

Abstract
Rationale
Coronary Artery Disease (CAD) is a critical determinant of morbidity and mortality. Previous studies have identified several cardiovascular disease (CVD) risk factors, which may partly arise from a shared genetic basis with CAD, and thus be useful for discovery of CAD genes.
Objective
We aimed to improve discovery of CAD genes, and inform the etiologic relationship between CAD and several CVD risk factors using a shared polygenic signal-informed statistical framework.
Methods and Results
Using genome-wide association studies (GWAS) summary statistics and shared polygenic pleiotropy-informed conditional and conjunctional false discovery rate (FDR) methodology, we systematically investigated genetic overlap between CAD and 8 traits related to CVD risk factors: low density lipoprotein (LDL) cholesterol, high density lipoprotein (HDL) cholesterol, triglycerides (TG), type 2 diabetes (T2D), C-reactive protein (CRP), body mass index (BMI), systolic blood pressure (SBP) and type 1 diabetes (T1D). We found significant enrichment of single nucleotide polymorphisms (SNPs) associated with CAD as a function of their association with LDL, HDL, TG, T2D, CRP, BMI, SBP and T1D. Applying the conditional FDR method to the enriched phenotypes, we identified 67 novel loci associated with CAD (overall conditional FDR < 0.01). Further, we identified 53 loci with significant effects in both CAD and at least one of LDL, HDL, TG, T2D, CRP, SBP and T1D.
Conclusions
The observed polygenic overlap between CAD and cardio-metabolic risk factors indicates an etiological relation that warrants further investigation. The new gene loci identified implicate novel genetic mechanisms related to CAD.
Keywords: Genome-wide association study, genetic pleiotropy, comorbid disorder, lipids, cardiovascular genomics, DNA polymorphisms, genetic epidemiology
Subject terms: Chronic Ischemic Heart Disease; Genetics, Association Studies; Hypertension
INTRODUCTION
Coronary artery disease (CAD) is a leading cause of death worldwide. The development of CAD is influenced by both genetic and environmental factors, as evident by its high heritability (40–50%), shown in twin and family studies1. Genome-wide association studies (GWAS) in CAD have identified a total of 46 genetic variants reaching genome-wide significance for CAD2. However, the identified genetic variants explain only a small proportion of estimated heritability2, i.e. only a small amount of the familial clustering of CAD. This apparent paradox is widely seen across GWAS for complex traits and is termed “the missing heritability problem”3, 4. However, recent discoveries suggest that existing GWAS can capture more of the heritability due to common variants if proper statistical tools are used5–7.
Hypertension8, obesity9, abdominal fat10, diabetes11, dyslipidemia12–14, inflammation as reflected by high levels of C-reactive protein (CRP)15, are associated with CAD. Several studies have found overlapping pathophysiology16, but the underlying shared genetic factors and the extent of the polygenic overlap across these phenotypes are mainly unknown. We have developed an analytical framework for complex traits building on the polygenic overlap17 between two or more phenotypes6. This method has the potential to capture more of the polygenic effects in complex traits18, and has successfully been applied to psychiatric6, cardiovascular19, neurological diseases20 and cancer21. This ‘shared polygenic signal’ method could be particularly informative in CAD, a disease with known co-morbidities and overlapping pathophysiology with related cardiovascular and metabolic disorders2, 22–25.
We used this approach to leverage the power of multiple large genomic studies to describe the extent of the polygenic overlap and identify overlapping SNPs between CAD and 8 associated traits and cardiovascular disease (CVD) risk factors where recent GWAS results are available: low density lipoprotein (LDL) cholesterol26, high density lipoprotein (HDL) cholesterol26, triglycerides (TG)26, type 2 diabetes (T2D)27, CRP28, body mass index (BMI)29, systolic blood pressure (SBP)30, 31 and type 1 diabetes (T1D)32. By combining data from these different GWAS, we hypothesized that the shared polygenic signal approach can improve discovery of CAD genes, and inform the etiologic relationship between CAD and CVD risk factors.
METHODS
Participant samples
We obtained summary statistics from large-scale genomic studies (p-values and risk allele when available) from public access websites or through collaboration with investigators. The summary statistics are based on the Metabochip33 for CAD2 (n=194,427 including 63,746 cases) and T2D27 (n=149,830), and standard GWAS for LDL26 (n=95,454), HDL26 (n=99,900), TG26 (n=96,568), BMI29 (n=123,865), SBP31 (n=203,056) and T1D32 (n=16,559), and CRP (n=66,185)28. Details on the inclusion criteria and phenotype characteristics of the different GWAS are described in the original publications.
There were some overlapping controls between CAD and T2D and also between CAD and T1D. In both instances this was mainly due to the inclusion of one or more sub-studies employing a shared control design (e.g. used by the Wellcome Trust Case Control Consortium and deCODE Genetics)34 (see Online Table I). There was also some sample overlap between CAD and LDL, HDL, TG, BMI and SBP (Online Table I). Note that even without raw data, an upper bound for the amount of sample overlap is obtainable from the original publications by comparing the sub-study definitions and samples sizes for CAD and each secondary trait (correlation of uncorrected test statistics due to sample overlap is given in Online Table I; see LeBlanc et al (in prep) for details).
The Women’s Genome Health Study (WGHS), initiated in 1992, is an ongoing prospective cohort including 23,294 initially healthy North American women of European ancestry with whole genome genotype data and follow-up formajor incident health events, including myocardial infarction (MI) and coronary heart disease (CHD; composed of MI, CHD death, and coronary revascularization) are recorded35. Over the approximately, 20 years of follow-up, there were 387 and 1007 cases respectively of incident MI and CHD among the 23,294 women.
The relevant institutional review boards or ethics committees approved the research protocol of the individual GWAS and all participants provided written informed consent.
Statistical analyses
We use Matlab (version R2013a) for all statistical analysis unless otherwise indicated. First, we looked for evidence of overlapping polygenic signal for CAD and each secondary trait. In the absence of an overlapping polygenic signal, the expectation is that the p-value distribution for CAD is independent from the p-values in the secondary trait. The dependency of the p-value distribution for CAD on each secondary trait can be visually explored using conditional quantile-quantile plots to evaluate genetic ‘enrichment’ in CAD as a function of a secondary phenotype. Quantile-quantile plots are a descriptive tool for visualizing the difference between an observed distribution and a theoretical distribution. With GWAS, quantiles of the observed (nominal) p-values, denoted by ‘p’, are plotted on the y-axis, with the quantiles of the theoretical null distribution (i.e. the uniform distribution), here denoted by ‘q’, on the x-axis. Conventionally, the -log10 transform is used to emphasize tail areas. If there is no deviation from the null distribution and thus no true genetic association present, a quantile-quantile plot falls on the 1:1 line. Leftward deflections of the observed distribution from the null line reflect increased tail probabilities in the distribution of the test statistics, and consequently an over-abundance of low p-values compared to that expected by chance, termed ‘enrichment’. Here, we constructed conditional quantile-quantile plots to investigate if enrichment in the primary phenotype (CAD) is related to significance in a given secondary phenotype, as visualized by a leftward deflection from the null line on the conditional quantile-quantile plot. A conditional quantile-quantile plot was separately constructed for CAD and each of the 8 secondary traits. To test for statistical significance associated with these conditional quantile-quantile plots, we used the Anderson-Darling test21. In brief, this is a statistical test of whether a given sample of data is drawn from a given probability distribution and allows us to determine if an observed leftward deflection is statistically significant (for additional details see 21). In this case, we used set of SNPs (GWAS p>0.1 in the secondary trait), i.e., SNPs that are signal depleted in the secondary trait, as the comparison set.
Second, once statistically significant enrichment was confirmed, we computed conditional False Discovery Rates (FDR), a statistical framework that leverages shared polygenic signal6, 18, to improve the discovery of SNPs for the primary trait of interest, CAD. The standard FDR is designed to control the expected proportion of incorrectly rejected null hypotheses, and is employed to correct for multiple comparisons. An extension of the standard FDR is the conditional FDR6, which in our application, is used to incorporate information from GWAS summary statistics of a second phenotype. The conditional FDR is defined as the probability of a SNP being null in the first phenotype given that the p-values in the first and second phenotype are as small as or smaller than the observed ones (see Supplemental Methods). Importantly, ranking SNPs according to conditional FDR re-orders SNPs compared to their raw CAD p-values, and this new ranking favors SNPs showing signal in both CAD and the given secondary trait. In contrast, the standard FDR does not re-rank the SNPs compared to their raw CAD p-values, but instead suggests a different significance cut-off compared to the Bonferroni correction.
In additional analysis, we computed the conjunctional FDR18 to detect loci showing strong evidence of association with both CAD and the given secondary trait. Low values in conditional FDR can be driven by association with both phenotypes or with the primary phenotype alone, whereas low values in conjunctional FDR are driven by association with both phenotypes.
