation strongly correlate with the functional properties of the genome. Histone H3 lysine 4 trimethylation (H3K4me3) marks active gene promoters and histone H3 lysine 36 (H3K36) methylation accumulates towards the 3′-ends of actively transcribed genes. In contrast, histone H3 lysine 9 trimethylation (H3K9me3), histone H3 lysine 27 trimethylation (H3K27me3) and loss of histone acetylation are associated with transcriptional repression. As a rule, histone marks are dynamic modifications that can be written, read and erased by dedicated proteins. The combinatorial interplay of the basal transcription factors, pol II recruitment, enhancer binding transcription factors and chromatin structure and modifications determines the mRNA expression signatures of cells and tissues. Disruption of mRNA expression, for example by mutations in genes encoding transcriptional regulators, is a major contributor for oncogenic transformation of cells into tumor cells [reviewed by (Bradner, et al. 2017)]. Below and in Figure 1, we give an overview of the published menin interactions with transcription factors and chromatin regulatory proteins.
Figure 1.
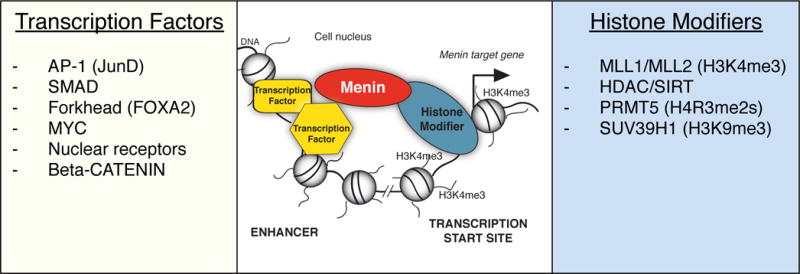
The MEN1 gene product menin connects transcription factors to histone modifying protein complexes. Menin can interact with several classes of transcription factors listed in the left panel. Transcription factors bind directly -or indirectly in the case of β-catenin- to specific DNA sequences at sites termed enhancers. Enhancers may be located far away from transcription start sites (TSS), however through DNA looping enhancer-bound proteins can interact directly with the TSS. Menin links transcription factors to chromatin modifying protein complexes, such as the MLL1 and MLL2 containing complexes that have H3K4me3 methyltransferase activity to activate transcription of target genes as shown in the middle panel. Other menin chromatin modifying interactors include HDACs, SIRT1, PRMT5 and SUV39H1. Histone deacetylation, H4R3me2s and H3K9me3 are associated with repression of transcription.
Abbreviations: multiple endocrine neoplasia type 1 (MEN1), mixed lineage leukemia (MLL), histone deacetylase (HDAC), sirtuin (SIRT), protein arginine N-methyltransferase (PRMT), suppressor of variegation 3–9 Homolog 1 (SUV39H1)
Menin binds to transcription factors
JunD
The first menin interacting protein that was identified is JunD by Agarwal et al. It was found in a yeast-two hybrid screen experiment (Agarwal, et al. 1999). JunD is a member of the activator protein 1 transcription (AP-1) factor family. AP-1 transcription factors act in a gene-specific manner and are downstream in the signaling cascade elicited by the RAS oncogene. The transcriptional activity of JunD is repressed by menin in reporter assays, putatively due to recruitment of histone deacetylase activity (Agarwal et al. 1999; Gobl, et al. 1999; Kim, et al. 2003). Menin interferes with Jun kinase (JNK)-mediated phosphorylation of JunD and also c-Jun, potentially interfering with RAS signaling (Gallo, et al. 2002). In 2012, menin was co-crystallized with a JunD peptide, which allowed detailed mapping of the interaction surface (Huang et al. 2012). Several independent research groups have confirmed the stable nature of the menin-JunD interaction. However, the pathogenic relevance for MEN1 remains elusive to date.
SMAD transcription factors
Transforming growth factor beta (TGF-β) and bone morphogenic protein (BMP) signaling is transduced to gene transcription via SMA and MAD (SMAD) gene-specific transcription factors. Genomic SMAD binding sites can be both at promoter-proximal and enhancer elements. Menin has been reported to interact with and co-activate SMAD1, SMAD3 and SMAD5 (Sowa, et al. 2003; Sowa, et al. 2004). In this fashion, menin can suppress prolactin and parathyroid hormone expression in murine cells, which is in line with the loss of function of menin in pituitary and parathyroid adenomas (Hendy, et al. 2005). The molecular mechanism of SMAD co-activation by menin is presently unknown.
Forkhead transcription factors
Forkhead box proteins are gene-specific transcription factors that bind to DNA-sequences and are downstream effectors of several signaling pathways. Menin has been reported to interact with several FOX family members including FOXN1/checkpoint suppressor 1 (CHES1) and FOXO1 (Busygina and Bale 2006; Wuescher, et al. 2011). In sporadic breast cancer cells, menin is enriched at enhancers that contain FOXA1 binding motifs (Dreijerink, et al. 2017). In mouse embryonic stem cell (mESC)-derived embryoid bodies, lower Foxa1 and Foxa2 mRNA levels were observed in the absence of Men1 gene expression (Zhang, et al. 2011). In addition, menin was found to be able to interact with Foxa2 and to regulate Foxa2 expression in mouse pancreatic cell lines indicating a potential role in MEN1-associated pNET tumorigenesis (Bonnavion, et al. 2017).
MYC
MYC (c-Myc) is a transcription factor that binds to E-boxes in the DNA to activate transcription of genes involved in tumorigenesis. Increased MYC expression and activity is correlated with cell proliferation rates and this is observed in many cancer types. Both activating and repressive effects of menin on MYC action have been reported. Menin has been suggested to interact with MYC and the SKI-interacting protein (SKIP) co-activator at the HIV-1 promoter (Bres, et al. 2009). In HEK293 embryonic kidney cells, menin and the mixed-lineage leukemia (MLL) complex were found to interact with the transcription factor FUSE binding protein 1 (FBP1/FUSBP1) to stimulate MYC expression (Zaman, et al. 2014). It was recently reported, that in fibrosarcoma cells menin can also interact directly with MYC and regulate MYC target genes in an H3K4me3-independent manner (Wu, et al. 2017). More in line with its tumor suppressive function, loss of menin was shown to result in upregulation of the c-Myc mRNA in mouse-derived pancreatic cells, possibly via increased Hedgehog signaling pathway activity (Gurung, et al. 2013a).
