

Abstract
Autophagy is a conserved cellular process that plays an important role in cardiovascular homeostasis. Basal levels of autophagy are required for the maintenance of organellar quality control. Autophagy is dynamically regulated in the heart in the fasting to re-feeding transition. Insulin signaling plays an important role in the regulation of myocardial fuel metabolism, mitochondrial function and cellular growth. Recent studies have suggested an important role for insulin signaling in the regulation of myocardial autophagy. This dynamic regulation of autophagy induction during fasting may contribute to organellar homeostasis and if perturbed under conditions of hyperinsulinemia could contribute to accelerated cardiac aging.
Keywords: Insulin, Autophagy, Diabetes, L.V. Dysfunction
The discovery of insulin in 1921 revolutionized the treatment and prognosis of individuals with diabetes. Subsequent development of reliable radioimmunoassays for insulin increased our understanding of the diverse pathophysiology of diabetes, such as Type 2 Diabetes, which is associated with insulin resistance, hyperinsulinemia and relative beta-cell dysfunction, versus Type 1 Diabetes, which is characterized by absolute insulin deficiency. The identification of the insulin receptor and the critical components of the insulin signaling pathways significantly advanced our understanding of the molecular mechanisms that govern insulin signal transduction. Classically, studies of insulin signal transduction pathways have focused on its major role in metabolic regulation, nutrient homeostasis in the pathophysiology of insulin resistance and diabetes, focusing on insulin responsive targets such as adipose tissue, liver, skeletal muscle and the brain. The ubiquitous expression of insulin receptors has underscored the pleiotropic effects of insulin signaling in multiple organs including those of the cardiovascular system1,2. Studies of cardiovascular insulin signaling have provided novel insights linking insulin signaling with cardiovascular physiology and cardiovascular complications of obesity and diabetes1–5.
Overview of Insulin Signaling in the Cardiovascular System
In endothelial cells, insulin signaling is an important regulator of the phosphorylation of endothelial nitric oxide contributing to vasorelaxation1, 3, 4, 6. Not only does this contribute to the maintenance of blood pressure, but also enhances the delivery of substrates to classical metabolic targets of insulin action such as skeletal muscle1, 7, 8. Impaired insulin action or nitric oxide bioavailability in the vasculature are believed to contribute to the pathophysiology of vascular dysfunction and hypertension that characterizes obesity, Type 2 Diabetes and other insulin resistant states and may also contribute to the pathophysiology of atherosclerosis3–6. Similarly in vascular smooth muscle cells, hyperinsulinemia and increased insulin signaling have been implicated in vascular smooth muscle hypertrophy and intimal hyperplasia in response to vascular injury. The heart is also an important target for insulin action2, 9. Insulin signaling has been shown to play an important role in the modulation of myocardial substrate metabolism, cardiac structure and the response of the heart to various stressors such as altered workload or ischemia9–15. Given the substantial and absolute requirement of the heart for ATP to sustain myocardial contraction, the heart maintains high levels of substrate utilization, deriving 50–70% of ATP from the metabolism of fatty acids and the remainder from the metabolism of glucose and lactate16. Glucose enters cardiomyocytes predominantly via glucose transporters GLUT4 and GLUT12, 9, 17–23. Myocardial contraction promotes translocation of GLUT4 transporters from intracellular compartments to the sarcolemma to sustain glucose uptake. Insulin stimulation further increases the trafficking of GLUT4 to the sarcolemma to further increase myocardial glucose uptake and subsequent glucose metabolism, which in turn reduces fatty acid metabolism via the Randle Cycle. Although, insulin also increases the translocation of the fatty acid translocase CD36 and targets fatty acids to the triglyceride pool24, 25, the net effect of insulin on myocardial metabolism is to increase glucose metabolism (glycolysis and glucose oxidation) and to reduce fatty acid oxidation13. These metabolic changes are associated with a reduction in myocardial oxygen consumption and provided the rationale for use of glucose-potassium and insulin in the management of acute ischemia26. In diabetes or insulin resistant states, the changes in myocardial substrate metabolism are largely the consequence not of direct effects of altered insulin signaling on cardiomyocytes but effects that are secondary to the impact of diabetes on the availability of metabolic substrates to the heart, specifically an increased delivery of fatty acids to the heart which leads to increased FA utilization and a reciprocal reduction in glucose utilization13, 27.
In addition to these direct metabolic actions, studies in gene-targeted mice have demonstrated an important role for insulin signaling in the constitutive regulation of myocardial mitochondrial oxidative capacity, and the mitochondrial adaptations to physiological cardiac hypertrophy10, 28–31. Moreover, insulin signaling is an important regulator of cardiomyocyte size by its effect on growth signaling pathways mediated by phosphoinositide-3-kinase (PI3 Kinase) and Akt/PKB9, 32 and is required for preserving myocardial function and structure in response to pressure overload hypertrophy and ischemia12, 14, 15. Conversely, excessive insulin signaling has also been implicated in accelerating left ventricular remodeling in heart failure33.
Molecular Architecture of Insulin Signaling
As summarized in Figure 1, insulin catalyzes its pleiotropic effects following binding to its cognate receptor (IR) and increasing its phosphorylation. Activated IRs subsequently engage a network of intracellular signaling intermediates that are engaged by insulin receptor substrates (IRS), which act as scaffolds that facilitate activation of divergent branches of the insulin signal transduction pathways such as activation of PI3K and Akt/PKB or the MAPK/ERK signaling pathways. The closely related insulin-like growth factor 1 (IGF-1) signaling pathway which is activated by binding of its cognate receptor, shares many signaling characteristics with insulin receptors and mediates partially overlapping, but also distinct signaling outputs34. Of relevance to cardiomyocytes is the observation that insulin and IGF-1 receptors exist as hybrid receptors that mediate significant crosstalk with their respective ligands28, 35. Indeed, genetic deletion of insulin or IGF-1 receptors reveals substantial crosstalk between insulin and IGF-1 signaling to activate intracellular signaling pathways28. Although these signaling pathways are often depicted as linear pathways, there are important nodes of crosstalk between these branches of the insulin signaling pathway36. In addition, recent studies have also underscored important interactions between insulin signal transduction pathways and other signaling pathways such as the beta-adrenergic signaling pathway37.
