

Abstract
In fluorescence microscopy, the serial acquisition of 2D images to form a 3D volume limits the maximum imaging speed. This is particularly evident when imaging adherent cells in a light-sheet fluorescence microscopy format, as their elongated morphologies require ~200 image planes per image volume. Here, by illuminating the specimen with three light-sheets, each independently detected, we present a light-efficient, crosstalk free, and volumetrically parallelized 3D microscopy technique that is optimized for high-speed (up to 14 Hz) subcellular (300 nm lateral, 600 nm axial resolution) imaging of adherent cells. We demonstrate 3D imaging of intracellular processes, including cytoskeletal dynamics in single cell migration and collective wound healing for 1500 and 1000 time points, respectively. Further, we capture rapid biological processes, including trafficking of early endosomes with velocities exceeding 10 microns per second and calcium signaling in primary neurons.
1. INTRODUCTION
Cells continuously sense and adapt to environmental cues by modulating biochemical, mechanical, and organellar processes that span orders of magnitude in both time and space, and a critical challenge in cell biology is understanding how these cues ultimately alter phenotypic outcomes [1]. Fluorescence microscopy, due to its resolution, molecular specificity, and capacity to obtain dynamic information across populations of cells, remains pivotal to this effort [2]. However, quantitative subcellular imaging requires Nyquist sampling in both space and time [3], and many biological processes occur too rapidly to be imaged three-dimensionally throughout entire cells. For example, ciliate dynamics in the model organism Tetrahymena thermophila cannot be accurately tracked, even when imaged at 3.2 volumes per second [4]. Similarly, models for the control and locomotion of motor-driven transport remain incomplete [5], in part due to challenges in accurately measuring their rapid 3D dynamics in vivo.
To date, microscopy techniques capable of sensitively imaging intracellular features in 3D have been limited to volumetric image acquisition rates of ~3 Hz [4, 6]. In part, this is due to bandwidth limitations of modern piezoelectric scanners that are used to rapidly reposition heavy microscope objectives. To mitigate these demands, several methods have been developed that operate in the absence of objective scanning. These include extended depth of focus, either through the introduction of spherical aberrations [7] or a cubic phase mask [8], oblique illumination [9, 10], confocally-aligned oblique illumination [11], rapidly defocusing the detected wavefront with an electrically tunable lens [12, 13], or remote focusing through wavefront engineering [14]. However, these methods typically sacrifice sensitivity (e.g., due to aberrations, incomplete use of the detection numerical aperture, and inevitably small pixel dwell times) and spatial resolution (usually on the order of a few microns), rendering them incompatible with imaging subcellular structures.
Another major challenge in improving the temporal resolution of fluorescence microscopy is that 2D image planes must be acquired serially to form a 3D volume [15]. To overcome the limitations of serial image acquisition, several optical strategies have been developed that enable simultaneous imaging of multiple planes throughout a 3D volume using refractive [16] or diffractive optics [17-20]. Nevertheless, in each case, the fluorescence signal imaged in each plane is reduced by the degree of parallelization, and most fluorescence is detected as out-of-focus blur. For example, when 3 planes are imaged simultaneously, 66% of the light in each plane is lost to adjacent planes as image blur. Further, diffractive systems suffer from additional losses due to the limited diffraction efficiencies of the beam-splitting and achromatic correction gratings (Note S1) [17]. Thus, to accommodate this drastic reduction in fluorescence intensity, such schemes inevitably require longer exposure times or increased illumination intensities, negating the advantages of parallelized image acquisition.
Here we introduce a parallelized and photon-efficient 3D subcellular imaging scheme, which we refer to as Parallelized Light-Sheet Fluorescence Microscopy (pLSFM). For shallow sample volumes, pLSFM achieves 3-fold spatial parallelization without crosstalk between image planes, and is thus 3 to 4.5-fold less lossy than refractive or diffractive methods, respectively [16-20]. Unlike techniques that introduce aberrations [8, 12], sacrifice numerical aperture [11], or rely on short dwell times [13, 14], resolution and sensitivity are not compromised. This allowed us to image 3D subcellular processes in whole adherent cells at 14 Hz, including cytoskeletal dynamics, endosomal trafficking, and neuronal action potentials.