Nuclear receptors
Based on the observation that the amino acid sequence of menin contains a putative nuclear receptor interaction motif, menin was identified as a co-activator of nuclear receptor (NR)-mediated transcription. Menin is able to interact with the estrogen receptor alpha (ERα) ligand-binding domain in an estradiol (E2)-dependent fashion (Dreijerink, et al. 2006). This interaction is important for transcription of ERα target genes through recruitment of H3K4me3 methyltransferase activity. Menin also regulates transcription of the ESR1 gene, which encodes ERα in breast cancer and pituitary cells (Dreijerink et al. 2017). Subsequent work showed that menin is a generic co-regulator for nuclear receptor-mediated transcription as it interacts with many other NR family members including the vitamin D receptor (VDR), the retinoid X receptor (RXR), the peroxisome proliferator-activated receptors (PPAR) alpha and gamma, the liver X receptor (LXR) alpha and the androgen receptor (AR) (Cheng, et al. 2015; Cheng, et al. 2011; Dreijerink et al. 2006; Dreijerink, et al. 2009a; Malik, et al. 2015). The clinical relevance of the menin-NR interaction was investigated in normal parathyroid and MEN1 patient-derived parathyroid adenoma tissues. In MEN1 tumors, lower levels of VDR target gene messenger RNAs are in line with the loss of the co-activator function of menin (Dreijerink, et al. 2009b).
Beta-CATENIN
The wingless-related integration site (Wnt)/β-catenin signaling route has important roles in development, but also in cancer biology. Menin has been reported to interact with the major effector of this pathway: β-catenin. In addition, menin was found to interact with one of β-catenin’s associated transcription factors, T-cell factor 3 (TCF3) and to have a role in the stimulation of Wnt signaling in murine islet cells (Chen, et al. 2008). Conversely, in mouse insulinoma cells lacking Men1, nuclear staining of β-catenin was present, a sign of active Wnt signaling in these tissues (Cao, et al. 2009). Inhibition of Wnt signaling resulted in reduced proliferation of Men1 deficient rodent beta cells. Accordingly, combining a beta-cell specific mouse knockout model of the Men1 gene with knockout of the Ctnnb1 gene (encoding β-catenin) resulted in lower tumor numbers and sizes in mice defective in Men1 and Ctnnb1 versus Men1 alone (Jiang, et al. 2014). These results suggest that activated Wnt signaling after loss of MEN1 gene expression could be a targetable mechanism in MEN1-related tumors.
Menin interacts with chromatin regulatory proteins
MLL1/MLL2
In 2004, using a combined approach of immunoprecipitation and mass spectrometry in 293T cells, Hughes et al. identified menin as part of a histone methyltransferase (HMT) complex that contained the trithorax family member mixed-lineage leukemia protein 2 [MLL2; also termed lysine methyltransferase 2B (KMT2B)] (Hughes, et al. 2004). The complex was found to methylate histone H3K4 and a direct interaction between menin and pol II was discovered. Yokoyama et al. used biochemical purification to identify a similar menin-containing complex that contained MLL1 (KMT2A) but not MLL2 in K562 erythroleukemia cells (Yokoyama, et al. 2004). MLL1 and MLL2 are proteins that contain su(var)3–9, enhancer-of-zeste and trithorax (SET) domains and are part of a family of H3K4 methylation writers that are derived from the yeast Set1 protein [reviewed in (Smith, et al. 2011)]. In humans, six distinct Set1-like complexes exist, each assembled around one of the SET1/MLL proteins (Figure 2). Using quantitative mass spectrometry, Van Nuland et al. determined the composition and stoichiometry of the human SET1/MLL complexes in HeLa cervical carcinoma cells and found that menin is exclusively present in the MLL1/MLL2 complexes in substoichiometric quantities but not in the other SET1/MLL complexes (van Nuland, et al. 2013).
Figure 2.
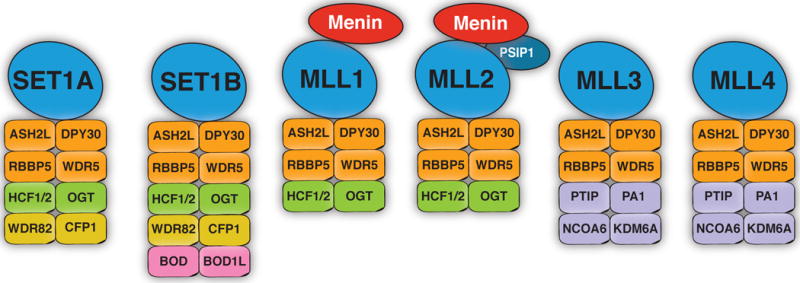
Composition of human SET1/MLL complexes in Hela cells based on immunoprecipitation experiments combined with mass spectrometry [adapted from (van Nuland et al. 2013)]. The SET1-2 (also termed KMT2F-G) and MLL1-4 (KMT2A-D) proteins are the catalytic subunits of six distinct human Set1-like complexes. These complexes contain four common subunits: ASH2L, DPY30, RBBP5 and WDR5, but also complex-specific components. Menin is exclusively found in MLL1 and MLL2-containing complexes. In MLL2 complexes in particular, menin has also been co-precipitated together with PSIP1.
Abbreviations: Absent, small, or homeotic-like (ASH2L), Bcl-2-related ovarian death gene (BOD), CXXC finger protein 1 (CFP1), host cell factor (HCF), lysine demethylase (KDM), lysine methyltransferase (KMT), mixed lineage leukemia (MLL), nuclear receptor coactivator (NCOA), O-Linked N-Acetylglucosamine transferase (OGT), PTIP associated protein (PA1), PC4 and SFRS1 interacting protein (PSIP1), Pax-interacting protein (PTIP), RB binding protein (RBBP), suvar enhancer-of-zeste, trithorax (SET), WD40 repeat (WDR)
In mouse embryonic fibroblasts (MEFs), the menin-MLL1 complex was found to be present at the Hoxc8 locus, both at the TSS and at an upstream enhancer site (Hughes et al. 2004). H3K4me3 is associated with active promoters and menin was found to be an activator of Hoxc8 transcription. Further evidence of menin-dependent H3K4me3 at target genes was provided by experiments using E2-induced transcription in MCF-7 breast cancer cells. After silencing of MEN1 expression, E2-induced H3K4me3 was significantly lower on the TSS of the TFF1 ERα target gene (Dreijerink et al. 2006). Genome-wide analysis of menin binding sites supported colocalization of menin, MLL1 and H3K4me3 in a variety of cell types (Scacheri, et al. 2006). The menin-MLL1 complex can be tethered to chromatin together with the lens epithelium-derived growth factor (LEDGF)/PC4 and SFRS1 interacting protein (PSIP1), a H3K36-binding protein (Yokoyama and Cleary 2008). In HeLa cells, PSIP1 was found to be mostly present in menin-MLL2 complexes (van Nuland et al. 2013).