The application and interpretation of FDR-based methodology is more challenging for post-GWAS specialized SNP panels such as the Metabochip33. The standard FDR is widely applied in GWAS where any given SNP is assumed to have the same prior probability of association as all other SNPs. The Metabochip (~200,000 SNPs) is designed to follow up SNPs of interest relating to metabolic and cardiovascular traits, including fine mapping around genome-wide significant SNPs. As such, the true positives (and the false positives) come in large dependent clumps. Large-scale dependence in the signal can lead to biased FDR36. To correct for this bias, we used an LD-pruned set of SNPs to estimate the conditional FDR distribution, which was then used for estimating the conditional FDR for the full SNP set (see the Supplemental Methods for details of this estimation procedure). To visualize the conditional and conjunctional FDR, we constructed Manhattan plots. Detailed information on conditional quantile-quantile plots, Manhattan plots, as well as conditional and conjunctional FDR can be found in earlier reports6, 18 and/or in the Supplement.
The conditional FDR assumes independent samples for CAD and each of the secondary traits. However, several of the participants were included in both a secondary trait GWAS and in the CAD study. Partially overlapping subjects between studies leads to dependencies between the test statistics for different traits for a given SNP under the null hypothesis37. We estimated the expected correlation of the cross-trait GWAS test statistics under the null hypothesis of no genetic associations using a similar method to the one described for GWAS meta-analysis37, 38 and corrected for the estimated correlation due to shared subjects using the Mahalanobis transformation (LeBlanc et al in prep). These corrected test statistics were used in all further analysis.
Stratified replication rate
As an internal validation of stratified enrichment, we performed a stratified replication rate analysis using methods described previously,18 where the contributing studies of the CARDIoGRAMplusC4D Consortium were repeatedly divided into independent discovery and validation sets. The purpose of this analysis is to show that an observed pattern of stratified enrichment is not due to spurious effects. In brief, we randomly selected half of the studies (24) for the discovery set, and used the remaining studies for replication, and repeated this procedure 200 times. For each SNP in the replication set and the discovery set, we computed a meta-analysis test statistic (Liptak’s method). For the discovery set, we calculated the associated two-tailed p-values, whereas for the replication samples they were converted to one-tailed p-values in order to preserve the direction of effect in the discovery sample. We then created a vector of -log10(p-value) cutoffs and binned SNPs according to their p-values in the discovery set SNPs. For each bin, we kept track of their respective p-values in the replication set. We can then calculate the replication rate for each bin as defined by the proportion of SNPs in that bin which has a replication p value < 0.05. We checked for stratified replication rates by plotting the replication rate curves for four strata based on significance in each secondary trait, using the same strata definitions as for the conditional quantile-quantile plots.
Independent validation
For all novel CAD SNPs identified in the conditional FDR analysis, we checked for nominal replication (p<0.05) in the WGHS. Since the WGHS data is collected prospectively, we conducted age-adjusted Cox regression over approximately 20 yrs of follow-up ending in 2013 for both MI and CHD.
Expression quantitative trait loci (eQTL) annotation
We tested whether the novel CAD SNPs discovered in the current study are associated with genotype-dependent gene expression in various tissue types. Such SNPs are known as eQTLs. To this end, we cross-referenced our novel findings from the conditional FDR analysis with three cis-eQTL databases: in whole blood39 (the most powerful eQTL database available), adipose tissue40 (relevant for metabolic disease) and lymphoblastoid cells (LCL)40. The whole blood eQTL data has been collected in a large collaborative effort n=5311 samples, the adipose and LCL eQTLs are from a sample size of approximately n=850. We considered a SNP to be an eQTL using an FDR q-value cutoff of 0.05. The FDR q-values were already available for whole blood, while for adipose tissue and LCL we downloaded the publically-available eQTL data and calculated q-values using the qvalue() package available from Bioconductor (version 2.14) in R (version 3.1.1).
Biological pathway analysis
To better understand the biological context of our results, we conducted an Ingenuity Pathway Analysis (IPA, QIAGEN Redwood City, www.qiagen.com/ingenuity) including all previously reported CAD genes and the nearest annotated gene for each novel SNP reported in our study. The available molecules and/or relationships in the IPA Knowledge Base for mammal (humans, mouse or rat) were considered. We set the confidence filter to relationships where the confidence is experimentally observed. We allowed a maximum size of 35 genes for generating networks and we allowed up to 25 networks in the overall analysis. IPA computes a score for each network according to the fit of that network to a set of focus gene and p-values are calculated using the right-tailed Fisher’s exact test.
RESULTS
We used a two-step analysis strategy. First we assessed overlapping polygenic enrichment for CAD and each of the other traits via conditional quantile-quantile plots, and applied the Anderson-Darling test to define which of the 8 secondary traits show significant polygenic overlap. This test requires the direction of the association and as this information was unavailable for SBP, we relied on a visual inspection of the conditional quantile-quantile plot for SBP. As illustrated in Online Table II, all testable traits showed significant enrichment after Bonferroni correction for 21 tests and SBP showed strong visual evidence for enrichment. Therefore all 8 secondary traits were retained for the second step of the analysis. Second we applied conditional and conjunctional FDR methods to identify new CAD risk loci and to identify overlapping loci between CAD and each of the 8 associated traits. Overall FDR thresholds of 0.01 and 0.05 were chosen for conditional and conjunctional FDR respectively. Conservatively adjusting for the 8 secondary traits being considered21 this translated to thresholds of 0.01/8 and 0.05/8 for conditional and conjunctional FDR.
Conditional quantile-quantile plots for CAD conditioned on nominal p-values of association with LDL, CRP, T1D and T2D showed significant enrichment across different levels of significance (Figure 1). Similar significant enrichment patterns were seen for HDL, TG, SBP and BMI (see Online Figure I). The increasing leftward shift with more strictly defined strata based on nominal p-values of associated phenotypes suggests a greater proportion of true associations for a given nominal CAD p-value. This is indicative of cross-trait polygenic enrichment. As illustrated in Figure 1, panel A: LDL, the proportion of SNPs in the −log10(pLDL) ≥ 3 category reaching a given significance level (e.g., −log10(pCAD) > 6) is much greater than for the all SNPs category, indicating a high level of enrichment (Figure 1).
Figure 1. Shared Polygenic Enrichment.
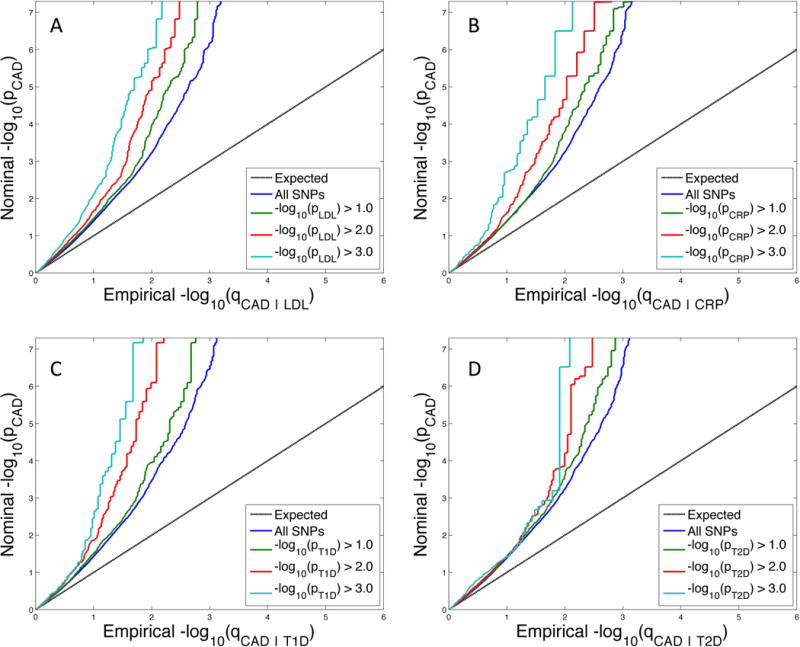
Conditional quantile-quantile plot of nominal versus empirical −log10 p-values in Coronary Artery Disease (CAD) as a function of significance of association with A) low density lipoprotein cholesterol (LDL), B) C-reactive protein (CRP), C) type 1 diabetes (T1D) and D) type 2 diabetes (T2D), at the level of −log10(p) > 0, −log10(p) > 1, −log10(p) > 2, −log10(p) > 3 corresponding to p < 1, p < 0.1, p < 0.01, p < 0.001, respectively. Due to the linkage disequilibrium structure on the Metabochip, a linkage disequilibrium-pruned set of SNPs was used for the quantile-quantile plots. Input p-values were adjusted for shared subjects, if present. Dotted lines indicate the null-hypothesis.
Stratified replication rates were observed for all secondary traits with the exception of BMI (Online Figure II), indicating that the observed enrichment in the conditional quantile-quantile plots is also associated with increased replication rates. The observed pattern of stratified enrichment does not result from spurious effects, and replication rate is increased by conditioning on significance in each of the secondary traits, with the possible exception of BMI.
Conditional and conjunctional FDR were calculated for CAD paired with each of the 8 secondary phenotypes showing enrichment. The results of each analysis were filtered as follows. First, we filtered the lists of significant SNPs by their linkage disequilibrium patterns as observed in the 1000 Genomes41 dataset and report only the most significant result per annotated gene. We considered a SNP to be an independent finding if the linkage disequilibrium, defined using r2, was less than 0.2 with all other SNPs in the filtered list. Second, we further filtered the list of significant SNPs for novelty with respect to previously published CAD SNPs. We filtered out any previously reported genes and SNPs, including SNPs in linkage disequilibrium (r2>0.2) with those previously reported SNPs. Thus, the list of significant SNPs presented in Table 1 represent, to the best of our knowledge, independent novel SNPs for CAD. The corresponding conditional Manhattan plot is given in Figure 2.