Figure 1. Schematic Representation of Insulin Signaling Pathways.
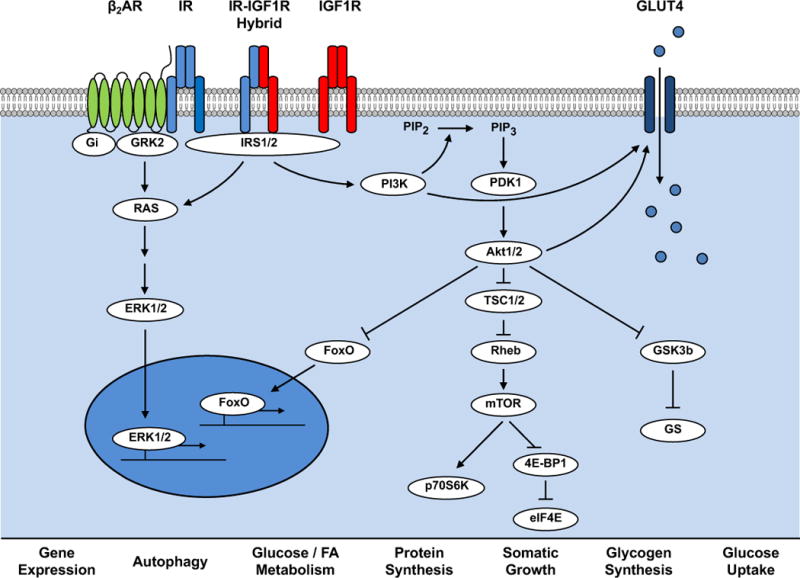
Insulin receptors (IR) and Insulin Like Growth Factor 1 receptors (IGF1R) are tyrosine kinase cell surface receptors that exist as homodimers or as hybrid heterodimers. Activation of these receptors leads to association with insulin receptor substrate isoforms (IRS1/2), which are signaling scaffolds that facilitate activation of intracellular signaling molecules such as phosphoinositide-3-kinase (PI3K), which converts phosphatidylinositol (3,4)-bisphosphate (PIP2) to phosphatidylinositol (3,4,5)-trisphosphate (PIP3) at the plasma membrane that interacts with the PH-domain containing protein Phosphoinositide Dependent Kinase (PDK1) that initiates activating phosphorylation of the serine threonine kinase Akt. Akt phosphorylates many intracellular targets that include: the Forkhead box O (FOXO1) transcription factor leading to its nuclear exclusion to reduce its transcriptional activity; tuberous sclerosis complex 2 (TSC2) which reverses its repression of Rheb leading to activation of mechanistic target of rapamycin (mTOR); Glycogen synthase kinase 3 beta (GSK3b) removing the repression of glycogen synthase thereby stimulating glycogen synthesis and by phosphorylating AS160 (not shown) leading to translocation of GLUT4 glucose transporters to increase glucose uptake. IRS activation also increases phosphorylation of the Mitogen Activated Protein Kinases (ERK1 and 2) that regulate gene expression. Insulin Receptors and IRS proteins also forms complexes with other receptor families as exemplified by beta-adrenergic receptors to modulate ERK and PI3K signaling.
Autophagy – Definition and Detection
Recent studies have underscored an important role for insulin signaling in the regulation of myocardial autophagy38, 39. These studies have not only provided insights into the physiological regulation myocardial autophagy by insulin signaling, but have also revealed important consequences on autophagy by conditions such as diabetes and obesity, which are associated with altered insulin signaling in the myocardium40. Before discussing these studies in detail a brief overview of autophagy and its regulation is provided. Autophagy is a conserved cellular process, which is characterized by the formation of double-membrane vesicular structures that engulf intracellular organelles that are subsequently targeted to lysosomes for enzymatic degradation of their cargo. The autophagic process is orchestrated by a family of autophagy regulating proteins (ATGs) that mediate specific steps (Figure 2). The non-specific or bulk engulfment of cellular cargo by autophagy is called macro-autophagy and in terms of insulin signaling, represents the pathway that has been most widely studied and for the purpose of this review will be described as “autophagy”. In addition, defined signaling pathways leading to autophagic engulfment of specific subcellular components exist and include mitophagy – the selective degradation of mitochondria, lipophagy – the selective degradation of lipid droplets, glycophagy – the specific degradation of glycogen, chaperone-mediated autophagy – which describes Lysosome-associated membrane protein 2 (LAMP2) mediated signaling that contributes to the lysosomal degradation of unfolded proteins and macropexophagy – the autophagosomal degradation of peroxisomes40–45.
Figure 2. Summary of Signaling Mediators of Autophagy.