The crystallization of menin both from Nematostella vectensis and subsequently a human protein construct has enabled the three-dimensional analysis of the menin MLL interaction (Huang et al. 2012; Murai, et al. 2011). The interaction surfaces of menin both with MLL1 and LEDGF and JunD were mapped to a central cavity of menin. However, these interactions cannot occur simultaneously, supporting differential roles for menin either as an interactor of JunD, or as a component of MLL1/MLL2 complexes. Strong genetic evidence for the contribution of loss of H3K4me3 to MEN1 tumorigenesis was provided in a study by Lin et al. who showed that additional genetic ablation of the retinoblastoma binding protein 2 [Rbp2; also known as Jarid1a or lysine demethylase 5a (Kdm5a)] H3K4 demethylase in Men1 deficient mice decreased pNET formation and prolonged survival (Lin, et al. 2011).
HDAC/SIRT
The silencing by menin of JunD-mediated transcription has been attributed to repressive chromatin modifications. Gobl et al. showed that repression of JunD-dependent transcription could be reversed using the histone deacetylase (HDAC) inhibitor trichostatin A (Gobl et al. 1999). Kim et al. found an association with SIN3A-HDAC complexes (Kim et al. 2003), which could not be confirmed by quantitative proteomic analysis of HeLa cells (van Nuland et al. 2013). In a more recent study, an interaction between menin and the histone deacetylase sirtuin 1 (SIRT1) was reported. Menin was shown to be required for the recruitment of SIRT to the Cd36 gene promoter in mouse hepatocytes (Cao, et al. 2013).
PRMT5
Work from the Hua group described menin as a repressor of Hedgehog signaling by histone H4 arginine 3 (R3) symmetric dimethylation (H4R3me2s). H4R3me2s is a repressive transcription mark. The protein arginine N-methyltransferase PRMT5 was identified as a menin interactor by mass spectrometry of purified flag-tagged menin bound proteins in 293T cells (Gurung, et al. 2013b). The Hedgehog signaling gene transcription factor GLI1 gene was found to be a target gene of the menin-PRMT5 mechanism. Administration of a Hedgehog signaling inhibitor resulted in reduced proliferation of insulinoma cells in a beta cell-specific MEN1 mouse model.
SUV39H1
In a Men1 knockout MEF cell line, global reduction of histone H3 lysine 9 trimethylation (H3K9me3) was observed. A candidate approach of overexpressing menin and several H3K9 methyltransferases in 293T cells resulted in the identification of the suppressor of variegation 3–9 Homolog 1 (SUV39H1) protein as a menin-interacting protein (Yang, et al. 2013). In addition to MEN1 gene mutations, sporadic pNETs often harbor loss-of-function mutations in the death domain-associated protein (DAXX) and alpha-thalassemia X-linked mental retardation protein (ATRX) tumor suppressor genes (Jiao et al. 2011). DAXX and ATRX are both involved in telomere lengthening and histone H3.3 exchange. In a recent report, Feng et al. reported a direct interaction between menin and DAXX after ectopic overexpression in 293T cells. In murine insulin-producing cells, Menin and DAXX enhanced H3K9me3 at the promoter of the membrane metallo-endopeptidase (Mme) gene coinciding with SUV39H1 recruitment at the same sites (Feng, et al. 2017).
Lessons from the menin cistrome
An experimental method named chromatin-immunoprecipitation (ChIP) allows for the detection of DNA protein interactions after formaldehyde crosslinking. This enables mapping of DNA binding motifs of transcriptional regulators or the presence of histone modifications on the DNA. This mapping can be carried out by testing candidate sites using targeted PCR, as has been reported for several of the target genes mentioned above. However, by combining ChIP with high throughput analyses of bound DNA, such as a DNA chip (ChIP on a chip) or more recently next generation sequencing (ChIP-seq) the presence of a given transcriptional regulator or a histone modification of interest can be studied on a genome-wide scale in an unbiased fashion. The combined set of genomic binding elements for a transcriptional regulator has been termed the cistrome (Lupien and Brown 2009). The first study addressing the menin cistrome by Agarwal et al. demonstrated binding of menin mostly at promoters and genes. However, intergenic loci comprised 33% of binding sites in HeLa S3 cervical carcinoma cells (Agarwal and Jothi 2012). Subsequent ChIP-chip studies of HeLa S3, HepG2 and pancreatic islet cells showed that menin, MLL1 and the SET1/MLL complex subunit RB binding protein 5 (Rbbp5) localized to the promoters of thousands of human genes but not always simultaneously. The data suggested that menin is present at sites of active transcription as menin occupancy generally correlated with high gene expression. Loss of menin did not result in significant changes in most transcript levels, except for the Motor Neuron And Pancreas Homeobox 1 (MNX1)/ Homeobox Protein HB9 (HLXB9) gene (Scacheri et al. 2006). In a comparative study of mESCs and pancreatic islet-like endocrine cells, average H3K4me3 levels at TSS did not differ between wild type and Men1 knockout cells. However, at a selected number of loci H3K4me3 was lower in the absence of Men1, in particular, H3K4me3 levels at the long non-coding RNA Meg3 locus and several Hox genes were decreased in islet cells (Agarwal and Jothi 2012). In a study in mouse pancreatic tissues, menin-dependent H3K4me3 loss was accompanied by gain of the repressive mark H3K27me3 at menin-target loci (Lin, et al. 2015).
While the MEN1 gene functions as a tumor suppressor gene in MEN1, it has an oncogenic role in sporadic breast cancer cells. A recent integrative analysis of the menin cistrome, H3K4me3 and gene expression in the well-established MCF-7 ER+ breast cancer cell line was carried out to further investigate this oncogenic mechanism of menin-MLL1/MLL2 target gene regulation. A menin-MLL1/MLL2 target gene was defined as a gene that has menin-dependent H3K4me3 and mRNA levels. In accordance with previous studies, only a limited number of genes fullfilled these requirements. Menin-MLL1/MLL2 target genes not only had menin bound at their TSS, but also showed an enrichment of menin binding to distal enhancer sites that showed looping to the target gene TSS (Dreijerink et al. 2017). From these observations it can be concluded that not only the presence of menin at TSS, but rather the combination of the presence of menin at TSS and enhancer sites, determines target gene specificity of menin-MLL1/MLL2 complexes. In the case of MCF-7 breast cancer cells, menin was present at breast cancer-specific FOXA1- and GATA3-bound enhancers. There was a large degree of overlap between menin-bound enhancers and known super-enhancers. Several master transcription factors for breast cancer were among the menin-MLL1/MLL2 target genes in MCF-7 cells. In fact, the ESR1 gene, encoding ERα, turned out to be a major target gene. Menin also co-activates the ERα protein at the protein level and simultaneous regulation of the ESR1 gene could be an auto-regulatory mechanism to control the core transcriptional regulatory circuitry in mammary cells. Similar regulatory circuitries could exist in MEN1-affected tissues, e.g. involving FOXA2 and MNX1/HLXB9 in pancreatic neuroendocrine cells. An obvious next step, which is unfortunately hampered by the current lack of appropriate model systems, would be to take a similar integrative ChIP-seq and RNA-seq approach in MEN1 tumor cells to identify the relevant transcription factors and target genes that are responsible for MEN1 tumorigenesis.