Table 1.
Conditional FDR (<0.01), after controlling for multiple testing across phenotypes.
| snp | gene | Chr | CAD|T2D | CAD|T1D | CAD|LDL | CAD|HDL | CAD|TG | CAD|BMI | CAD|CRP | CAD|SBP | Min cond FDR | CAD p-value |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs10747342* | HS2ST1 | 1 | 2.59E-03 | 1.16E-03 | 3.11E-03 | 1.67E-03 | 1.27E-03 | 2.45E-03 | 2.94E-03 | 3.00E-03 | T1D | 6.57E-06 |
| rs1418458* | BC067883 | 1 | 6.55E-03 | 1.21E-03 | 4.43E-03 | 6.45E-03 | 6.63E-03 | 5.72E-03 | 6.97E-03 | 6.19E-03 | T1D | 1.81E-05 |
| rs4268379 | SARS | 1 | 6.76E-05 | 3.20E-04 | 8.30E-04 | 5.69E-04 | 7.20E-04 | 2.95E-04 | 2.63E-04 | 5.81E-04 | T2D | 7.46E-07 |
| rs11806316 | NGF | 1 | 9.55E-04 | 2.11E-04 | 9.30E-04 | 9.70E-04 | 5.07E-04 | 6.88E-04 | 9.78E-04 | 1.02E-03 | T1D | 1.54E-06 |
| rs10788792* | GOLPH3L | 1 | 8.12E-03 | NA | 1.01E-03 | 4.44E-03 | 4.99E-03 | 8.66E-03 | 8.30E-03 | 7.38E-03 | LDL | 3.35E-05 |
| rs10800418 | NME7 | 1 | 1.11E-03 | 1.06E-04 | 1.77E-04 | 9.12E-04 | 5.83E-04 | 6.23E-04 | 8.58E-04 | 5.96E-04 | T1D | 1.19E-06 |
| rs6700559 | DDX59 | 1 | 8.19E-04 | 5.55E-05 | 6.76E-04 | 6.78E-04 | 7.20E-04 | 4.39E-04 | 6.60E-04 | 6.00E-04 | T1D | 7.79E-07 |
| rs2820315 | LMOD1 | 1 | 1.15E-03 | NA | 4.83E-04 | 1.31E-03 | 1.28E-03 | 3.81E-04 | 8.68E-04 | 9.08E-04 | BMI | 1.70E-06 |
| rs6663784* | CAPN9 | 1 | 4.03E-03 | 3.50E-03 | 3.11E-03 | 1.26E-03 | 1.20E-03 | 2.51E-03 | 3.62E-03 | 2.55E-03 | TG | 7.35E-06 |
| rs16986953 | AK097927 | 2 | 8.33E-04 | NA | 3.67E-04 | 8.64E-04 | 6.80E-04 | 5.19E-04 | 7.53E-04 | 7.44E-04 | LDL | 9.12E-07 |
| rs10186133 | IL1F10 | 2 | 7.23E-03 | 5.93E-03 | 4.81E-03 | 6.45E-03 | 7.16E-03 | 6.75E-03 | 7.28E-04 | 6.19E-03 | CRP | 2.21E-05 |
| rs2322864* | CXCR4 | 2 | 8.60E-03 | NA | 6.07E-04 | 5.84E-03 | 5.19E-03 | 6.21E-03 | 6.41E-03 | 7.48E-03 | LDL | 2.26E-05 |
| rs6435757* | IKZF2 | 2 | 2.25E-03 | 1.19E-03 | 1.67E-03 | 2.19E-03 | 2.01E-03 | 1.50E-03 | 1.31E-03 | 1.71E-03 | T1D | 3.13E-06 |
| rs13423088* | BC017935 | 2 | NA | NA | NA | NA | NA | NA | 1.25E-03 | 6.97E-04 | SBP | 2.48E-06 |
| rs7419961* | AX748264 | 2 | 4.15E-03 | NA | 8.96E-04 | 2.24E-03 | 2.40E-03 | 4.09E-03 | 3.61E-03 | 3.13E-03 | LDL | 9.36E-06 |
| rs748431* | FGD5 | 3 | 2.88E-03 | 2.79E-03 | 1.66E-03 | 3.01E-03 | 4.43E-03 | 3.13E-03 | 4.68E-03 | 4.95E-04 | SBP | 1.18E-05 |
| rs11715915* | AMT | 3 | 3.76E-03 | NA | 2.68E-03 | 3.35E-03 | 1.83E-03 | 2.41E-03 | 1.12E-03 | 2.79E-04 | SBP | 6.29E-06 |
| rs1512301* | GNPDA2 | 4 | 5.05E-03 | 3.79E-03 | 1.15E-03 | 4.24E-03 | 4.44E-03 | 3.55E-03 | 4.21E-03 | 4.04E-03 | LDL | 1.09E-05 |
| rs4690974* | MAP9 | 4 | 8.50E-03 | NA | 6.62E-03 | 8.56E-03 | 8.54E-03 | 7.94E-03 | 7.46E-03 | 8.62E-04 | SBP | 2.52E-05 |
| rs2736100* | TERT | 5 | 2.47E-03 | 5.57E-04 | 1.17E-03 | 7.17E-04 | 9.36E-04 | 2.11E-03 | 1.00E-03 | 1.86E-03 | T1D | 4.33E-06 |
| rs12916* | HMGCR | 5 | 3.54E-03 | NA | 1.40E-02 | 3.01E-03 | 5.18E-03 | 1.13E-03 | 4.21E-03 | 4.04E-03 | BMI | 1.10E-05 |
| rs246600 | ARHGAP26 | 5 | 9.47E-05 | 1.07E-04 | 8.11E-05 | 1.06E-04 | 1.36E-04 | 4.80E-05 | 5.72E-05 | 8.63E-05 | BMI | 7.84E-08 |
| rs2814982* | C6orf106 | 6 | 3.47E-03 | 3.35E-03 | 5.20E-04 | 3.03E-03 | 4.21E-03 | 3.35E-03 | 2.54E-03 | 3.46E-03 | LDL | 7.83E-06 |
| rs1321309* | CDKN1A | 6 | 2.25E-03 | NA | 1.63E-03 | 2.46E-03 | 1.86E-03 | 2.09E-03 | 1.08E-03 | 2.07E-03 | CRP | 4.18E-06 |
| rs6905288* | VEGFA | 6 | 1.85E-03 | 2.95E-03 | 1.46E-03 | 2.56E-03 | 3.46E-03 | 1.66E-03 | 2.94E-03 | 2.79E-04 | SBP | 6.21E-06 |
| rs9367716* | PRIM2 | 6 | 1.98E-03 | 2.74E-04 | 2.44E-03 | 2.06E-03 | 2.91E-03 | 2.04E-03 | 1.93E-03 | 3.42E-03 | T1D | 7.48E-06 |
| rs4613862 | BC038576 | 6 | 1.49E-03 | 8.76E-04 | 1.30E-03 | 4.99E-04 | 6.22E-04 | 5.35E-04 | 1.04E-03 | 1.09E-03 | HDL | 1.73E-06 |
| rs12663498* | PLEKHG1 | 6 | 5.67E-03 | 4.25E-03 | 4.44E-03 | 2.63E-03 | 1.86E-03 | 3.03E-03 | 1.90E-03 | 6.73E-04 | SBP | 1.29E-05 |
| rs1247351 | MAP3K4 | 6 | 9.58E-04 | 7.75E-04 | 2.16E-04 | 8.64E-04 | 7.20E-04 | 2.96E-04 | 8.00E-04 | 5.92E-04 | LDL | 9.01E-07 |
| rs10278591* | MAD1 | 7 | 2.25E-03 | 1.67E-03 | 1.82E-03 | 1.35E-03 | 1.21E-03 | 1.65E-03 | 2.00E-03 | 1.49E-03 | TG | 3.78E-06 |
| rs11204085 | SLC18A1 | 8 | 2.32E-04 | NA | 1.79E-04 | 9.24E-04 | 1.50E-03 | 1.58E-04 | 1.38E-04 | 1.63E-04 | CRP | 1.69E-07 |
| rs6984210 | BMP1 | 8 | NA | NA | NA | NA | NA | NA | 9.67E-04 | 8.66E-04 | SBP | 1.22E-06 |
| rs12343412* | SLC44A1 | 9 | 2.55E-03 | 3.33E-04 | 1.34E-03 | 3.02E-03 | 2.63E-03 | 1.47E-03 | 2.65E-03 | 2.37E-03 | T1D | 4.78E-06 |
| rs867764* | CAMK1D | 10 | 2.32E-03 | 1.67E-03 | 1.97E-03 | 1.41E-03 | 1.61E-03 | 9.86E-04 | 1.79E-03 | 1.44E-03 | BMI | 3.22E-06 |
| rs3748242 | ANXA11 | 10 | 5.54E-04 | 4.40E-04 | 4.98E-04 | 4.65E-04 | 4.88E-04 | 4.32E-04 | 2.83E-04 | 3.69E-04 | CRP | 5.29E-07 |
| rs1980653 | OBFC1 | 10 | 1.43E-03 | 1.08E-03 | 4.54E-04 | 9.12E-04 | 8.15E-04 | 8.21E-04 | 1.10E-03 | 1.06E-03 | LDL | 1.59E-06 |
| rs425325* | PLEKHA7 | 11 | 5.77E-03 | NA | 3.86E-03 | 3.64E-03 | 4.69E-03 | 4.53E-03 | 4.46E-03 | 6.73E-04 | SBP | 1.29E-05 |