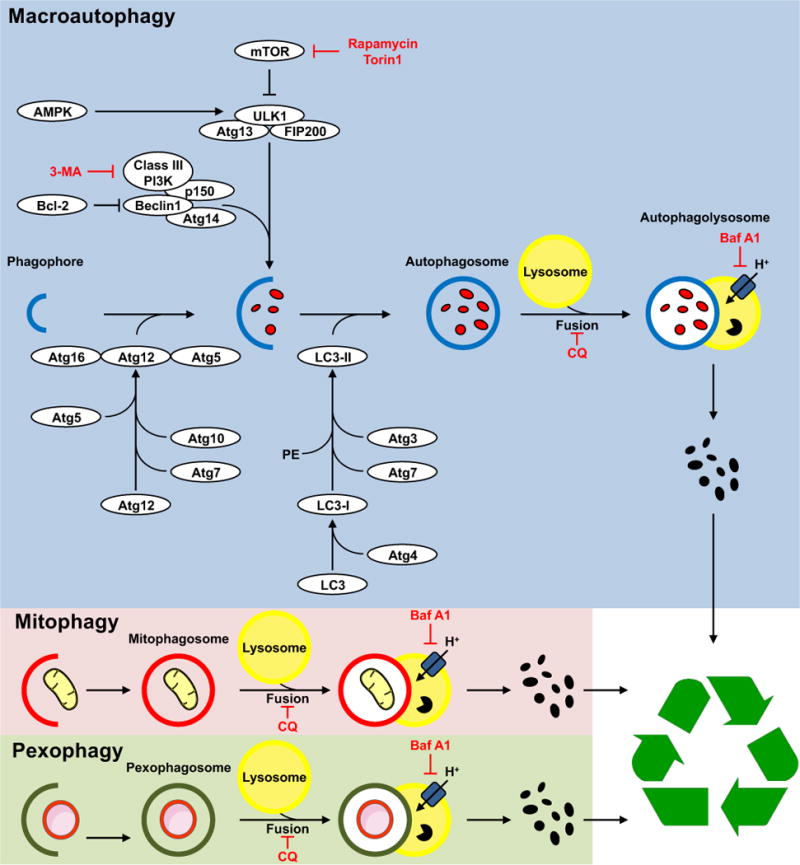
A family of autophagy regulating proteins (Atg) are involved in specific steps in autophagy including biogenesis of autophagosome membranes, membrane expansion and genesis of autophagosomes and sequestration of cargo. Reagents in red represent pharmacological inhibitors that are commonly used to block specific autophagic steps. 3-MA – 3 methyl adenine; CQ – Chloroquine; BafA – Bafilomycin A. The insets provide illustrations of examples of two organelle-specific pathways of autophagy, namely mitophagy and pexophagy in which mitochondria and peroxisomes respectively, are specifically targeted to autophagosomes. Signaling molecules that regulate mitophagy are summarized in the text. Once formed autophagosomes fuse with lysosomes and their content is digested and recycled.
Various tools are used to measure autophagy40, 46. Commonly used approaches include ultrastructural evaluation of tissues for autophagosome-like structures, immunohistochemical approaches that detect specific proteins that insert into autophagosomes (particularly microtubule-associated protein 1A/1B-light chain 3 (LC3 II)), co-localization of these structures with lysosomes or increased levels of LC3II by immunoblot. Reporters that label LC3 with fluorescence proteins such as green or red fluorescence protein (GFP or RFP) respectively can be transfected into cells or expressed in transgenic mice to provide a tool for quantifying autophagy by quantifying the number of labeled puncta that represent autophagosomes. An increase in the number of autophagosomes could be due to an increase in the generation of new autophagosomes or a reduction in lysosomal clearance. To determine the mechanism for changes in autophagosome number, approaches to measure autophagic turnover or flux have been designed. Exposing cells or treating animals with inhibitors of autophagosome/lysosome fusion will further increase the content of autophagosomes if autophagy induction is increased, whereas if an increase in autophagosome number is secondary to a defect in autophagosome clearance, then no further increase would be observed. Likewise, the use of dual-labeled LC3 with GFP and RFP has been used to estimate autophagic turnover on the basis of the quenching of GFP fluorescence in the acidic environment of the lysosome, in which RFP remains stable. Thus increased flux will result in increased RFP fluorescence and decreased turnover will be associated with persistence and overlap of GFP and RFP fluorescence. Finally, an important autophagosome cargo p62 is degraded within the lysosome, thus a reduction in p62 is also commonly used to indicate increased autophagic flux.
Signaling Pathways that Regulate Autophagy
Multiple upstream signaling pathways regulate the induction of autophagy in all cell types40, 47, 48. As summarized in Figure 3, macro-autophagy is activated by diverse upstream signaling pathways that largely respond to nutrient availability. An important integrator of nutrient sensing and substrate availability is the mechanistic target of rapamycin (mTOR). Activation of mTOR potently suppresses autophagy, and in the face of nutrient deprivation or pharmacological inhibition, mTOR inhibition leads to the initiation of signaling pathways that promote autophagosome formation in part by reduction of an inhibitory phosphorylation of the autophagy regulator unc-51 like autophagy activating kinase 1 (ULK1)49. Under more severe conditions of nutrient stress, activation of the energy sensor AMP activated protein kinase (AMPK) will lead to an inhibitory phosphorylation of mTOR and a stimulatory phosphorylation of ULK1 leading to autophagy activation50, 51. In addition to direct activation of signaling intermediates, nutrient deprivation can also modulate autophagy by transcriptional mechanisms that induce the expression of genes that encode for autophagy inducing proteins. One such transcriptional mechanism is mediated by the forkhead box O1 transcription factor (FOXO1) that is reversibly acetylated by sirtuin 1 in conditions of nutrient deprivation leading to nuclear occupancy and transcriptional activation of autophagy regulatory proteins that harbor FOXO binding sites52. It is clear that growth factor signaling pathways as exemplified by insulin signaling interacts importantly with many of these canonical autophagy regulatory signals. Thus by activating PI3K and Akt/PKB signaling, insulin will induce mTOR activation and autophagy inhibition. Similarly, insulin/Akt mediated phosphorylation of FOXO1, may induce nuclear exclusion of FOXO1 leading to reduced expression of autophagy related proteins. Thus insulin, which is an important sensor of systemic nutrient availability, will suppress autophagy via various molecular mechanisms. It is important to note that mTOR independent regulation of autophagy has been described, and organelle-restricted autophagy such as mitophagy, are regulated by signaling mechanisms that are in part distinct from that of macro-autophagy40, 45, 53, 54. The role of insulin signaling in the regulation of these novel alternate pathways is incompletely understood.
Figure 3. Regulation of Autophagy by Nutrient Status.