Menin-dependent gene expression signatures
The fate and function of a cell are determined by its gene expression signature. As menin is a transcriptional regulator, MEN1-related tumorigenesis is likely to be the result of aberrant tumor suppressive gene expression due to the loss of menin. Restoration of expression of menin target genes in MEN1-affected tissues could therefore have therapeutic consequences.
Among the first gene sets described to be regulated by menin were the cyclin-dependent kinase inhibiting (CDKI) genes CDKN2C and CDKN1B encoding the p18INK4c and p27KIP1 proteins respectively. CDKI’s are targets of cyclin-dependent kinases and have important roles in cell cycle control (Milne, et al. 2005). Genetic knock-out mouse models of the Cdkn2c and Cdkn1b genes develop a mix of MEN1 and multiple endocrine neoplasia type 2 (MEN2)-type endocrine tumors, underscoring the potential importance of these genes for endocrine tumorigenesis (Franklin, et al. 1998). Moreover, multiple endocrine neoplasia type 4 (MEN4) is a rare inherited condition characterized by pituitary and parathyroid adenomas and caused by germ line mutations in the CDKN1B gene (Molatore, et al. 2010). Using a candidate approach, menin and MLL1 were found to be present at the Cdkn2c and Cdkn1b promoters in MEFs and later also in mouse pancreatic tumors (Karnik, et al. 2005; Milne et al. 2005). Activation of these genes could be an important tumor suppressive function of menin. CDK inhibition could be explored as a therapeutic strategy for MEN1 tumors. However, a recent integrative genomics study in Men1-depleted pancreatic islets from 2-month-old mice, the CDK genes were not among the menin-MLL1/MLL2 target genes (Lin et al. 2015).
An experimental approach using H3K4me3 ChIP-seq and microarrays in menin-null versus wild type mESCs identified the long non-coding RNA Meg3 as a potential menin-MLL1/MLL2 target. A similar analysis in pancreatic islet-like endocrine cells revealed several Hox genes, which are the classic MLL target genes, as menin-MLL1/MLL2 target genes (Agarwal and Jothi 2012). Loss of MEG3 has been reported in sporadic pituitary adenomas (Zhang, et al. 2003). In a follow-up study in mouse pancreatic islet cells, overexpression of Meg3 coincided with down regulation of the c-Met oncogene. Compared with normal islets, human MEN1-associated PNETs expressed lower levels of MEG3 and higher levels of c-MET, suggesting that c-Met could be an interesting future treatment target in MEN1-related pNETs (Modali, et al. 2015).
Genome-wide analysis of menin-dependent H3K4me3 combined with gene expression data revealed that the MNX1/HLXB9 gene is a gene that is activated by the menin-MLL1/MLL2 complex (Scacheri et al. 2006). The MNX1/HLXB9 protein is a transcription factor that is important for pancreas and beta cell development. In addition, MNX1/HLXB9 was found to interact with menin in overexpression experiments in MIN6 mouse insulin producing cells and to induce apoptosis and reduced proliferation in this cell model (Shi, et al. 2013). In a broader sense, differential Men1-dependent expression of pancreatic transcription factors, such as MNX1/HLXB9, Foxa2, MafA, MafB and Pdx1 has been described in several reports (Fontaniere, et al. 2006; Hamze, et al. 2013; Lu, et al. 2012; Scacheri et al. 2006; Zhang et al. 2011). In fact, the Men1 gene appears to be essential for normal development of the endocrine but not of the exocrine pancreas in mice. In ex vivo culturing experiments of the pancreatic bud, in the absence of menin, the number of cells expressing the pancreatic endocrine precursor transcription factor Neurog3 was markedly reduced (Fontaniere, et al. 2008). Beta cell-specific Men1 knockout has also been shown to be able to initiate the development of trans differentiated glucagon expressing tumors, further highlighting the critical role of menin in endocrine pancreatic development (Li, et al. 2015).
In tissues that are not affected in MEN1, menin was found not to be involved in anti-proliferative gene expression and to have different functions. For example, menin regulates a gene signature in pro-B cells that affects B cell differentiation but not proliferation (Li, et al. 2013). Moreover, in contrast to its tumor suppressive role in MEN1-related tumors, in sporadic breast cancer cells menin regulates a growth promoting gene expression signature (Dreijerink et al. 2017). These observations should be kept in mind when considering approaches aimed at restoring the loss of menin function.
Relevance of menin research for MEN1-related tumors and potential therapeutic opportunities
The current view of the molecular function of menin is that menin links gene-specific transcription factors to chromatin modification in a cell-specific context. Menin is able to bind to MYC, nuclear receptors and β-catenin as well as AP-1, SMAD and Forkhead transcription factors. Menin is a stable component of the MLL1 and MLL2 histone methyltransferase complexes and possibly also interacts with histone deacetylases, PRMT5 and SUV39H1. In MEN1-associated tissues, loss of menin leads to the disruption of anti-proliferative gene expression programs and to the development of endocrine tumors. Restoration of the epigenetic perturbations or correction of the function of aberrantly expressed genes in the absence of menin hold promise for molecular mechanism-based means to treat or prevent MEN1-related tumors. The most compelling evidence supporting this strategy was reported by Lin et al. in 2011 and 2015 (Lin et al. 2011; Lin et al. 2015). In mice with beta cell-specific menin depletion, combined genetic knockout of the Rbp2/Jarid1a/Kdm5a gene, resulted in reduced tumor formation and increased survival. Rbp2 is a histone demethylase for H3K4me2/3. At the TSS of the insulin-like growth factor 2 mRNA binding protein 2 (Igf2bp2) gene, the menin-dependent absence of H3K4me3 in Men1 fl/fl, RIP-Cre mice could be reversed by simultaneously knocking out Rbp2 (Lin et al. 2015).
These observations indicate that compensation of the loss of H3K4me3 mark by the loss of menin at certain target genes may restore the function of menin in pancreatic tumors. Members of the JARID/lysine demethylase 5 (KDM5) family have H3K4me3 demethylase acitivity. The enzymatic activity of the KDM5A-D proteins is dependent on iron and alpha-ketoglutarate (αKG). KDM inhibitors that occupy the αKG binding site are a promising new class of compounds that is currently under development and could be used to rescue the loss of H3K4me3 in MEN1 tumors. (Horton, et al. 2016; Vinogradova, et al. 2016). Conversely, as the loss of H3K4me3 at menin target genes was found to coincide with gain of H3K27me3, compounds inhibiting H3K27 methyltransferase activity could have similar effects on menin target genes affected by the loss of menin (Lin et al. 2015).