2. RESULTS
A. Parallelizing 3D Image Acquisition by Staggered Illumination and Detection
To achieve 3D parallelization, we tilt a cover slip mounted specimen and illuminate it with three laterally and axially displaced 1D Gaussian light-sheets (Figs. 1A and B). Because the light-sheets are staggered in sample space, fluorescence from each light-sheet can be imaged orthogonally in a light-sheet fluorescence microscopy (LSFM) format with a second objective, separated in image space with knife-edge mirrors, and independently detected on separate cameras (Figs. 1C, S1, and S2). The three planes are thus imaged independently, and a complete 3D volume is acquired by scanning the sample with a piezoelectric actuator through the three light-sheets. The illumination duty cycle for each 1D Gaussian light-sheet is 100%, which allows for both sensitive and delicate imaging of subcellular architectures [6].
Fig. 1.

Optical principle and imaging performance of pLSFM. (A) Three axially and laterally staggered light-sheets (blue) illuminate different sections of a specimen. Fluorescence (green), is only generated within the beam waist of each light-sheet. (B) Image of staggered light-sheets in a solution of fluorescein. Scale bar 20 microns. (C) Simplified diagram of detection light path. Owing to their lateral and axial displacement, the fluorescence from each section can be independently detected in image space. (D) 3D rendering of volumetric pLSFM data before and (E) after stitching. (F) Lateral and axial point spread functions for the three cameras before and after 10 iterations of Richardson-Lucy deconvolution. Scale bar 1 μm. (G). Sagittal cross-section of cell shown in F. Scale bar 10 μm. (H-K) Cross-sections from G, shown normal to the coverslip. Scale bar 10 μm.
Since image planes I and III are located 25 μm away from the nominal focal plane of the detection objective, spherical aberrations decrease the fluorescence intensity by ~25%. Although not used on the data shown in this manuscript, these aberrations and losses can be corrected optically (Fig. S3). Imaging is cross-talk free, and ~95% of the light is properly picked off by the knife-edge mirrors, for a field of view of ~13 microns in the laser propagation direction (Note S2, Figs. S4 and S5). This corresponds to a height normal to the coverslip of ~8.5 microns for views II and III (view I is not fundamentally limited in its FOV), which is sufficiently large for imaging most adherent mammalian cell types. Imaging beyond that range is possible, however at the cost of gradual increase in cross-talk and light loss. Thus, in its current implementation, pLSFM is optimized for parallelized 3D imaging of adherent cells and monolayers with elongated, thin morphologies.
The decreased image size afforded by the confined Y-dimension maximizes the image acquisition rate of scientific CMOS cameras (1603 frames per second for 128 pixels in the Y-dimension). Following registration of each sub-volume with fluorescent nanospheres (Fig. S6), the data from each camera is deconvolved, fused, and de-skewed, resulting in a ~3-fold increased image volume encompassing ~130 × 20 × 100 microns (Figs. 1D and E). The lateral resolution is close to diffraction limited (~300 nm, full width half maximum, FWHM) for all three views after 10 iterations of Richardson-Lucy deconvolution (Fig. 1F and Table S1). The deconvolved axial resolution is ~600 nm FWHM for each view, which is determined by the thickness of the illumination beam waist and the optical transfer function of the detection objective (Fig. S7) [21]. The final 3D image (Figs. 1E and G) exhibits excellent optical sectioning and individual image slices are free of out-of-focus image blur (Figs. 1H-K).