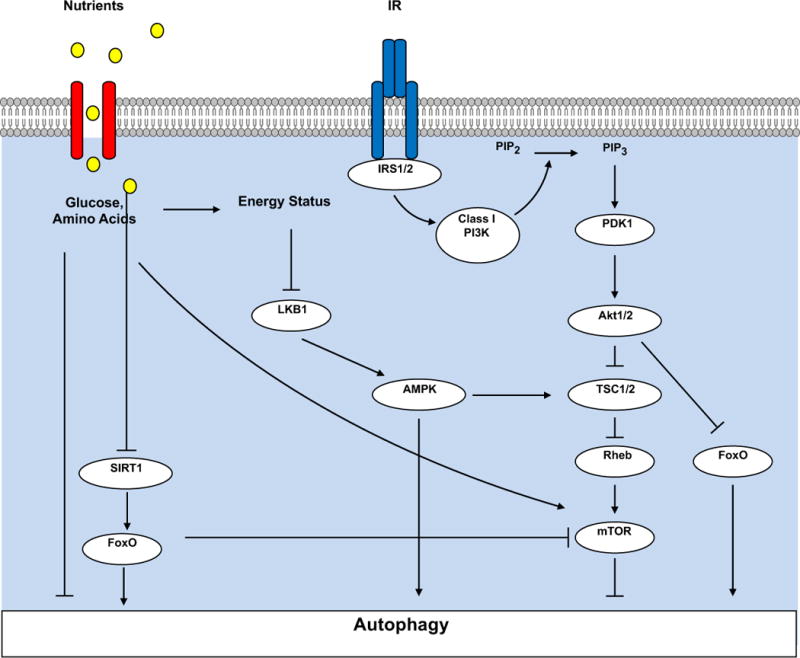
Nutrients such as amino acids or glucose can directly activate mTOR, which represses autophagy. Under conditions of nutrient deficiency, activation of AMP activated protein kinase (AMPK) by binding to AMP and promoting its phosphorylation by Liver kinase B1 (LKB1).
Activated AMPK will phosphorylate and inhibit mTOR leading to autophagy activation. AMPK may also directly activate autophagy by phosphorylating the autophagy mediator unc-51 like autophagy activating kinase 1 (ULK1)(not shown). Nutrient deficiency increases NAD+, which activates the NAD –dependent deactylase Sirtuin 1 (SIRT1), which will de-acetylate FOXO1, which is preferentially retained in the nucleus to increase its transcriptional activity on the promoters of genes encoding proteins that regulate autophagy. Increased nutrient availability promotes insulin release and activation of insulin receptors, which as summarized in Figure 1 leads to activation of mTOR, which suppresses autophagy and to phosphorylation of FOXO1, which excludes it from the nucleus, to lower its transcriptional activity on the promoters of genes that regulate autophagy.
Regulation of Myocardial Autophagy by Insulin Signaling
Many studies have revealed that constitutive levels of autophagy in the heart may play an important role in organellar quality control and the response of the heart to hemodynamic stress51, 55–58. Moreover, a suppression of cardiac autophagy has been associated with aging-associated cardiac dysfunction55, 59, 60. Studies in rodents revealed that fasting or caloric restriction is associated with a profound induction of myocardial autophagy in the adult heart61, 62. Moreover, these changes are reversible upon re-feeding. Also, in the immediate perinatal period of starvation, before breast feeding is established, autophagy is markedly induced within the heart and prevention of this process limits the ability of the fetus to survive brief periods of starvation63. Thus autophagy plays an essential role not only in maintaining cardiomyocyte viability during periods of nutrient deprivation, but also plays an important role in organellar quality control. Short-term, nutrient deprivation is associated with multiple changes in systemic metabolic homeostasis, which could impact autophagy. For example nutrient deprivation can induce sirtuin activity or reduce the availability of substrates such as amino acids or glucose that could activate mTOR. In addition, protracted or more severe nutrient deprivation can also activate autophagy by increasing the activity of AMPK. However, an important mediator of the fasting-associated induction of autophagy in the heart could be the fall in circulating insulin concentrations. To test this hypothesis we completely disrupted insulin signaling in cardiomyocytes by deleting the IRS1 and IRS1 genes. Loss of IRS1 and IRS2 resulted in unrestrained autophagy within the heart, which ultimately led to significant myocyte loss, heart failure and early mortality. These changes were also accompanied by mitochondrial dysfunction and increased apoptosis. To prove that the unrestrained autophagy could contribute to heart failure in this context we genetically inhibited autophagy in IRS1/2 deficient animals and restored cardiac function and structure39. Given that IRS1and IRS2 mediate insulin and IGF-1 signaling it is possible that the dramatic induction of autophagy in IRS1/2 deficient hearts could be the consequence of loss of signaling via both hormones.
Single knockouts of the insulin or the IGF-1 receptors respectively do not lead to catastrophic phenotypes in non-stressed hearts and the impact of loss of these signaling pathways in cardiomyocytes on autophagy remain to be determined9, 29. However, it was observed that in mice with deficiency of circulating IGF-1 on the basis of loss of hepatic IGF-1 synthesis, nutrient deprivation was associated with an increase in myocardial autophagy, relative to fasted wildtype mice64. Conversely, acute insulin administration to rats rendered insulinopenic with streptozotocin and to cultured cardiomyocytes in vitro following nutrient withdrawal acutely suppressed autophagic signaling38, 39. Changes in insulin concentrations in the fasting to re-feeding transition are more marked than are changes in circulating concentration of IGF-139, 64. As such it is likely that insulin might represent an important mediator of the physiological induction of autophagy following short term nutrient deprivation as occurs during overnight fasting or caloric restriction but the autophagic set point is modulated by ambient concentrations of IGF-1.
Given the potential beneficial effects of modest caloric restriction on longevity and cardiovascular health it is tempting to speculate that a modest increase in autophagy on the basis of reduced insulin signaling could increase basal autophagy sufficiently to enhance organellar quality control. Support for this notion comes from the observations that systemic insulin resistance, particularly when mild is associated with enhanced insulin signaling in the myocardium, which could lead to suppressed autophagy22, 33, 59, 60, 65. Aging is associated with generalized insulin resistance and hyperinsulinemia and studies in the hearts of aging rodents reveal that autophagy is suppressed59, 60. Indeed, the hearts of mice with reduced PI3K/Akt signaling on the basis of expression of a dominant negative PI3K transgene, exhibited reduced cardiac aging in concert with increased indices of autophagy and reduced accumulation of damaged organelles59. The opposite was observed in the hearts of animals with constitutive activation of PI3K/Akt signaling, which exacerbated aging-related cardiac dysfunction in concert with inhibition of basal autophagic flux59, 60. Our studies in mice with complete deficiency of IRS1 and IRS2, and studies in mice with constitutive or inducible loss of mTOR signaling in cardiomyocytes in which autophagy is over-activated and that ultimately develop heart failure, underscores the delicate balance between modest increases in autophagy that may promote cellular homeostasis and excessive autophagy that may promote cell death39, 66, 67.