A recent candidate compound screen in neuroendocrine tumor cell lines and in a pancreatic beta cell-specific Men1 knock-out mouse model by Lines et al. identified JQ1 as a potential treatment for neuroendocrine tumors (Lines, et al. 2017). Of note, no H3K4 demethylase inhibitors were tested in this study. JQ1 is a compound that targets the bromodomain and extraterminal domain (BET) protein family members that can bind active histone acetyl marks as well as other acetylated proteins through their bromodomains. The BET family includes the bromodomain-containing BRD2, BRD3, BRD4 and BRDT proteins. BET inhibition has been shown to be particularly effective in cancers that express high levels of the oncogenic transcription factor MYC by inhibiting MYC expression [reviewed by (Shu and Polyak 2017)]. JQ1 reduced the proliferation rate and increased apoptosis in a MEN1 pNET mouse model. However, a MYC-dependent mechanism could not be confirmed in the NET cell lines (Lines et al. 2017).
Apart from mouse models, there are no established experimental model systems based on human cells for MEN1-associated tumors. A large portion of the basic research on menin has been performed in cell types that are not affected in MEN1 [reviewed in (Pieterman, et al. 2014)]. The widely-used neuroendocrine cell lines BON-1 and QGP-1 do not harbor MEN1 gene mutations (Boora, et al. 2015). There is an urgent need for cell lines derived from MEN1-associated tumors. The MEN1 mouse models that have been developed mimic MEN1 to a large extent except for the fact that MEN1 mice do not develop non-functioning pNETs, which constitute the most important threat to the life expectancy of MEN1 patients. This poses an important problem for MEN1 research. In order to be able to properly test and validate future epigenetic treatments for MEN1-related tumors, novel preclinical models will have to be developed. Xenograft models, in which human tumor material is implanted and propagated in mice, may hold promise for the future. In addition, the establishment of three-dimensional tumor cell organoid cultures could enable large-scale experiments and drug screens aimed at restoring transcriptional regulation in MEN1-associated tumors (van de Wetering, et al. 2015).
Acknowledgments
Funding: MB is supported by the National Cancer Institute, grant number: P01 CA080111.
Footnotes
Declaration of interest: The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- Agarwal SK. Exploring the tumors of multiple endocrine neoplasia type 1 in mouse models for basic and preclinical studies. Int J Endocr Oncol. 2014;1:153–161. doi: 10.2217/ije.14.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal SK, Guru SC, Heppner C, Erdos MR, Collins RM, Park SY, Saggar S, Chandrasekharappa SC, Collins FS, Spiegel AM, et al. Menin interacts with the AP1 transcription factor JunD and represses JunD-activated transcription. Cell. 1999;96:143–152. doi: 10.1016/s0092-8674(00)80967-8. [DOI] [PubMed] [Google Scholar]
- Agarwal SK, Jothi R. Genome-wide characterization of menin-dependent H3K4me3 reveals a specific role for menin in the regulation of genes implicated in MEN1-like tumors. PLoS One. 2012;7:e37952. doi: 10.1371/journal.pone.0037952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnavion R, Teinturier R, Gherardi S, Leteurtre E, Yu R, Cordier-Bussat M, Du R, Pattou F, Vantyghem MC, Bertolino P, et al. Foxa2, a novel protein partner of the tumour suppressor menin, is deregulated in mouse and human MEN1 glucagonomas. J Pathol. 2017;242:90–101. doi: 10.1002/path.4885. [DOI] [PubMed] [Google Scholar]
- Boora GK, Kanwar R, Kulkarni AA, Pleticha J, Ames M, Schroth G, Beutler AS, Banck MS. Exome-level comparison of primary well-differentiated neuroendocrine tumors and their cell lines. Cancer Genet. 2015;208:374–381. doi: 10.1016/j.cancergen.2015.04.002. [DOI] [PubMed] [Google Scholar]
- Bradner JE, Hnisz D, Young RA. Transcriptional Addiction in Cancer. Cell. 2017;168:629–643. doi: 10.1016/j.cell.2016.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bres V, Yoshida T, Pickle L, Jones KA. SKIP interacts with c-Myc and Menin to promote HIV-1 Tat transactivation. Mol Cell. 2009;36:75–87. doi: 10.1016/j.molcel.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busygina V, Bale AE. Multiple endocrine neoplasia type 1 (MEN1) as a cancer predisposition syndrome: clues into the mechanisms of MEN1-related carcinogenesis. Yale J Biol Med. 2006;79:105–114. [PMC free article] [PubMed] [Google Scholar]
- Canaff L, Vanbellinghen JF, Kanazawa I, Kwak H, Garfield N, Vautour L, Hendy GN. Menin missense mutants encoded by the MEN1 gene that are targeted to the proteasome: restoration of expression and activity by CHIP siRNA. J Clin Endocrinol Metab. 2012;97:E282–291. doi: 10.1210/jc.2011-0241. [DOI] [PubMed] [Google Scholar]
- Cao Y, Liu R, Jiang X, Lu J, Jiang J, Zhang C, Li X, Ning G. Nuclear-cytoplasmic shuttling of menin regulates nuclear translocation of {beta}-catenin. Mol Cell Biol. 2009;29:5477–5487. doi: 10.1128/MCB.00335-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Xue Y, Xue L, Jiang X, Wang X, Zhang Z, Yang J, Lu J, Zhang C, Wang W, et al. Hepatic menin recruits SIRT1 to control liver steatosis through histone deacetylation. J Hepatol. 2013;59:1299–1306. doi: 10.1016/j.jhep.2013.07.011. [DOI] [PubMed] [Google Scholar]
- Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276:404–407. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- Chen G, A J, Wang M, Farley S, Lee LY, Lee LC, Sawicki MP. Menin promotes the Wnt signaling pathway in pancreatic endocrine cells. Mol Cancer Res. 2008;6:1894–1907. doi: 10.1158/1541-7786.MCR-07-2206. [DOI] [PubMed] [Google Scholar]
- Cheng P, Li G, Yang SS, Liu R, Jin G, Zhou XY, Hu XG. Tumor suppressor Menin acts as a corepressor of LXRalpha to inhibit hepatic lipogenesis. FEBS Lett. 2015;589:3079–3084. doi: 10.1016/j.febslet.2015.04.049. [DOI] [PubMed] [Google Scholar]