B. Sensitive Time Lapse Imaging of Cytoskeletal Dynamics
In theory, due to its light-efficient and spatially parallelized image acquisition, pLSFM improves 3D microscopy by enabling 3-fold longer dwell times for a given volumetric image acquisition rate. In practice, this factor is reduced to ~2.5-2.7 due to intentional overlap between the image sub-volumes. In the case of no overlap, which we refer to for simplicity, pLSFM can thus operate with 3-fold lower peak illumination intensities, and 3-fold longer camera integration times, respectively, compared to conventional serial 3D light-sheet acquisition. As peak illumination intensity is one of the principal determinants in photobleaching and phototoxicity [4], this provides gentler illumination without sacrificing signal-to-noise or overall imaging speed. This allowed us to image a migrating MDA-MB-231 breast cancer cell expressing GFP-Tractin for 1500 Z-stacks, comprising 306,000 individual images, without stereotypic signs of phototoxicity, which are often evident as alterations in cell morphology or initiation of cellular retraction (Figs. 2A-C, and Visualization 1) [4, 22]. We also imaged vimentin dynamics in retinal pigment epithelial (RPE) cells for 1000 Z-stacks at the leading edge of a wound healing response (Figs. 2D-E, and Visualization 2). Due to the large field of view, and uncompromised resolution, polarization of vimentin filament networks can be observed simultaneously in multiple cells as they coordinate collective cell migration throughout the monolayer.
Fig. 2.

pLSFM enables sensitive time-lapse imaging. (A) 3D rendering of migrating MDA-MB-231 breast cancer cell as imaged by pLSFM, labeled with GFP-Tractin. (B) 3D rendering of same cell after 750 and (C) 1500 time points. (D) Wound healing response in collectively migrating and genome edited RPE cells, heterozygously expressing GFP-vimentin. Lateral (XY) maximum intensity projection. (E) Single plane cross-sections normal to the coverslip show distribution of vimentin at both the dorsal and ventral surfaces of migrating cells. Each plane taken from different positions relative to the leading of the migrating epithelial sheet. (F) Lateral maximum intensity projection of filopodial dynamics in an RPE cell, labeled with LifeAct-mNeonGreen. (G) Montage of filopodia formation from sub-region in f. (H) Dynamics of microtubule plus tips (+TIPs), labeled with EB3-mNeonGreen, in a confluent layer of U2OS cells, shown as a lateral maximum intensity projection. (I) Photobleaching comparison between pLSFM (N = 6) and single plane SPIM (N = 8). Mean normalized intensity shown, and error bars report the standard deviation. For each mode, camera integration time and illumination intensity remained constant. pLSFM achieves 2.7-fold faster volumetric image acquisition rate than single plane SPIM, without increasing the rate of photobleaching. All scale bars 10 μm.
C. Faster Volumetric Image Acquisition Rates without Increased Photobleaching
Alternatively, for a given camera framerate, the volumetric image acquisition rate of pLSFM increases linearly with the degree of parallelization. This allows fast timescale biological events, including filopodial dynamics in an RPE cell, to be imaged with high spatiotemporal sampling (Figs. 2F, G, and Visualization 3). Despite a 2.7-fold increase in volumetric image acquisition rate (~10% overlap between image sub-volumes), photobleaching is unaffected. To demonstrate this, we imaged microtubule plus tips (+TIPs) with EB3-mNeonGreen in a layer of confluent U2OS osteosarcoma cells (Fig. 2H, and Visualization 4), and compared imaging performance to traditional single plane LSFM using the same exposure duration and illumination intensity. To quantify photobleaching, EB3 comets were automatically detected and their intensities measured through time in 3D with u-track [23]. Indeed, pLSFM allowed us to achieve a 2.7-fold increase in the volumetric image acquisition rate, while maintaining the same level of photobleaching as single plane LSFM (Fig. 2I). Thus, our approach overcomes both technological (maximum piezo scanning speed) and photophysical (finite fluorescence photon flux) limitations of existing 3D microscopy, without compromising resolution or introducing out-of-focus image blur.