| rs12801636 | PCNXL3 | 11 | 5.49E-04 | 9.15E-04 | 2.63E-04 | 6.50E-04 | 9.87E-04 | 6.02E-04 | 9.33E-05 | 1.77E-04 | CRP | 1.15E-06 |
| rs590121 | SERPINH1 | 11 | 6.67E-04 | NA | 5.97E-04 | 7.20E-04 | 6.80E-04 | 5.66E-04 | 5.52E-04 | 6.42E-04 | CRP | 7.76E-07 |
| rs7306455* | NDUFA12 | 12 | 2.64E-03 | 9.15E-04 | 1.95E-03 | 2.44E-03 | 2.49E-03 | 1.23E-03 | 2.00E-03 | 2.07E-03 | T1D | 3.70E-06 |
| rs10774613 | CUX2 | 12 | 3.63E-04 | 1.97E-05 | 5.87E-05 | 3.96E-04 | 5.12E-04 | 1.91E-04 | 2.93E-04 | 2.10E-04 | T1D | 3.96E-07 |
| rs7296651* | ALDH2 | 12 | NA | 1.25E-04 | NA | NA | NA | NA | 1.83E-04 | 1.54E-04 | T1D | 3.15E-06 |
| rs11066320 | PTPN11 | 12 | 1.23E-04 | 4.05E-06 | 1.28E-05 | 8.40E-05 | 1.10E-04 | 1.14E-05 | 6.92E-05 | 5.55E-06 | T1D | 6.70E-08 |
| rs1015249* | RPH3A | 12 | 5.01E-03 | 3.32E-04 | 4.25E-04 | 4.09E-03 | 2.15E-03 | 2.29E-03 | 4.41E-03 | 1.58E-03 | T1D | 9.64E-06 |
| rs2708081 | OASL | 12 | 6.75E-05 | 6.86E-05 | 1.22E-04 | 1.14E-04 | 3.56E-04 | 2.34E-04 | 3.41E-05 | 2.70E-04 | CRP | 3.08E-07 |
| rs825483* | ZNF664 | 12 | 8.27E-04 | 3.08E-03 | 3.11E-03 | 2.56E-03 | 2.48E-03 | 1.49E-03 | 2.94E-03 | 2.15E-03 | T2D | 5.81E-06 |
| rs11057830 | SCARB1 | 12 | 1.73E-04 | NA | 7.08E-05 | 9.07E-05 | 3.17E-04 | 8.59E-05 | 1.51E-04 | 1.04E-04 | LDL | 1.48E-07 |
| rs9603710* | TTL/TEL | 13 | 1.98E-03 | NA | 1.22E-03 | 2.01E-03 | 2.01E-03 | 1.62E-03 | 1.90E-03 | 1.80E-03 | LDL | 3.21E-06 |
| rs9316753 | BC044614 | 13 | 1.01E-03 | 7.33E-04 | 3.92E-04 | 4.08E-04 | 7.33E-04 | 5.35E-04 | 7.53E-04 | 6.80E-04 | LDL | 9.12E-07 |
| rs2273996* | LMO7 | 13 | 1.40E-02 | 1.22E-03 | 1.20E-02 | 9.47E-03 | 1.36E-02 | 1.16E-02 | 1.26E-02 | 1.13E-02 | T1D | 5.13E-05 |
| rs2146238 | CYP46A1 | 14 | 8.55E-04 | NA | 1.00E-04 | 8.60E-04 | 7.20E-04 | 5.19E-04 | 8.00E-04 | 7.22E-04 | LDL | 9.54E-07 |
| rs6494488* | RBPMS2 | 15 | 1.58E-03 | 1.17E-03 | 1.42E-03 | 1.56E-03 | 1.30E-03 | 6.60E-04 | 1.25E-03 | 1.22E-03 | BMI | 1.99E-06 |
| rs7202877* | CTRB1 | 16 | 5.30E-03 | 7.07E-04 | 6.99E-03 | 7.59E-03 | 8.89E-03 | 5.07E-03 | 7.12E-03 | 6.82E-03 | T1D | 2.65E-05 |
| rs4888378* | CFDP1 | 16 | 7.52E-03 | 1.99E-03 | 2.72E-03 | 3.62E-03 | 3.65E-03 | 4.41E-03 | 6.09E-03 | 7.17E-04 | SBP | 1.80E-05 |
| rs4299203 | LRRC48 | 17 | 2.14E-04 | NA | 1.90E-04 | 1.94E-04 | 1.67E-04 | 7.01E-05 | 1.67E-04 | 1.51E-04 | BMI | 1.59E-07 |
| rs17608766* | GOSR2 | 17 | 1.74E-03 | 9.00E-04 | 1.30E-03 | 2.72E-04 | 1.50E-03 | 6.52E-04 | 1.50E-03 | 1.26E-04 | SBP | 2.37E-06 |
| rs3179840* | ZNF652 | 17 | 8.77E-03 | 7.73E-03 | 5.47E-03 | 4.25E-03 | 4.61E-03 | 7.15E-03 | 7.46E-03 | 9.99E-04 | SBP | 2.43E-05 |
| rs1867624* | AX746971 | 17 | 1.66E-03 | 1.40E-03 | 4.46E-04 | 8.74E-04 | 1.58E-03 | 6.70E-04 | 1.68E-03 | 1.26E-04 | SBP | 2.25E-06 |
| rs13465* | ILF3 | 19 | 1.56E-03 | 4.71E-04 | 1.62E-03 | 1.55E-03 | 1.61E-03 | 1.06E-03 | 1.33E-03 | 1.13E-03 | T1D | 2.12E-06 |
| rs17616661* | KANK2 | 19 | 3.08E-03 | 1.38E-03 | 7.09E-04 | 1.55E-03 | 2.35E-03 | 1.97E-03 | 1.19E-03 | 2.48E-03 | LDL | 5.03E-06 |
| rs12459996* | CYP2F1 | 19 | 1.67E-03 | 1.82E-03 | 5.87E-04 | 6.31E-04 | 2.23E-03 | 9.48E-04 | 1.69E-03 | 7.70E-04 | LDL | 3.35E-06 |
| rs12460848 | MARK4 | 19 | 1.61E-03 | NA | 1.04E-03 | 1.13E-03 | 1.42E-03 | 8.05E-04 | 5.76E-04 | 1.02E-03 | CRP | 1.68E-06 |
| rs4802322* | STRN4 | 19 | 1.01E-02 | 8.50E-04 | 5.33E-03 | 8.59E-03 | 9.51E-03 | 9.74E-03 | 7.49E-03 | 8.05E-03 | T1D | 3.00E-05 |
| rs867186 | EDEM2 | 20 | 4.54E-05 | 1.46E-04 | 1.56E-04 | 1.15E-04 | 1.05E-04 | 1.21E-04 | 1.21E-04 | 1.21E-04 | T2D | 1.36E-07 |
| rs3827066* | ZNF335 | 20 | 5.77E-03 | NA | 1.46E-03 | 1.55E-03 | 1.22E-03 | 5.07E-03 | 2.52E-03 | 4.72E-03 | TG | 1.35E-05 |
| rs1882961* | NRIP1 | 21 | 7.63E-03 | 9.09E-03 | 4.64E-03 | 9.43E-03 | 1.10E-02 | 1.04E-02 | 3.48E-03 | 1.04E-03 | SBP | 3.39E-05 |
| rs9608859* | OSM | 22 | 1.76E-03 | NA | 1.81E-03 | 8.06E-04 | 1.79E-03 | 1.48E-03 | 1.41E-03 | 1.43E-03 | HDL | 2.68E-06 |
Independent (r2 < 0.2) SNP(s) with a conditional FDR (condFDR) < 0.01 (after Bonferoni correction for 8 traits) in Coronary Artery Disease (CAD) given the significance level in the associated phenotype. We defined the most significant CAD SNP in each LD block based on the minimum conditional FDR for each associated phenotype. The most significant SNPs in each gene of the LD block are listed along with the associated phenotype that provided the signal. Coronary artery disease (CAD), low density lipoprotein (LDL) cholesterol, high density lipoprotein (HDL) cholesterol, triglycerides (TG), type 2 diabetes (T2D), C-reactive protein (CRP), body mass index (BMI), systolic blood pressure (SBP), type 1 diabetes (T1D), chromosome number (Chr). Conditional FDR values < 0.01, after adjusting for multiple testing across phenotypes are in bold. The most significant phenotype association per gene is shown (min condFDR). NA indicates that a given SNP was not available for a given trait.
SNP would not have been detected using standard (unconditioned) FDR methodology at the same threshold.
Figure 2.