- Cheng P, Yang SS, Hu XG, Zhou XY, Zhang YJ, Jin G, Zhou YQ. Menin prevents liver steatosis through co-activation of peroxisome proliferator-activated receptor alpha. FEBS Lett. 2011;585:3403–3408. doi: 10.1016/j.febslet.2011.09.043. [DOI] [PubMed] [Google Scholar]
- Conemans EB, Nell S, Pieterman CR, de Herder WW, Dekkers OM, Hermus AR, van der Horst-Schrivers AN, Bisschop PH, Havekes B, Drent ML, et al. Prognostic Factors for Survival of Men 1 Patients with Duodenopancreatic Tumors Metastatic to the Liver: Results from the Dmsg Study Group. Endocr Pract. 2017 doi: 10.4158/EP161639.OR. [DOI] [PubMed] [Google Scholar]
- Dreijerink KM, Goudet P, Burgess JR, Valk GD. Breast-cancer predisposition in multiple endocrine neoplasia type 1. N Engl J Med. 2014;371:583–584. doi: 10.1056/NEJMc1406028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreijerink KM, Groner AC, Vos ES, Font-Tello A, Gu L, Chi D, Reyes J, Cook J, Lim E, Lin CY, et al. Enhancer-Mediated Oncogenic Function of the Menin Tumor Suppressor in Breast Cancer. Cell Rep. 2017;18:2359–2372. doi: 10.1016/j.celrep.2017.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreijerink KM, Mulder KW, Winkler GS, Hoppener JW, Lips CJ, Timmers HT. Menin links estrogen receptor activation to histone H3K4 trimethylation. Cancer Res. 2006;66:4929–4935. doi: 10.1158/0008-5472.CAN-05-4461. [DOI] [PubMed] [Google Scholar]
- Dreijerink KM, Varier RA, van Beekum O, Jeninga EH, Hoppener JW, Lips CJ, Kummer JA, Kalkhoven E, Timmers HT. The multiple endocrine neoplasia type 1 (MEN1) tumor suppressor regulates peroxisome proliferator-activated receptor gamma-dependent adipocyte differentiation. Mol Cell Biol. 2009a;29:5060–5069. doi: 10.1128/MCB.01001-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreijerink KM, Varier RA, van Nuland R, Broekhuizen R, Valk GD, van der Wal JE, Lips CJ, Kummer JA, Timmers HT. Regulation of vitamin D receptor function in MEN1-related parathyroid adenomas. Mol Cell Endocrinol. 2009b;313:1–8. doi: 10.1016/j.mce.2009.08.020. [DOI] [PubMed] [Google Scholar]
- Feng Z, Wang L, Sun Y, Jiang Z, Domsic J, An C, Xing B, Tian J, Liu X, Metz DC, et al. Menin and Daxx Interact to Suppress Neuroendocrine Tumors through Epigenetic Control of the Membrane Metallo-Endopeptidase. Cancer Res. 2017;77:401–411. doi: 10.1158/0008-5472.CAN-16-1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaniere S, Duvillie B, Scharfmann R, Carreira C, Wang ZQ, Zhang CX. Tumour suppressor menin is essential for development of the pancreatic endocrine cells. J Endocrinol. 2008;199:287–298. doi: 10.1677/JOE-08-0289. [DOI] [PubMed] [Google Scholar]
- Fontaniere S, Tost J, Wierinckx A, Lachuer J, Lu J, Hussein N, Busato F, Gut I, Wang ZQ, Zhang CX. Gene expression profiling in insulinomas of Men1 beta-cell mutant mice reveals early genetic and epigenetic events involved in pancreatic beta-cell tumorigenesis. Endocr Relat Cancer. 2006;13:1223–1236. doi: 10.1677/erc.1.01294. [DOI] [PubMed] [Google Scholar]
- Franklin DS, Godfrey VL, Lee H, Kovalev GI, Schoonhoven R, Chen-Kiang S, Su L, Xiong Y. CDK inhibitors p18(INK4c) and p27(Kip1) mediate two separate pathways to collaboratively suppress pituitary tumorigenesis. Genes Dev. 1998;12:2899–2911. doi: 10.1101/gad.12.18.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo A, Cuozzo C, Esposito I, Maggiolini M, Bonofiglio D, Vivacqua A, Garramone M, Weiss C, Bohmann D, Musti AM. Menin uncouples Elk-1, JunD and c-Jun phosphorylation from MAP kinase activation. Oncogene. 2002;21:6434–6445. doi: 10.1038/sj.onc.1205822. [DOI] [PubMed] [Google Scholar]
- Gobl AE, Berg M, Lopez-Egido JR, Oberg K, Skogseid B, Westin G. Menin represses JunD-activated transcription by a histone deacetylase-dependent mechanism. Biochim Biophys Acta. 1999;1447:51–56. doi: 10.1016/s0167-4781(99)00132-3. [DOI] [PubMed] [Google Scholar]
- Guru SC, Goldsmith PK, Burns AL, Marx SJ, Spiegel AM, Collins FS, Chandrasekharappa SC. Menin, the product of the MEN1 gene, is a nuclear protein. Proc Natl Acad Sci U S A. 1998;95:1630–1634. doi: 10.1073/pnas.95.4.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurung B, Feng Z, Hua X. Menin directly represses Gli1 expression independent of canonical Hedgehog signaling. Mol Cancer Res. 2013a;11:1215–1222. doi: 10.1158/1541-7786.MCR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurung B, Feng Z, Iwamoto DV, Thiel A, Jin G, Fan CM, Ng JM, Curran T, Hua X. Menin epigenetically represses Hedgehog signaling in MEN1 tumor syndrome. Cancer Res. 2013b;73:2650–2658. doi: 10.1158/0008-5472.CAN-12-3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamze Z, Vercherat C, Bernigaud-Lacheretz A, Bazzi W, Bonnavion R, Lu J, Calender A, Pouponnot C, Bertolino P, Roche C, et al. Altered MENIN expression disrupts the MAFA differentiation pathway in insulinoma. Endocr Relat Cancer. 2013;20:833–848. doi: 10.1530/ERC-13-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendy GN, Kaji H, Sowa H, Lebrun JJ, Canaff L. Menin and TGF-beta superfamily member signaling via the Smad pathway in pituitary, parathyroid and osteoblast. Horm Metab Res. 2005;37:375–379. doi: 10.1055/s-2005-870152. [DOI] [PubMed] [Google Scholar]
- Heppner C, Kester MB, Agarwal SK, Debelenko LV, Emmert-Buck MR, Guru SC, Manickam P, Olufemi SE, Skarulis MC, Doppman JL, et al. Somatic mutation of the MEN1 gene in parathyroid tumours. Nat Genet. 1997;16:375–378. doi: 10.1038/ng0897-375. [DOI] [PubMed] [Google Scholar]