Mitophagy
Whereas insulin signaling is likely an important physiological regulator of myocardial autophagy, a role for insulin in the regulation of mitophagy remains to be established. Studies in muscle cell lines have provided evidence that whereas nutrient deficiency might promote bulk autophagy, mitophagy might actually be decreased68. Mitophagy is activated in part by mitochondrial depolarization leading to stabilization and activation of PINK1 (phosphatase and tensin homolog-induced putative kinase) on the outer mitochondrial membrane. PINK1 phosphorylates various mitochondrial proteins such as mitofusin2 (mfn2), which recruits the E3 ubiquitin ligase Parkin, which in turn is activated by PINK1. Parkin ubiquinates mitochondrial target proteins such as mfn1, mfn2 and the voltage-dependent anion channel (VDAC) leading to their interaction with the adaptor protein, which drives interaction with the autophagosome membrane via its LC3 binding domain. Other signaling intermediates such as NIX, BNIP3 ((BCL2/adenovirus E1B 19kDa interacting protein 3), FUNDC1 (UN14 domain-containing protein 1), which may act as autophagy receptors and the E3 ubiquitin ligase SMAD-specific E3 ubiquitin ligase 1(SMURF1) play important roles in promoting mitophagy and these processes have been reviewed in detail elsewhere40, 45. Asymmetrical mitochondrial fission often yields a daughter mitochondrion with reduced membrane potential that is ultimately targeted for mitophagic degradation69. We recently observed that in cardiac muscle cells, insulin promotes mitochondrial fusion, which is required for its ability to activate mitochondrial substrate utilization70. Whether or not the converse is true, i.e. that reduced insulin signaling may lead to mitochondrial fission thereby potentially promoting mitophagy remains to be established.
Diabetes and Insulin Resistance
Given the role of insulin signaling in the regulation of myocardial autophagy and the association of diabetes with increased propensity for LV dysfunction, many investigators have evaluated the impact of diabetes on myocardial autophagy40. Diabetes is associated with complex systemic changes in the metabolic milieu. Type 1 Diabetes is associated with insulinopenia and a relative paucity of insulin action in the myocardium. By contrast, in Type 2 Diabetes, that is invariably associated with hyperinsulinemia and where this has been evaluated, is associated with reduced myocardial insulin-stimulated glucose uptake, but in many cases with intact proximal insulin signaling to the level of Akt2, 22, 65. In both of these circumstances uncontrolled diabetes is associated with increased circulating concentrations of glucose and fatty acids. These metabolic disturbances are therefore likely to influence myocardial autophagy in directions that might be difficult to predict. Based on the known effects of insulin to regulate autophagy as described above, then hypoinsulinemia in Type 1 Diabetes might be predicted to increase autophagic flux, whereas hyperinsulinemia in Type 2 Diabetes might be predicted to reduce autophagic flux. However, in animal models of Type 1 (insulinopenic) diabetes, autophagic flux has been reported to be reduced, via mechanisms that might involve activation of mTOR and repression of AMPK40, 71–73. As recently reviewed40, it remains to be established if the repression of autophagy in the hearts of animal models of Type 1 Diabetes is an adaptive or a maladaptive response given conflicting data. Pharmacological strategies that are associated with increased autophagy ameliorate cardiac dysfunction, whereas genetic strategies that may increase autophagy may worsen cardiac function while genetically lowering autophagic flux could be beneficial. Data in animal models of Type 2 Diabetes, usually induced by high-fat or high-fructose feeding are also variable40. This is due in part to limitations in determining autophagic flux. Where autophagic flux has been determined in models of insulin resistance and Type 2 Diabetes even in contexts where autophagosome number might be increased, there is supporting evidence that autophagic flux might be decreased. Indeed in a rodent study in which obesity and diabetes was induced by high-fat feeding the repression of autophagy was associated with increased myocardial injury following ischemia and reperfusion and treatment with rapamycin, which increased autophagic flux was associated with increased cardioprotection74. Taken together, diabetes leads to complex changes in autophagy in the heart that likely occurs on the basis of the confluence of multiple inciting pathophysiological mechanisms, which may or not be correlated with changes in myocardial insulin signaling. Additional studies will be required to elucidate the pathophysiology and functional consequences of altered myocardial autophagy in diabetes and to identify targets that can be therapeutically modulated.
Conclusion
Autophagy is a dynamic process that undergoes physiological regulation in the heart. In the transition from the fed state to the fasting state autophagy is activated within cardiac muscle and this might be related to falling concentrations of insulin. We posit that this dynamic regulation of autophagy might play an important role in organellar quality control. Aging is associated with generalized insulin resistance and the associated persistent hyperinsulinemia might constitutively suppress myocardial autophagy thereby contributing to age-related LV dysfunction. Additional studies are therefore required to determined if caloric restriction or other approaches to reduce myocardial insulin signaling might ameliorate LV dysfunction in the context of aging. The impact of diabetes on myocardial autophagy is complex and the specific contribution of insulin signaling is more difficult to discern. However, strategies to increase autophagic flux in the Type 2 Diabetes might be of benefit in the context of ischemia and reperfusion injury. This raises important questions regarding the impact of timing of insulin administration in susceptible patients with the metabolic syndrome in the context of reperfusion, which remains to be resolved.
Acknowledgments
Studies in the Abel Laboratory have been supported by RO1 HL108379 from the National Institutes of Health. CR was supported by a post-doctoral fellowship from the German Research Foundation (DFG).
Footnotes
Conflict of Interest Statement
The authors have no conflicts of interest to declare.