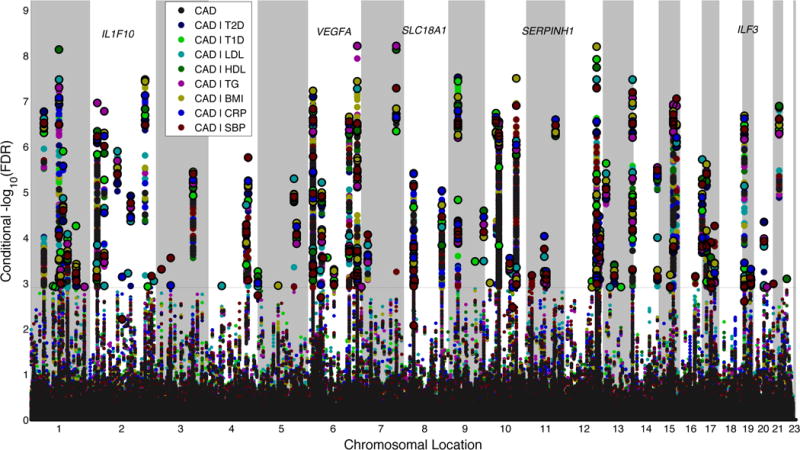
‘Conditional FDR Manhattan plot’ of −log10 (FDR)* values for Coronary Artery Disease (CAD) alone (black), and −log10 (conditional FDR) for CAD given type 2 diabetes (T2D; CAD|T2D; navy blue), CAD given type 1 diabetes (T1D; CAD|T1D; light green), CAD given low density lipoprotein (LDL; CAD|LDL; aqua). CAD given high density lipoprotein (HDL; CAD|HDL; dark green), CAD given triglycerides (TG; CAD|TG; fuchsia), CAD given body mass index (BMI; CAD|BMI; mustard yellow). CAD given C-reactive protein (CRP; CAD|CRP; royal blue) and CAD given systolic blood pressure (SBP; CAD|SBP; maroon). SNPs with −log10 (conditional FDR) > 2.9 (i.e. overall FDR < 0.01 after Bonferroni correction for eight traits) are shown with large points. A black circle around the large points indicates the most significant SNP in each linkage disequilibrium block and this SNP was annotated with the closest gene which is listed above the symbols in each locus, except for the HLA region on chromosome 6, which was excluded from the analysis. Details for the novel loci with −log10 (conditional FDR) > 2.9 are given in Table 1.* For the –log10 (FDR) for CAD alone the maximum value displayed in this figure is 6.5. This is done purely for display purposes and as such should be interpreted as >6.5.
Over all 8 secondary traits, we identify 101 SNPs associated with CAD, 67 of which have not previously associated with CAD (previously reported SNPs not shown). Many of these new loci are located in regions with borderline significant association with CAD in previous studies42 as is evident by the CAD association p-value column given in Table 1. Of interest, several of the identified loci are found across the conditional analysis from several risk factors. These loci are not found using standard methods applying a genome-wide Bonferroni correction.
We looked to the WGHS for independent validation of these 67 new CAD SNPs and 12 of these show nominal replication for at least one endpoint (CHD or MI); see Online Table III.
Of the 67 novel CAD loci, 32 show genotype-dependent gene expression in whole blood regulating the expression of 57 unique genes and 42 of these 67 SNPs would not have been detected using the standard (unconditioned) FDR. We found evidence for 16 and 18 loci having an eQTL effect in adipose tissue and LCL respectively (Table 2). For six of these loci we observed an eQTL effect on the same gene in both whole blood and adipose tissue. Interestingly, 18 loci show an effect on the gene expression of more than one gene.
Table 2.
Novel CAD SNPs that are also eQTLs in blood and/or adipose tissue and/or lymphoblastoid cells.
| SNP | Chr | Nearest Gene | Blood eQTL for | Blood eQTL p-value | Adipose eQTL for | Adipose eQTL p-value | LCL eQTL for | LCL eQTL p-value |
|---|---|---|---|---|---|---|---|---|
| rs10747342 | 1 | HS2ST1 | LMO4 | 2.63E-05 | LMO4 | 6.08E-05 | ||
| rs10788792 | 1 | GOLPPH3L | ARNT | 2.38E-05 | CTSS | 3.01E-04 | CTSS | 6.35E-36 |
| CTSK | 4.98E-32 | LASS2 | 4.72E-06 | |||||
| CTSS | 3.86E-150 | CTSK | 2.19E-16 | |||||
| rs2820315 | 1 | LMOD1 | IPO9 | 4.10E-06 | LMOD1 RNPEP | 1.43E-11 7.19E-05 | ||
| rs6700559 | 1 | DDX59 | DDX59 | 4.10E-106 | DDX59 | 6.27E-08 | ||
| rs10800418 | 1 | NME7 | NME7 | 2.58E-12 | NME7 | 5.12E-18 | ||
| rs6663784 | 1 | CAPN9 | AGT | 6.65E-06 | ||||
| rs10186133 | 2 | IL1F10 | PSD4 | 9.43E-07 | ||||
| rs6435757 | 2 | IKZF2 | IKZF2 | 4.96E-06 | ||||
| rs748431 | 3 | FGD5 | FGD5 | 2.31E-04 | ||||
| rs11715915 | 3 | AMT | USP4 | 1.86E-24 | RBM6 | 1.24E-04 | KLHDC8B | 1.08E-04 |
| NICN1 | 9.50E-29 | APEH | 2.51E-05 | |||||
| rs1321309 | 6 | CDKN1A | CDKN1A | 4.12E-23 | CDKN1A | 6.74E-19 | ||
| Z85996.1-1 | 1.04E-42 | |||||||
| rs2814982 | 6 | C6orf106 | c6orf106 | 1.56E-11 | ||||
| rs9367716 | 6 | PRIM2 | RAB23 | 6.47E-15 | ||||
| rs10278591 | 7 | MAD1 | MAD1L1 | 1.05E-06 | MAD1L1 | 2.46E-04 | ||
| rs11204085 | 8 | SLC18A1 | LPL | 1.00E-04 | ||||
| rs3748242 | 10 | ANXA11 | AL512662.8-2,SFTPD | 7.62E-05 | c10orf58 | 3.45E-05 | ||
| ANXA11 | 2.24E-19 | ANXA11 | 1.93E-05 | |||||
| rs12801636 | 11 | PCNXL3 | SIPA1 | 1.19E-06 | ||||
| rs590121 | 11 | SERPINH1 | GDPD5 | 8.69E-10 | ||||
| rs1015249 | 12 | RPH3A | OAS1 | 2.77E-13 | ||||
| rs2708081 | 12 | OASL | CAMKK2 | 1.83E-10 | c12orf43 | 1.15E-05 | CAMKK2 | 2.86E-04 |
| c12orf43 | 5.55E-20 | |||||||
| P2RX4 | 1.95E-10 | |||||||
| OASL | 7.32E-10 | |||||||
| rs10774613 | 12 | CUX2 | IFT81 | 2.92E-04 | ||||
| rs7296651 | 12 | ALDH2 | ERP29 | 1.96E-21 | c12orf30 | 3.97E-05 | c12orf30 | 3.84E-04 |
| TMEM116 | 1.49E-67 | TMEM116 | 1.12E-08 | TMEM116 | 7.17E-12 | |||
| ACAD10 | 4.64E-04 | |||||||
| FLJ30092 | 4.62E-08 | |||||||
| rs7306455 | 12 | NDUFA12 | NDUFA12 | 8.57E-06 | ||||
| rs825483 | 12 | ZNF664 | CCDC92 | 9.29E-06 | CCDC92 | 2.48E-16 | ||
| rs6494488 | 15 | RBPMS2 | ANKDD1A | 2.21E-18 | TRIP4 | 1.55E-04 | TRIP4 | 1.53E-09 |
| RBPMS2 | 1.29E-72 | |||||||
| rs4888378 | 16 | CFDP1 | CFDP1 | 1.63E-10 | CHST6 | 1.50E-04 | ||
| rs7202877 | 16 | CTRB1 | CFDP1 | 6.33E-12 | ||||
| rs17608766 | 17 | GOSR2 | GOSR2 | 6.42E-06 | ||||
| rs3179840 | 17 | ZNF652 | GNGT2 | 1.23E-36 | GNGT2 | 8.18E-10 | ||
| PHOSPHO1 | 2.62E-08 | |||||||
| rs4299203 | 17 | LRRC48 | ATPAF2 | 3.97E-09 | DRG2 | 3.11E-07 | ||
| c17orf39 | 1.73E-19 | |||||||
| DRG2 | 1.73E-09 | |||||||
| TOM1L2 | 7.10E-08 | |||||||
| SREBF1 | 6.45E-51 | |||||||
| rs1867624 | 17 | AX746971 | PECAM1 | 5.97E-10 | ||||
| rs12459996 | 19 | CYP2F1 | HNRNPUL1 | 3.86E-16 | BLVRB | 5.69E-04 | ||
| SNRPA | 4.01E-04 | |||||||
| B9D2 | 2.97E-04 | |||||||
| rs12460848 | 19 | MARK4 | CKM | 7.91E-08 | ||||
| KLC3 | 4.92E-12 | |||||||
| VASP | 5.67E-14 | |||||||
| rs17616661 | 19 | KANK2 | KANK2 | 5.60E-17 | KANK2 | 5.26E-06 | ||
| rs4802322 | 19 | STRN4 | CALM3 | 7.14E-06 | FKRP | 2.53E-07 | ||
| PRKD2 | 1.10E-23 | |||||||
| FKRP | 3.11E-31 | |||||||
| SLC1A5 | 6.12E-10 | |||||||
| rs3827066 | 20 | ZNF335 | AL162458.10-3,MMP9 | 1.13E-04 | WFDC3 | 1.78E-04 | PLTP | 1.20E-06 |
| CD40 | 1.27E-05 | NEURL2 | 6.50E-09 | |||||
| DNTTIP1 | 2.52E-14 | PLTP | 1.60E-06 | |||||
| TNNC2 | 3.98E-19 | |||||||
| rs867186 | 20 | EDEM2 | ACSS2 | 2.99E-07 | PROCR | 3.24E-15 | ||
| EIF6 | 2.25E-101 | |||||||
| TRPC4AP | 8.04E-108 | |||||||
| rs9608859 | 22 | OSM | SF3A1 | 4.29E-94 | THOC5 | 6.97E-05 | ||
| MTP18 | 1.63E-08 |
Chromosome number (chr), expression quantitative trait locus (eQTL), lymphoblastoid cell (LCL). A SNP was considered to be an eQTL using an FDR threshold of 0.05.