- Horton JR, Liu X, Gale M, Wu L, Shanks JR, Zhang X, Webber PJ, Bell JS, Kales SC, Mott BT, et al. Structural Basis for KDM5A Histone Lysine Demethylase Inhibition by Diverse Compounds. Cell Chem Biol. 2016;23:769–781. doi: 10.1016/j.chembiol.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Gurung B, Wan B, Matkar S, Veniaminova NA, Wan K, Merchant JL, Hua X, Lei M. The same pocket in menin binds both MLL and JUND but has opposite effects on transcription. Nature. 2012;482:542–546. doi: 10.1038/nature10806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes CM, Rozenblatt-Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC, Hayes DN, Shanmugam KS, Bhattacharjee A, Biondi CA, et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell. 2004;13:587–597. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- Jiang X, Cao Y, Li F, Su Y, Li Y, Peng Y, Cheng Y, Zhang C, Wang W, Ning G. Targeting beta-catenin signaling for therapeutic intervention in MEN1-deficient pancreatic neuroendocrine tumours. Nat Commun. 2014;5:5809. doi: 10.1038/ncomms6809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, Schulick RD, Tang LH, Wolfgang CL, Choti MA, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331:1199–1203. doi: 10.1126/science.1200609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnik SK, Hughes CM, Gu X, Rozenblatt-Rosen O, McLean GW, Xiong Y, Meyerson M, Kim SK. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc Natl Acad Sci U S A. 2005;102:14659–14664. doi: 10.1073/pnas.0503484102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Lee JE, Cho EJ, Liu JO, Youn HD. Menin, a tumor suppressor, represses JunD-mediated transcriptional activity by association with an mSin3A-histone deacetylase complex. Cancer Res. 2003;63:6135–6139. [PubMed] [Google Scholar]
- Larsson C, Skogseid B, Oberg K, Nakamura Y, Nordenskjold M. Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma. Nature. 1988;332:85–87. doi: 10.1038/332085a0. [DOI] [PubMed] [Google Scholar]
- Lemmens I, Van de Ven WJ, Kas K, Zhang CX, Giraud S, Wautot V, Buisson N, De Witte K, Salandre J, Lenoir G, et al. Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. The European Consortium on MEN1. Hum Mol Genet. 1997;6:1177–1183. doi: 10.1093/hmg/6.7.1177. [DOI] [PubMed] [Google Scholar]
- Lemos MC, Thakker RV. Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat. 2008;29:22–32. doi: 10.1002/humu.20605. [DOI] [PubMed] [Google Scholar]
- Li BE, Gan T, Meyerson M, Rabbitts TH, Ernst P. Distinct pathways regulated by menin and by MLL1 in hematopoietic stem cells and developing B cells. Blood. 2013;122:2039–2046. doi: 10.1182/blood-2013-03-486647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Su Y, Cheng Y, Jiang X, Peng Y, Li Y, Lu J, Gu Y, Zhang C, Cao Y, et al. Conditional deletion of Men1 in the pancreatic beta-cell leads to glucagon-expressing tumor development. Endocrinology. 2015;156:48–57. doi: 10.1210/en.2014-1433. [DOI] [PubMed] [Google Scholar]
- Lin W, Cao J, Liu J, Beshiri ML, Fujiwara Y, Francis J, Cherniack AD, Geisen C, Blair LP, Zou MR, et al. Loss of the retinoblastoma binding protein 2 (RBP2) histone demethylase suppresses tumorigenesis in mice lacking Rb1 or Men1. Proc Natl Acad Sci U S A. 2011;108:13379–13386. doi: 10.1073/pnas.1110104108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Watanabe H, Peng S, Francis JM, Kaplan N, Pedamallu CS, Ramachandran A, Agoston A, Bass AJ, Meyerson M. Dynamic epigenetic regulation by menin during pancreatic islet tumor formation. Mol Cancer Res. 2015;13:689–698. doi: 10.1158/1541-7786.MCR-14-0457. [DOI] [PubMed] [Google Scholar]
- Lines KE, Stevenson M, Filippakopoulos P, Muller S, Lockstone HE, Wright B, Grozinsky-Glasberg S, Grossman AB, Knapp S, Buck D, et al. Epigenetic pathway inhibitors represent potential drugs for treating pancreatic and bronchial neuroendocrine tumors. Oncogenesis. 2017;6:e332. doi: 10.1038/oncsis.2017.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lips CJ, Dreijerink KM, Hoppener JW. Variable clinical expression in patients with a germline MEN1 disease gene mutation: clues to a genotype-phenotype correlation. Clinics (Sao Paulo) 2012;67(Suppl 1):49–56. doi: 10.6061/clinics/2012(Sup01)10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Hamze Z, Bonnavion R, Herath N, Pouponnot C, Assade F, Fontaniere S, Bertolino P, Cordier-Bussat M, Zhang CX. Reexpression of oncoprotein MafB in proliferative beta-cells and Men1 insulinomas in mouse. Oncogene. 2012;31:3647–3654. doi: 10.1038/onc.2011.538. [DOI] [PubMed] [Google Scholar]
- Lupien M, Brown M. Cistromics of hormone-dependent cancer. Endocr Relat Cancer. 2009;16:381–389. doi: 10.1677/ERC-09-0038. [DOI] [PubMed] [Google Scholar]
- Malik R, Khan AP, Asangani IA, Cieslik M, Prensner JR, Wang X, Iyer MK, Jiang X, Borkin D, Escara-Wilke J, et al. Targeting the MLL complex in castration-resistant prostate cancer. Nat Med. 2015;21:344–352. doi: 10.1038/nm.3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matkar S, Thiel A, Hua X. Menin: a scaffold protein that controls gene expression and cell signaling. Trends Biochem Sci. 2013;38:394–402. doi: 10.1016/j.tibs.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne TA, Hughes CM, Lloyd R, Yang Z, Rozenblatt-Rosen O, Dou Y, Schnepp RW, Krankel C, Livolsi VA, Gibbs D, et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc Natl Acad Sci U S A. 2005;102:749–754. doi: 10.1073/pnas.0408836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modali SD, Parekh VI, Kebebew E, Agarwal SK. Epigenetic regulation of the lncRNA MEG3 and its target c-MET in pancreatic neuroendocrine tumors. Mol Endocrinol. 2015;29:224–237. doi: 10.1210/me.2014-1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molatore S, Liyanarachchi S, Irmler M, Perren A, Mannelli M, Ercolino T, Beuschlein F, Jarzab B, Wloch J, Ziaja J, et al. Pheochromocytoma in rats with multiple endocrine neoplasia (MENX) shares gene expression patterns with human pheochromocytoma. Proc Natl Acad Sci U S A. 2010;107:18493–18498. doi: 10.1073/pnas.1003956107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai MJ, Chruszcz M, Reddy G, Grembecka J, Cierpicki T. Crystal structure of menin reveals binding site for mixed lineage leukemia (MLL) protein. J Biol Chem. 2011;286:31742–31748. doi: 10.1074/jbc.M111.258186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieterman CR, Conemans EB, Dreijerink KM, de Laat JM, Timmers HT, Vriens MR, Valk GD. Thoracic and duodenopancreatic neuroendocrine tumors in multiple endocrine neoplasia type 1: natural history and function of menin in tumorigenesis. Endocr Relat Cancer. 2014;21:R121–142. doi: 10.1530/ERC-13-0482. [DOI] [PubMed] [Google Scholar]