References
- 1.Muniyappa R, Montagnani M, Koh KK, Quon MJ. Cardiovascular actions of insulin. Endocr Rev. 2007;28:463–491. doi: 10.1210/er.2007-0006. [DOI] [PubMed] [Google Scholar]
- 2.Abel ED, O’Shea KM, Ramasamy R. Insulin resistance: metabolic mechanisms and consequences in the heart. Arterioscler Thromb Vasc Biol. 2012;32:2068–2076. doi: 10.1161/ATVBAHA.111.241984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wende AR, Symons JD, Abel ED. Mechanisms of lipotoxicity in the cardiovascular system. Curr Hypertens Rep. 2012;14:517–531. doi: 10.1007/s11906-012-0307-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Symons JD, McMillin SL, Riehle C, Tanner J, Palionyte M, Hillas E, et al. Contribution of insulin and Akt1 signaling to endothelial nitric oxide synthase in the regulation of endothelial function and blood pressure. Circ Res. 2009;104:1085–1094. doi: 10.1161/CIRCRESAHA.108.189316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rask-Madsen C, King GL. Vascular complications of diabetes: mechanisms of injury and protective factors. Cell Metab. 2013;17:20–33. doi: 10.1016/j.cmet.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang QJ, Holland WL, Wilson L, Tanner JM, Kearns D, Cahoon JM, et al. Ceramide mediates vascular dysfunction in diet-induced obesity by PP2A-mediated dephosphorylation of the eNOS-Akt complex. Diabetes. 2012;61:1848–1859. doi: 10.2337/db11-1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abe H, Yamada N, Kamata K, Kuwaki T, Shimada M, Osuga J, et al. Hypertension, hypertriglyceridemia, and impaired endothelium-dependent vascular relaxation in mice lacking insulin receptor substrate-1. J Clin Invest. 1998;101:1784–1788. doi: 10.1172/JCI1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kubota T, Kubota N, Kumagai H, Yamaguchi S, Kozono H, Takahashi T, et al. Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell Metab. 2011;13:294–307. doi: 10.1016/j.cmet.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 9.Belke DD, Betuing S, Tuttle MJ, Graveleau C, Young ME, Pham M, et al. Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. J Clin Invest. 2002;109:629–639. doi: 10.1172/JCI13946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boudina S, Bugger H, Sena S, O’Neill BT, Zaha VG, Ilkun O, et al. Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation. 2009;119:1272–1283. doi: 10.1161/CIRCULATIONAHA.108.792101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bugger H, Riehle C, Jaishy B, Wende AR, Tuinei J, Chen D, et al. Genetic loss of insulin receptors worsens cardiac efficiency in diabetes. J Mol Cell Cardiol. 2012;52:1019–1026. doi: 10.1016/j.yjmcc.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu P, Zhang D, Swenson L, Chakrabarti G, Abel ED, Litwin SE. Minimally invasive aortic banding in mice: effects of altered cardiomyocyte insulin signaling during pressure overload. Am J Physiol Heart Circ Physiol. 2003;285:H1261–1269. doi: 10.1152/ajpheart.00108.2003. [DOI] [PubMed] [Google Scholar]
- 13.Mazumder PK, O’Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, et al. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes. 2004;53:2366–2374. doi: 10.2337/diabetes.53.9.2366. [DOI] [PubMed] [Google Scholar]
- 14.McQueen AP, Zhang D, Hu P, Swenson L, Yang Y, Zaha VG, et al. Contractile dysfunction in hypertrophied hearts with deficient insulin receptor signaling: possible role of reduced capillary density. J Mol Cell Cardiol. 2005;39:882–892. doi: 10.1016/j.yjmcc.2005.07.017. [DOI] [PubMed] [Google Scholar]
- 15.Sena S, Hu P, Zhang D, Wang X, Wayment B, Olsen C, et al. Impaired insulin signaling accelerates cardiac mitochondrial dysfunction after myocardial infarction. J Mol Cell Cardiol. 2009;46:910–918. doi: 10.1016/j.yjmcc.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res. 2013;113:709–724. doi: 10.1161/CIRCRESAHA.113.300376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abel ED. Glucose transport in the heart. Front Biosci. 2004;9:201–215. doi: 10.2741/1216. [DOI] [PubMed] [Google Scholar]
- 18.Abel ED, Kaulbach HC, Tian R, Hopkins JC, Duffy J, Doetschman T, et al. Cardiac hypertrophy with preserved contractile function after selective deletion of GLUT4 from the heart. J Clin Invest. 1999;104:1703–1714. doi: 10.1172/JCI7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pereira RO, Wende AR, Olsen C, Soto J, Rawlings T, Zhu Y, et al. Inducible overexpression of GLUT1 prevents mitochondrial dysfunction and attenuates structural remodeling in pressure overload but does not prevent left ventricular dysfunction. J Am Heart Assoc. 2013;2:e000301. doi: 10.1161/JAHA.113.000301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pereira RO, Wende AR, Olsen C, Soto J, Rawlings T, Zhu Y, et al. GLUT1 deficiency in cardiomyocytes does not accelerate the transition from compensated hypertrophy to heart failure. J Mol Cell Cardiol. 2014;72:95–103. doi: 10.1016/j.yjmcc.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tian R, Abel ED. Responses of GLUT4-deficient hearts to ischemia underscore the importance of glycolysis. Circulation. 2001;103:2961–2966. doi: 10.1161/01.cir.103.24.2961. [DOI] [PubMed] [Google Scholar]
- 22.Wright JJ, Kim J, Buchanan J, Boudina S, Sena S, Bakirtzi K, et al. Mechanisms for increased myocardial fatty acid utilization following short-term high-fat feeding. Cardiovasc Res. 2009;82:351–360. doi: 10.1093/cvr/cvp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu Y, Pereira RO, O’Neill BT, Riehle C, Ilkun O, Wende AR, et al. Cardiac PI3K-Akt impairs insulin-stimulated glucose uptake independent of mTORC1 and GLUT4 translocation. Mol Endocrinol. 2013;27:172–184. doi: 10.1210/me.2012-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glatz JF, Luiken JJ, Bonen A. Membrane fatty acid transporters as regulators of lipid metabolism: implications for metabolic disease. Physiol Rev. 2010;90:367–417. doi: 10.1152/physrev.00003.2009. [DOI] [PubMed] [Google Scholar]