To further evaluate genetic overlap, we used the conjunctional FDR to identify SNPs with significant effects in both CAD and its associated risk factors. The conjunctional Manhattan plot for CAD is shown in Online Figure II. We identified 53 loci achieving conjunctional FDR<0.05, after adjustment for using multiple risk factors and pruning the results in the same manner as for the conditional FDR (Online Table IV; corresponding z-scores in Online Table V).
Follow-up Ingenuity Pathways Analysis (IPA) identified highly significantly associated “Top Canonical Pathways” relevant to CAD (e.g. LXR (liver X receptor)/RXR (retinoid X receptor) as well as FXR (Farnesoid X Receptor)/RXR Activation and Atherosclerosis Signaling); (Online Table V). Additionally, in “Top Diseases and Bio Function” CAD relevant diseases and functions are on top (Cardiovascular Disease and Lipid Metabolism) in the subgroups “Diseases and Disorders” and “Molecular and Cellular Functions”.
DISCUSSION
Combining data from large-scale genomic studies from different phenotypes in a conditional FDR framework, we show polygenic overlap between CAD and several CVD risk factor phenotypes and identify 67 novel CAD susceptibility loci. Further, conjunctional FDR analysis identified 53 novel loci associated with both CAD and the CVD risk factors LDL, HDL, TG, T1D, T2D, CRP and SBP. Importantly, we validated the conditional FDR approach by showing that replication rates in independent CAD sub-studies increase as a function of p-value in each secondary trait, with the possible exception of BMI. Further, we see nominal replication for 12/67 SNPs in the WGHS. Overall, these results suggest that a proportion of the clinically and epidemiologically observed association between these phenotypes can be explained by overlapping genetic loci (pleiotropy) and not simply shared environmental risk factors. Further, the findings provide further evidence that CAD is a highly polygenic disease.
Our findings of polygenic overlap provide novel insights into the relationship between CAD and major CVD risk factors. We demonstrate an interesting genetic dissociation among these risk factors and CAD, with strong enrichment for lipids, inflammation and metabolic disorders. The combination of dyslipidemia (i.e., high TG and LDL cholesterol and low HDL cholesterol), T2D, and high blood pressure forms the metabolic syndrome12–14, 43, 44, and all of these factors (particularly LDL) showed strong genetic overlap with CAD. This is in agreement with a recent reports suggesting a common genetic basis for regulation of lipid and glucose homeostasis45, while previous studies did not show common genes for the different components of the metabolic syndrome46, but revealed strong lipid gene contribution. It is further supported by the pathway analysis that identified “Atherosclerosis Signaling” and “FXR/RXR Activation” among the three most relevant pathways. Genes activated by the FXR has been shown to influence vascular tension and regulate the unloading of cholesterol from foam cells47. Another important finding is the overlap between CAD and T2D. Based on conditional analysis of these two phenotypes, 21 novel loci were identified. This is in line with previous single gene studies suggesting a genetic link between T2D and CAD48.
The strong shared polygenic signal between LDL and CAD emphasizes the important role of LDL in CAD development, and support the notion that risk genes for atherosclerosis, such as LDL genes, are causal for CAD as recently suggested49. Finally, two of the phenotypes most strongly overlapping with CAD were CRP and T1D, two immune related phenotypes. CRP is regarded as a reliable marker of systemic inflammation and its role as a biomarker in CAD has been attributed to its ability to reflect up-stream inflammatory pathways. However, the finding in the present study suggests that the link between CRP and CAD may also reflect overlapping genetic loci. T1D is related to auto-immune mechanisms and its genetic overlap with CAD underlines the important role of the immune system in CAD, and could be due to a large number of overlapping genes between immune and lipid phenotypes50. In fact, the bidirectional interaction between inflammation and lipids is regarded as a phenotypic hallmark of atherosclerosis, and our findings suggest that this phenotype could reflect overlapping genes between these two interacting pathophysiological arms of atherogenesis. The pathway analysis revealed “LXR/RXR Activation”, as the top ranked canonical pathway. LXR/RXR are heterodimer nuclear receptors/transcription factors. LXR acts as a cholesterol sensor, and LXR pathway activation has been shown to stimulate lipogenesis and hypertriglyceridemia51. LXR/RXR can also modulate inflammatory responses to cholesterol exposure and could represent a regulator of the interaction between lipids and inflammation, being the most important pathway in the pathogenesis of CAD.
In the original CAD GWAS and follow-up Metabochip study, 46 loci were identified2. By combining the original CAD results with the CVD risk factor phenotypes GWAS, we identified 101 significant loci associated with CAD, of which 67 are novel, using the conditional FDR approach. Even though the original CAD study was quite large2, the increased power provided by additional GWAS of associated phenotypes together with the conditional FDR method more than doubled gene discovery. The novel SNPs discovered here contribute to explaining more of the missing heritability for CAD, but we cannot quantify how much more is explained since we are working at the summary-statistic level. These findings underline the cost-effectiveness of the current statistical methods and highlight several interesting genes in CAD pathology. IL1F10 (interleukin 1 family, member 10 (theta)) was identified in the pathway analysis of the CAD GWAS, it is known to bind IL1R and stimulate NF-kB pathway. VEGFA (vascular endothelial growth factor A) is well known in the CVD field, but to the best of our knowledge, this has never been shown in genetic studies. SLC18A1 (solute carrier family 18 (vesicular monoamine transporter), member 1) has been implicated in neuropsychiatric disorders, but not previously in CVD. SERPINH1 (serpin peptidase inhibitor, clade H (heat shock protein 47), member 1, (collagen binding protein 1)) is a heat shock protein, known to be involved in atherosclerosis. ILF3 (interleukin enhancer binding factor 3, 90kDa) is a matrix metallo proteinase, well studied in the CVD research field, and the findings of ILF10 and ILF3 underscore the role of the IL-1 cytokine family in CAD.
Although nearest-gene annotation can be informative, the vast majority of discovered SNPs are located outside coded DNA regions52. Therefore, annotating the identified genetic variants to the correct causal genes for the phenotype of interest often remains challenging52. One of the potential mechanisms whereby SNPs may affect phenotype variations is through altered gene expression. We successfully identified eQTL effects in whole blood, LCL and adipose tissue, suggesting these genes as potential causal candidates. Of interest, some of the genetic variants showed an effect on the gene expression of more than one gene. We speculate that the shared effect of the genetic variants on the phenotypes under study might be explained by the regulation of several different genes, but further studies would be necessary connect the genes with altered gene expression seen in Table 2 to the clinical phenotypes. Moreover, the majority of the genes regulated by the genetic variants were different from the nearest annotated gene. Given that the original whole blood has markedly different power and used different statistical eQTL definitions than the LCL and adipose tissue eQTL studies, a detailed cross-tissue comparison is not possible. Further studies are needed to determine the functional mechanisms involved in the novel CAD loci identified here.
There are certain limitations associated with the present results. Due to the overlap in some of the GWAS samples examined, we cannot completely exclude the contribution from environmental or behavioral factors. The shared participants between genomic studies could also affect the findings. However, we did adjust for overlapping subjects, and used strict FDR thresholds to account for the 8 secondary traits. Although clinical comorbidity and shared pathophysiology between these phenotypes poses a challenge for the interpretation of the basis of the shared polygenetic signals, their utility for increasing the power to detect new loci for CAD is not affected. The question remains if the identified shared genes are independent of other phenotypes (biological pleiotropy), or that the current findings are results of overlapping phenotypes (mediated by other phenotypes), as several of these risk factors can be co-occurring (mediated pleiotropy)53. However, it appears reasonable to interpret our findings as reflecting the existence of shared genetically determined pathophysiological processes across CAD and the associated phenotypes. In general, FDR methodology is a less conservative approach to multiple testing than Bonferroni correction. However, by using the conditional FDR, we are not simply relaxing the significance threshold, but are increasing power and incorporating useful information from a second trait into the analysis, allowing us to identify the SNPs more likely to replicate. We have not strictly replicated all of these findings in independent samples but we have shown that replication rates increase by conditioning on significance in the secondary traits, and have shown that 12 SNPs nominally replicate in the WGS. While the prospective design of the WGHS makes it suitable for validation of the candidate CAD associations, the numbers of incident events of MI and CAD were much smaller than in the discovery sample, which was composed of a preponderance of men compared with the all-female composition of the WGHS. However, in spite of much lower power and possibility of differences according to sex, the WGHS is the largest and most relevant independent dataset we were able to access and we found nominal association for novel CAD 12 loci.