- Scacheri PC, Davis S, Odom DT, Crawford GE, Perkins S, Halawi MJ, Agarwal SK, Marx SJ, Spiegel AM, Meltzer PS, et al. Genome-wide analysis of menin binding provides insights into MEN1 tumorigenesis. PLoS Genet. 2006;2:e51. doi: 10.1371/journal.pgen.0020051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpa A, Chang DK, Nones K, Corbo V, Patch AM, Bailey P, Lawlor RT, Johns AL, Miller DK, Mafficini A, et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature. 2017;543:65–71. doi: 10.1038/nature21063. [DOI] [PubMed] [Google Scholar]
- Shi K, Parekh VI, Roy S, Desai SS, Agarwal SK. The embryonic transcription factor Hlxb9 is a menin interacting partner that controls pancreatic beta-cell proliferation and the expression of insulin regulators. Endocr Relat Cancer. 2013;20:111–122. doi: 10.1530/ERC-12-0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu S, Polyak K. BET Bromodomain Proteins as Cancer Therapeutic Targets. Cold Spring Harb Symp Quant Biol. 2017 doi: 10.1101/sqb.2016.81.030908. [DOI] [PubMed] [Google Scholar]
- Smith E, Lin C, Shilatifard A. The super elongation complex (SEC) and MLL in development and disease. Genes Dev. 2011;25:661–672. doi: 10.1101/gad.2015411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowa H, Kaji H, Canaff L, Hendy GN, Tsukamoto T, Yamaguchi T, Miyazono K, Sugimoto T, Chihara K. Inactivation of menin, the product of the multiple endocrine neoplasia type 1 gene, inhibits the commitment of multipotential mesenchymal stem cells into the osteoblast lineage. J Biol Chem. 2003;278:21058–21069. doi: 10.1074/jbc.M302044200. [DOI] [PubMed] [Google Scholar]
- Sowa H, Kaji H, Hendy GN, Canaff L, Komori T, Sugimoto T, Chihara K. Menin is required for bone morphogenetic protein 2- and transforming growth factor beta-regulated osteoblastic differentiation through interaction with Smads and Runx2. J Biol Chem. 2004;279:40267–40275. doi: 10.1074/jbc.M401312200. [DOI] [PubMed] [Google Scholar]
- Thakker RV, Newey PJ, Walls GV, Bilezikian J, Dralle H, Ebeling PR, Melmed S, Sakurai A, Tonelli F, Brandi ML. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1) J Clin Endocrinol Metab. 2012;97:2990–3011. doi: 10.1210/jc.2012-1230. [DOI] [PubMed] [Google Scholar]
- van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, van Houdt W, van Gorp J, Taylor-Weiner A, Kester L, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015;161:933–945. doi: 10.1016/j.cell.2015.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Nuland R, Smits AH, Pallaki P, Jansen PW, Vermeulen M, Timmers HT. Quantitative dissection and stoichiometry determination of the human SET1/MLL histone methyltransferase complexes. Mol Cell Biol. 2013;33:2067–2077. doi: 10.1128/MCB.01742-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradova M, Gehling VS, Gustafson A, Arora S, Tindell CA, Wilson C, Williamson KE, Guler GD, Gangurde P, Manieri W, et al. An inhibitor of KDM5 demethylases reduces survival of drug-tolerant cancer cells. Nat Chem Biol. 2016;12:531–538. doi: 10.1038/nchembio.2085. [DOI] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Yuan M, Shen S, Ma X, Fang J, Zhu L, Sun L, Liu Z, He X, Huang, et al. Menin enhances c-Myc-mediated transcription to promote cancer progression. Nat Commun. 2017;8:15278. doi: 10.1038/ncomms15278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuescher L, Angevine K, Hinds T, Ramakrishnan S, Najjar SM, Mensah-Osman EJ. Insulin regulates menin expression, cytoplasmic localization, and interaction with FOXO1. Am J Physiol Endocrinol Metab. 2011;301:E474–483. doi: 10.1152/ajpendo.00022.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaguchi H, Ohkura N, Takahashi M, Nagamura Y, Kitabayashi I, Tsukada T. Menin missense mutants associated with multiple endocrine neoplasia type 1 are rapidly degraded via the ubiquitin-proteasome pathway. Mol Cell Biol. 2004;24:6569–6580. doi: 10.1128/MCB.24.15.6569-6580.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YJ, Song TY, Park J, Lee J, Lim J, Jang H, Kim YN, Yang JH, Song Y, Choi A, et al. Menin mediates epigenetic regulation via histone H3 lysine 9 methylation. Cell Death Dis. 2013;4:e583. doi: 10.1038/cddis.2013.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A, Cleary ML. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell. 2008;14:36–46. doi: 10.1016/j.ccr.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A, Wang Z, Wysocka J, Sanyal M, Aufiero DJ, Kitabayashi I, Herr W, Cleary ML. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol. 2004;24:5639–5649. doi: 10.1128/MCB.24.13.5639-5649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman S, Sukhodolets K, Wang P, Qin J, Levens D, Agarwal SK, Marx SJ. FBP1 Is an Interacting Partner of Menin. Int J Endocrinol. 2014;2014:535401. doi: 10.1155/2014/535401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetoune AB, Fontaniere S, Magnin D, Anczukow O, Buisson M, Zhang CX, Mazoyer S. Comparison of nonsense-mediated mRNA decay efficiency in various murine tissues. BMC Genet. 2008;9:83. doi: 10.1186/1471-2156-9-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HL, Li WY, Zhang CP, Zhu YX, Wu L, Long HM, Li G, Luo M. Differentially expressed genes in Men1 knockout and wildtype embryoid bodies for pancreatic islet development. Mol Med Rep. 2011;4:301–305. doi: 10.3892/mmr.2011.409. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhou Y, Mehta KR, Danila DC, Scolavino S, Johnson SR, Klibanski A. A pituitary-derived MEG3 isoform functions as a growth suppressor in tumor cells. J Clin Endocrinol Metab. 2003;88:5119–5126. doi: 10.1210/jc.2003-030222. [DOI] [PubMed] [Google Scholar]