- 25.Schwenk RW, Luiken JJ, Bonen A, Glatz JF. Regulation of sarcolemmal glucose and fatty acid transporters in cardiac disease. Cardiovasc Res. 2008;79:249–258. doi: 10.1093/cvr/cvn116. [DOI] [PubMed] [Google Scholar]
- 26.Grossman AN, Opie LH, Beshansky JR, Ingwall JS, Rackley CE, Selker HP. Glucose-insulin-potassium revived: current status in acute coronary syndromes and the energy-depleted heart. Circulation. 2013;127:1040–1048. doi: 10.1161/CIRCULATIONAHA.112.130625. [DOI] [PubMed] [Google Scholar]
- 27.Abel ED, Litwin SE, Sweeney G. Cardiac remodeling in obesity. Physiol Rev. 2008;88:389–419. doi: 10.1152/physrev.00017.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ikeda H, Shiojima I, Ozasa Y, Yoshida M, Holzenberger M, Kahn CR, et al. Interaction of myocardial insulin receptor and IGF receptor signaling in exercise-induced cardiac hypertrophy. J Mol Cell Cardiol. 2009;47:664–675. doi: 10.1016/j.yjmcc.2009.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim J, Wende AR, Sena S, Theobald HA, Soto J, Sloan C, et al. Insulin-like growth factor I receptor signaling is required for exercise-induced cardiac hypertrophy. Mol Endocrinol. 2008;22:2531–2543. doi: 10.1210/me.2008-0265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Neill BT, Kim J, Wende AR, Theobald HA, Tuinei J, Buchanan J, et al. A conserved role for phosphatidylinositol 3-kinase but not Akt signaling in mitochondrial adaptations that accompany physiological cardiac hypertrophy. Cell Metab. 2007;6:294–306. doi: 10.1016/j.cmet.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Riehle C, Wende AR, Zhu Y, Oliveira KJ, Pereira RO, Jaishy BP, et al. Insulin receptor substrates are essential for the bioenergetic and hypertrophic response of the heart to exercise training. Mol Cell Biol. 2014;34:3450–3460. doi: 10.1128/MCB.00426-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shiojima I, Yefremashvili M, Luo Z, Kureishi Y, Takahashi A, Tao J, et al. Akt signaling mediates postnatal heart growth in response to insulin and nutritional status. J Biol Chem. 2002;277:37670–37677. doi: 10.1074/jbc.M204572200. [DOI] [PubMed] [Google Scholar]
- 33.Shimizu I, Minamino T, Toko H, Okada S, Ikeda H, Yasuda N, et al. Excessive cardiac insulin signaling exacerbates systolic dysfunction induced by pressure overload in rodents. J Clin Invest. 2010;120:1506–1514. doi: 10.1172/JCI40096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Werner H, Weinstein D, Bentov I. Similarities and differences between insulin and IGF-I: structures, receptors, and signalling pathways. Arch Physiol Biochem. 2008;114:17–22. doi: 10.1080/13813450801900694. [DOI] [PubMed] [Google Scholar]
- 35.Blanquart C, Gonzalez-Yanes C, Issad T. Monitoring the activation state of insulin/insulin-like growth factor-1 hybrid receptors using bioluminescence resonance energy transfer. Mol Pharmacol. 2006;70:1802–1811. doi: 10.1124/mol.106.026989. [DOI] [PubMed] [Google Scholar]
- 36.Begum N, Ragolia L, Rienzie J, McCarthy M, Duddy N. Regulation of mitogen-activated protein kinase phosphatase-1 induction by insulin in vascular smooth muscle cells. Evaluation of the role of the nitric oxide signaling pathway and potential defects in hypertension. J Biol Chem. 1998;273:25164–25170. doi: 10.1074/jbc.273.39.25164. [DOI] [PubMed] [Google Scholar]
- 37.Fu Q, Xu B, Liu Y, Parikh D, Li J, Li Y, et al. Insulin inhibits cardiac contractility by inducing a Gi-biased beta2-adrenergic signaling in hearts. Diabetes. 2014;63:2676–2689. doi: 10.2337/db13-1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paula-Gomes S, Goncalves DA, Baviera AM, Zanon NM, Navegantes LC, Kettelhut IC. Insulin suppresses atrophy- and autophagy-related genes in heart tissue and cardiomyocytes through AKT/FOXO signaling. Horm Metab Res. 2013;45:849–855. doi: 10.1055/s-0033-1347209. [DOI] [PubMed] [Google Scholar]
- 39.Riehle C, Wende AR, Sena S, Pires KM, Pereira RO, Zhu Y, et al. Insulin receptor substrate signaling suppresses neonatal autophagy in the heart. J Clin Invest. 2013;123:5319–5333. doi: 10.1172/JCI71171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kobayashi S, Liang Q. Autophagy and mitophagy in diabetic cardiomyopathy. Biochim Biophys Acta. 2014 doi: 10.1016/j.bbadis.2014.05.020. [DOI] [PubMed] [Google Scholar]
- 41.Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. 2012;22:407–417. doi: 10.1016/j.tcb.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh R, Cuervo AM. Lipophagy: connecting autophagy and lipid metabolism. Int J Cell Biol. 2012;2012:282041. doi: 10.1155/2012/282041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res. 2014;24:24–41. doi: 10.1038/cr.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mellor KM, Varma U, Stapleton DI, Delbridge LM. Cardiomyocyte glycophagy is regulated by insulin and exposure to high extracellular glucose. Am J Physiol Heart Circ Physiol. 2014;306:H1240–1245. doi: 10.1152/ajpheart.00059.2014. [DOI] [PubMed] [Google Scholar]