In conclusion, we found substantial polygenic overlap between CAD and several related conditions, importantly LDL, T2D and CRP, providing more evidence for fundamental etiological relationship between these phenotypes that cannot be explained by lifestyle factors. The 67 novel CAD loci identified here provide new insight into genetic mechanisms of CAD and may form the basis for earlier diagnosis and new prevention and treatment strategies.
Supplementary Material
Novelty and Significance.
What Is Known?
Previous work has identified 46 genetic risk variants associated with coronary artery disease (CAD)
Genetic data for traits with overlapping pathophysiology can be combined to improve power for identifying novel genetic risk variants.
What New Information Does This Article Contribute?
We identified 67 new genetic risk variants for CAD.
CAD and several cardio-metabolic traits share a large number of genetic risk factors.
Clinical and epidemiological evidence suggests a relationship between CAD and cardio-metabolic traits. In the presence of a shared polygenic signal (i.e., a large number of shared risk variants each with a small effect), traits with overlapping pathophysiology with CAD can be used in combination with novel statistical methodology to improve discovery of variants associated with CAD. Using large-scale genetic data from CAD and genetic data from hypertension, obesity, abdominal fat, diabetes, dyslipidemia, and inflammation (C-reactive protein), we found a polygenic overlap between CAD and each of these related traits. We identified 67 novel CAD risk variants and 53 risk variants jointly associated with CAD and at least one other related trait. These results highlight the importance of shared polygenic risk factors between coronary artery disease and cardiovascular risk factors. Our findings provide important insights into molecular mechanisms underlying coronary artery disease and have potential implications for prevention and treatment strategies.
Acknowledgments
The authors would like to thank the DIAGRAM Consortium, the CARDIoGRAMplusC4D Consortium, the Global Lipids Genetics Consortium, The International Consortium for Blood Pressure GWAS, the GEFOS Consortium, the Giant Consortium, the Type 1 Diabetes Consortium and the CHARGE Consortium Inflammation Working Group for the summary statistics GWAS data.
SOURCES OF FUNDING
The present project was supported by the Research Council of Norway (213837, 223273); the South East Norway Health Authority (2013–123); the Kristian Gerhard Jebsen Foundation; in addition to the National Institutes of Health grants R01AG031224, R01EB000790, and RC2DA29475, and the European Union project CVgenes@target; a grant from the Leducq Foundation, CADgenomics; and BMBF, Forschungskonsortien zur Systemmedizin, e:AtheroSysMED; DFG, SFB 1123. The WGHS is supported by HL043851, HL080467, and HL099355 from the National Heart, Lung, and Blood Institute and CA047988 from the National Cancer Institute with collaborative scientific support and funding for genotyping provided by Amgen. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Nonstandard Abbreviations and Acronyms
- BMI
body mass index
- CAD
coronary artery disease
- CRP
C-reactive protein
- CVD
cardiovascular disease
- eQTL
expression quantitative trait locus
- FDR
false discovery rate
- GWAS
genome-wide association study
- HDL
high density lipoprotein
- LCL
lymphoblastoid cells
- LDL
low density lipoprotein
- SBP
systolic blood pressure
- SNP
single nucleotide polymorphism
- T1D
type 1 diabetes
- T2D
type 2 diabetes
- TG
triglycerides
Footnotes
This manuscript was sent to Elizabeth M. McNally, Consulting Editor, for review by expert referees, editorial decision, and final disposition.
DISCLOSURES
None reported.
References
- 1.Peden JF, Farrall M. Thirty-five common variants for coronary artery disease: The fruits of much collaborative labour. Hum Mol Genet. 2011;20:R198–205. doi: 10.1093/hmg/ddr384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.CAD Consortium. Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, Thompson JR, Ingelsson E, Saleheen D, Erdmann J, Goldstein BA, Stirrups K, Konig IR, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45:25–33. doi: 10.1038/ng.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eichler EE, Flint J, Gibson G, Kong A, Leal SM, Moore JH, Nadeau JH. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010;11:446–450. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li C, Yang C, Gelernter J, Zhao H. Improving genetic risk prediction by leveraging pleiotropy. Hum Genet. 2014;133:639–650. doi: 10.1007/s00439-013-1401-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andreassen OA, Thompson WK, Schork AJ, Ripke S, Mattingsdal M, Kelsoe JR, Kendler KS, O’Donovan MC, Rujescu D, Werge T, Sklar P, Roddey JC, Chen C-H, et al. Improved detection of common variants associated with schizophrenia and bipolar disorder using pleiotropy-informed conditional false discovery rate. PLoS Genet. 2013;9:e1003455. doi: 10.1371/journal.pgen.1003455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang J, Manolio TA, Pasquale LR, Boerwinkle E, Caporaso N, Cunningham JM, de Andrade M, Feenstra B, Feingold E, Hayes MG, Hill WG, Landi MT, Alonso A, et al. Genome partitioning of genetic variation for complex traits using common snps. Nat Genet. 2011;43:519–525. doi: 10.1038/ng.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van den Hoogen PC, Feskens EJ, Nagelkerke NJ, Menotti A, Nissinen A, Kromhout D. The relation between blood pressure and mortality due to coronary heart disease among men in different parts of the world. Seven countries study research group. N Engl J Med. 2000;342:1–8. doi: 10.1056/NEJM200001063420101. [DOI] [PubMed] [Google Scholar]
- 9.The Global Burden of Metabolic Risk Factors for Chronic Diseases Collaboration. Metabolic mediators of the effects of body-mass index, overweight, and obesity on coronary heart disease and stroke: A pooled analysis of 97 prospective cohorts with 1.8 million participants. Lancet. 2014 doi: 10.1016/S0140-6736(13)61836-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romero-Corral A, Montori VM, Somers VK, Korinek J, Thomas RJ, Allison TG, Mookadam F, Lopez-Jimenez F. Association of bodyweight with total mortality and with cardiovascular events in coronary artery disease: A systematic review of cohort studies. Lancet. 2006;368:666–678. doi: 10.1016/S0140-6736(06)69251-9. [DOI] [PubMed] [Google Scholar]
- 11.Group AS. Cushman WC, Evans GW, Byington RP, Goff DC, Jr, Grimm RH, Jr, Cutler JA, Simons-Morton DG, Basile JN, Corson MA, Probstfield JL, Katz L, Peterson KA, et al. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med. 2010;362:1575–1585. doi: 10.1056/NEJMoa1001286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation. 2005;111:3481–3488. doi: 10.1161/CIRCULATIONAHA.105.537878. [DOI] [PubMed] [Google Scholar]
- 13.Messerli FH, Williams B, Ritz E. Essential hypertension. Lancet. 2007;370:591–603. doi: 10.1016/S0140-6736(07)61299-9. [DOI] [PubMed] [Google Scholar]
- 14.Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005;365:1415–1428. doi: 10.1016/S0140-6736(05)66378-7. [DOI] [PubMed] [Google Scholar]
- 15.Lagrand WK, Visser CA, Hermens WT, Niessen HW, Verheugt FW, Wolbink GJ, Hack CE. C-reactive protein as a cardiovascular risk factor: More than an epiphenomenon? Circulation. 1999;100:96–102. doi: 10.1161/01.cir.100.1.96. [DOI] [PubMed] [Google Scholar]
- 16.Coffman TM. Under pressure: The search for the essential mechanisms of hypertension. Nat Med. 2011;17:1402–1409. doi: 10.1038/nm.2541. [DOI] [PubMed] [Google Scholar]
- 17.Wagner GP, Zhang J. The pleiotropic structure of the genotype-phenotype map: The evolvability of complex organisms. Nat Rev Genet. 2011;12:204–213. doi: 10.1038/nrg2949. [DOI] [PubMed] [Google Scholar]
- 18.Andreassen OA, Djurovic S, Thompson WK, Schork AJ, Kendler KS, O’Donovan MC, Rujescu D, Werge T, van de Bunt M, Morris AP, McCarthy MI, International Consortium for Blood Pressure Group, Diabetes Genetics Replicaton and Meta-analysis Consortium, Psychiatric Genomics Consortium Schizophrenia Working Group et al. Improved detection of common variants associated with schizophrenia by leveraging pleiotropy with cardiovascular-disease risk factors. Am J Hum Genet. 2013;92:197–209. doi: 10.1016/j.ajhg.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andreassen OA, McEvoy LK, Thompson WK, Wang Y, Reppe S, Schork AJ, Zuber V, Barrett-Connor E, Gautvik K, Aukrust P, Karlsen TH, Djurovic S, Desikan RS, et al. Identifying common genetic variants in blood pressure due to polygenic pleiotropy with associated phenotypes. Hypertension. 2014;63:819–826. doi: 10.1161/HYPERTENSIONAHA.113.02077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andreassen OA, Harbo HF, Wang Y, Thompson WK, Schork AJ, Mattingsdal M, Zuber V, Bettella F, Ripke S, Kelsoe JR, Kendler KS, O’Donovan MC, Sklar P, et al. Genetic pleiotropy between multiple sclerosis and schizophrenia but not bipolar disorder: Differential involvement of immune-related gene loci. Mol Psychiatry. 2014 doi: 10.1038/mp.2013.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andreassen OA, Zuber V, Thompson WK, Schork AJ, Bettella F, Djurovic S, Desikan RS, Mills IG, Dale AM. Shared common variants in prostate cancer and blood lipids. Int J Epidemiol. 2014 doi: 10.1093/ije/dyu090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilson PW, D’Agostino RB, Levy D, Belanger AM, Silbershatz H, Kannel WB. Prediction of coronary heart disease using risk factor categories. Circulation. 1998;97:1837–1847. doi: 10.1161/01.cir.97.18.1837. [DOI] [PubMed] [Google Scholar]
- 23.Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998;339:229–234. doi: 10.1056/NEJM199807233390404. [DOI] [PubMed] [Google Scholar]
- 24.Malik S, Wong ND, Franklin SS, Kamath TV, L’Italien GJ, Pio JR, Williams GR. Impact of the metabolic syndrome on mortality from coronary heart disease, cardiovascular disease, and all causes in united states adults. Circulation. 2004;110:1245–1250. doi: 10.1161/01.CIR.0000140677.20606.0E. [DOI] [PubMed] [Google Scholar]
- 25.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Eng J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 26.Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, Johansen CT, Fouchier SW, Isaacs A, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morris AP, Voight BF, Teslovich TM, Ferreira T, Segre AV, Steinthorsdottir V, Strawbridge RJ, Khan H, Grallert H, Mahajan A, Prokopenko I, Kang HM, Dina C, Esko T, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44:981–990. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dehghan A, Dupuis J, Barbalic M, Bis JC, Eiriksdottir G, Lu C, Pellikka N, Wallaschofski H, Kettunen J, Henneman P, Baumert J, Strachan DP, Fuchsberger C, et al. Meta-analysis of genome-wide association studies in >80 000 subjects identifies multiple loci for c-reactive protein levels. Circulation. 2011;123:731–738. doi: 10.1161/CIRCULATIONAHA.110.948570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, Allen HL, Lindgren CM, Luan J, Magi R, Randall JC, Vedantam S, Winkler TW, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42:937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heid IM, Jackson AU, Randall JC, Winkler TW, Qi L, Steinthorsdottir V, Thorleifsson G, Zillikens MC, Speliotes EK, Magi R, Workalemahu T, White CC, Bouatia-Naji N, et al. Meta-analysis identifies 13 new loci associated with waist-hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nat Genet. 2010;42:949–960. doi: 10.1038/ng.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, Chasman DI, Smith AV, Tobin MD, Verwoert GC, Hwang SJ, Pihur V, Vollenweider P, O’Reilly PF, et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478:103–109. doi: 10.1038/nature10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, Julier C, Morahan G, Nerup J, Nierras C, Plagnol V, Pociot F, Schuilenburg H, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41:703–707. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Voight BF, Kang HM, Ding J, Palmer CD, Sidore C, Chines PS, Burtt NP, Fuchsberger C, Li Y, Erdmann J, Frayling TM, Heid IM, Jackson AU, et al. The metabochip, a custom genotyping array for genetic studies of metabolic, cardiovascular, and anthropometric traits. PLoS Genet. 2012;8:e1002793. doi: 10.1371/journal.pgen.1002793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Witte JS. Genome-wide association studies and beyond. Annu Rev Public Health. 2010;31:9–20. doi: 10.1146/annurev.publhealth.012809.103723. 24 p following 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ridker PM, Chasman DI, Zee RY, Parker A, Rose L, Cook NR, Buring JE. Rationale, design, and methodology of the women’s genome health study: A genome-wide association study of more than 25,000 initially healthy american women. Clin Chem. 2008;54:249–255. doi: 10.1373/clinchem.2007.099366. [DOI] [PubMed] [Google Scholar]
- 36.Chen L, Storey JD. Relaxed significance criteria for linkage analysis. Genetics. 2006;173:2371–2381. doi: 10.1534/genetics.105.052506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin DY, Sullivan PF. Meta-analysis of genome-wide association studies with overlapping subjects. Am J Hum Genet. 2009;85:862–872. doi: 10.1016/j.ajhg.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zaykin DV, Kozbur DO. P-value based analysis for shared controls design in genome-wide association studies. Genet Epidemiol. 2010;34:725–738. doi: 10.1002/gepi.20536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Westra HJ, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, Christiansen MW, Fairfax BP, Schramm K, Powell JE, Zhernakova A, Zhernakova DV, Veldink JH, et al. Systematic identification of trans eqtls as putative drivers of known disease associations. Nat Genet. 2013;45:1238–1243. doi: 10.1038/ng.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grundberg E, Meduri E, Sandling JK, Hedman AK, Keildson S, Buil A, Busche S, Yuan W, Nisbet J, Sekowska M, Wilk A, Barrett A, Small KS, et al. Global analysis of DNA methylation variation in adipose tissue from twins reveals links to disease-associated variants in distal regulatory elements. Am J Hum Genet. 2013;93:876–890. doi: 10.1016/j.ajhg.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ehret GB. Genome-wide association studies: Contribution of genomics to understanding blood pressure and essential hypertension. Curr Hypertens Rep. 2010;12:17–25. doi: 10.1007/s11906-009-0086-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.D’Agostino RB, Sr, Vasan RS, Pencina MJ, Wolf PA, Cobain M, Massaro JM, Kannel WB. General cardiovascular risk profile for use in primary care: The framingham heart study. Circulation. 2008;117:743–753. doi: 10.1161/CIRCULATIONAHA.107.699579. [DOI] [PubMed] [Google Scholar]
- 44.Conroy RM, Pyorala K, Fitzgerald AP, Sans S, Menotti A, De Backer G, De Bacquer D, Ducimetiere P, Jousilahti P, Keil U, Njolstad I, Oganov RG, Thomsen T, et al. Estimation of ten-year risk of fatal cardiovascular disease in europe: The score project. Eur Heart J. 2003;24:987–1003. doi: 10.1016/s0195-668x(03)00114-3. [DOI] [PubMed] [Google Scholar]
- 45.Albert JS, Yerges-Armstrong LM, Horenstein RB, Pollin TI, Sreenivasan UT, Chai S, Blaner WS, Snitker S, O’Connell JR, Gong DW, Breyer RJ, 3rd, Ryan AS, McLenithan JC, Shuldiner AR, et al. Null mutation in hormone-sensitive lipase gene and risk of type 2 diabetes. N Eng J Med. 2014;370:2307–2315. doi: 10.1056/NEJMoa1315496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kristiansson K, Perola M, Tikkanen E, Kettunen J, Surakka I, Havulinna AS, Stancakova A, Barnes C, Widen E, Kajantie E, Eriksson JG, Viikari J, Kahonen M, Lehtimaki T, et al. Genome-wide screen for metabolic syndrome susceptibility loci reveals strong lipid gene contribution but no evidence for common genetic basis for clustering of metabolic syndrome traits. Circ Cardiovasc Genet. 2012;5:242–249. doi: 10.1161/CIRCGENETICS.111.961482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Porez G, Prawitt J, Gross B, Staels B. Bile acid receptors as targets for the treatment of dyslipidemia and cardiovascular disease. J Lipid Res. 2012;53:1723–1737. doi: 10.1194/jlr.R024794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qi L, Qi Q, Prudente S, Mendonca C, Andreozzi F, di Pietro N, Sturma M, Novelli V, Mannino GC, Formoso G, Gervino EV, Hauser TH, Muehlschlegel JD, et al. Association between a genetic variant related to glutamic acid metabolism and coronary heart disease in individuals with type 2 diabetes. JAMA. 2013;310:821–828. doi: 10.1001/jama.2013.276305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Do R, Willer CJ, Schmidt EM, Sengupta S, Gao C, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, Beckmann JS, et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet. 2013;45:1345–1352. doi: 10.1038/ng.2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Andreassen OA, Desikan RS, Wang Y, Thompson WK, Schork AJ, Zuber V, Doncheva NT, Ellinghaus E, Albrecht M, Mattingsdal M, Franke A, Lie BA, Mills I, et al. Abundant genetic overlap between blood lipids and immune-mediated diseases indicates shared molecular genetic mechanisms. PLoS One. 2015;10:e0123057. doi: 10.1371/journal.pone.0123057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Parikh M, Patel K, Soni S, Gandhi T. Liver x receptor: A cardinal target for atherosclerosis and beyond. J Atheroscler Thromb. 2014;21:519–531. [PubMed] [Google Scholar]
- 52.Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Solovieff N, Cotsapas C, Lee PH, Purcell SM, Smoller JW. Pleiotropy in complex traits: Challenges and strategies. Nat Rev Genet. 2013;14:483–495. doi: 10.1038/nrg3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.