- 45.Hammerling BC, Gustafsson AB. Mitochondrial quality control in the myocardium: Cooperation between protein degradation and mitophagy. J Mol Cell Cardiol. 2014;75C:122–130. doi: 10.1016/j.yjmcc.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Webster BR, Scott I, Traba J, Han K, Sack MN. Regulation of autophagy and mitophagy by nutrient availability and acetylation. Biochim Biophys Acta. 2014;1841:525–534. doi: 10.1016/j.bbalip.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stipanuk MH. Macroautophagy and its role in nutrient homeostasis. Nutr Rev. 2009;67:677–689. doi: 10.1111/j.1753-4887.2009.00252.x. [DOI] [PubMed] [Google Scholar]
- 49.Dunlop EA, Tee AR. mTOR and autophagy: A dynamic relationship governed by nutrients and energy. Semin Cell Dev Biol. 2014 doi: 10.1016/j.semcdb.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 50.Kubli DA, Gustafsson AB. Cardiomyocyte health: adapting to metabolic changes through autophagy. Trends Endocrinol Metab. 2014;25:156–164. doi: 10.1016/j.tem.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hariharan N, Maejima Y, Nakae J, Paik J, Depinho RA, Sadoshima J. Deacetylation of FoxO by Sirt1 Plays an Essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes. Circ Res. 2010;107:1470–1482. doi: 10.1161/CIRCRESAHA.110.227371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, et al. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009;461:654–658. doi: 10.1038/nature08455. [DOI] [PubMed] [Google Scholar]
- 54.Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2009;16:46–56. doi: 10.1038/cdd.2008.110. [DOI] [PubMed] [Google Scholar]
- 55.De Meyer GR, Martinet W. Autophagy in the cardiovascular system. Biochim Biophys Acta. 2009;1793:1485–1495. doi: 10.1016/j.bbamcr.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 56.Takagi H, Matsui Y, Sadoshima J. The role of autophagy in mediating cell survival and death during ischemia and reperfusion in the heart. Antioxid Redox Signal. 2007;9:1373–1381. doi: 10.1089/ars.2007.1689. [DOI] [PubMed] [Google Scholar]
- 57.Taneike M, Yamaguchi O, Nakai A, Hikoso S, Takeda T, Mizote I, et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy. 2010;6:600–606. doi: 10.4161/auto.6.5.11947. [DOI] [PubMed] [Google Scholar]
- 58.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 59.Inuzuka Y, Okuda J, Kawashima T, Kato T, Niizuma S, Tamaki Y, et al. Suppression of phosphoinositide 3-kinase prevents cardiac aging in mice. Circulation. 2009;120:1695–1703. doi: 10.1161/CIRCULATIONAHA.109.871137. [DOI] [PubMed] [Google Scholar]
- 60.Hua Y, Zhang Y, Ceylan-Isik AF, Wold LE, Nunn JM, Ren J. Chronic Akt activation accentuates aging-induced cardiac hypertrophy and myocardial contractile dysfunction: role of autophagy. Basic Res Cardiol. 2011;106:1173–1191. doi: 10.1007/s00395-011-0222-8. [DOI] [PubMed] [Google Scholar]
- 61.Kanamori H, Takemura G, Maruyama R, Goto K, Tsujimoto A, Ogino A, et al. Functional significance and morphological characterization of starvation-induced autophagy in the adult heart. Am J Pathol. 2009;174:1705–1714. doi: 10.2353/ajpath.2009.080875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takemura G, Kanamori H, Goto K, Maruyama R, Tsujimoto A, Fujiwara H, et al. Autophagy maintains cardiac function in the starved adult. Autophagy. 2009;5:1034–1036. doi: 10.4161/auto.5.7.9297. [DOI] [PubMed] [Google Scholar]
- 63.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 64.Troncoso R, Vicencio JM, Parra V, Nemchenko A, Kawashima Y, Del Campo A, et al. Energy-preserving effects of IGF-1 antagonize starvation-induced cardiac autophagy. Cardiovasc Res. 2012;93:320–329. doi: 10.1093/cvr/cvr321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cook SA, Varela-Carver A, Mongillo M, Kleinert C, Khan MT, Leccisotti L, et al. Abnormal myocardial insulin signalling in type 2 diabetes and left-ventricular dysfunction. Eur Heart J. 2010;31:100–111. doi: 10.1093/eurheartj/ehp396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang D, Contu R, Latronico MV, Zhang J, Rizzi R, Catalucci D, et al. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J Clin Invest. 2010;120:2805–2816. doi: 10.1172/JCI43008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shende P, Plaisance I, Morandi C, Pellieux C, Berthonneche C, Zorzato F, et al. Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation. 2011;123:1073–1082. doi: 10.1161/CIRCULATIONAHA.110.977066. [DOI] [PubMed] [Google Scholar]
- 68.Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13:589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Parra V, Verdejo HE, Iglewski M, Del Campo A, Troncoso R, Jones D, et al. Insulin stimulates mitochondrial fusion and function in cardiomyocytes via the Akt-mTO-RNFkappaB-Opa-1 signaling pathway. Diabetes. 2014;63:75–88. doi: 10.2337/db13-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes. 2011;60:1770–1778. doi: 10.2337/db10-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xu X, Kobayashi S, Chen K, Timm D, Volden P, Huang Y, et al. Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem. 2013;288:18077–18092. doi: 10.1074/jbc.M113.474650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhao Y, Zhang L, Qiao Y, Zhou X, Wu G, Wang L, et al. Heme oxygenase-1 prevents cardiac dysfunction in streptozotocin-diabetic mice by reducing inflammation, oxidative stress, apoptosis and enhancing autophagy. PLoS One. 2013;8:e75927. doi: 10.1371/journal.pone.0075927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sciarretta S, Zhai P, Shao D, Maejima Y, Robbins J, Volpe M, et al. Rheb is a critical regulator of autophagy during myocardial ischemia: pathophysiological implications in obesity and metabolic syndrome. Circulation. 2012;125:1134–1146. doi: 10.1161/CIRCULATIONAHA.111.078212. [DOI] [PMC free article] [PubMed] [Google Scholar]