D. Endosomal Trafficking at 10 Microns per Second
To highlight the potential for rapid volumetric imaging, we used pLSFM to observe early endosomal trafficking in Rab5a-labeled cells [24]. Retrograde trafficking of early endosomal Rab5a-positive vesicles is carried out by cytoplasmic dynein, a motor protein which in vitro achieves a processive motion of ~1 micron per second [25]. In vivo, early endosomes have been observed to translocate in short but linear bursts with velocities greater than 7 microns per second [26]. However, these events have not been observed in 3D, since faithful measurement of active transport necessitates that temporal sampling increases with particle velocity, acceleration, and density [6]. In ChoK1 cells, imaged at 2.92 Hz for 500 time points, rapid vesicular dynamics were observed (Fig. 3a and Visualization 5). These included the fast translocation of vesicles from the basal to dorsal surface of the cell (Fig. 3B), which would otherwise be missed with two-dimensional widefield or confocal imaging.
Fig. 3.
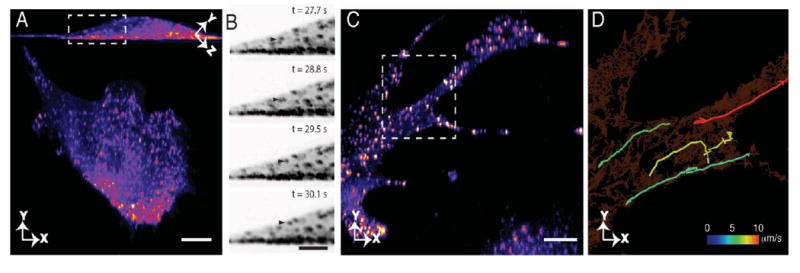
pLSFM enables the observation of rapid vesicular dynamics in 3D. (A) Dynamics of Rab5a vesicles, visualized at a volumetric image acquisition rate of 2.92 Hz over 500 time points. Maximum intensity projection parallel to the coverslip and in lateral imaging plane, for top and bottom panels, respectively. Scale bar 10 μm. (B) Montage of vesicle translocation towards the dorsal surface of the cell, from the sub-region in a. Image contrast has been inverted to better highlight weak features. Scale bar 5 μm. (C) Rapid Rab5a-vesicle movement in an RPE cell, volumetrically imaged at 7.26 Hz, for 400 time points, shown as lateral maximum intensity projection. Scale bar 10 μm. (D) Selected tracks of Rab5a-vesicle undergoing directed transport pseudocolored according to their maximum velocity shown on top of all cumulative tracks detected in this area, from the sub-region in C.
This highlights the importance of imaging a cell in its entirety, as limitations in 2D imaging could lead to inaccurate biological conclusions. We further imaged vesicle translocation in an RPE cell for 400 time points at a volumetric image acquisition rate of 7.26 Hz (Fig. 3C, Visualization 6). At this volumetric image acquisition rate, using automated particle detection and tracking, we observed Rab5a-positive vesicles that exhibited retrograde transport with peak velocities beyond 10 microns per second (Fig. 3D). To our knowledge, such fast vesicular movements have never been observed in 3D, possibly due to limitations in the volumetric image acquisition rate. At this sample scanning speed, some oscillations and distortions became visually noticeable. However, 95% of the oscillations observed were below 50 nm in amplitude (Fig S8), which is significantly smaller than the inter-particle distance (~450 nm), and therefore do not result in ambiguities in particle tracking.
Biochemically, the activity and velocity of dynein is modulated through its association with adapter proteins and its cargo, perhaps pointing towards unknown effectors that ‘supercharge’ retrograde transport [5]. This clearly illustrates the potential of pLSFM to measure rapid intracellular dynamics and to study biophysical phenomena in situ at the single particle level. Further, this points to challenges in in vitro reconstitution systems, where critical or unknown components may be omitted.
E. Action Potentials in Primary Neurons Imaged at a 14 Hz Volumetric Acquisition Rate
Secondary messenger waves (e.g., Ca2+) propagate at even greater speeds through cells and organisms, thus also necessitating faster image acquisition rates for proper temporal resolution [27]. By overcoming the technical limitations associated with piezoelectric scanners (improved bandwidth at decreased scan amplitudes) and camera readout times (efficient use of CMOS parallelization), pLSFM is capable of volumetrically imaging at 14 Hz, or ~2800 image planes per second. This enabled us to observe calcium signals in dissociated primary cortical neurons labeled with GCaMP-6f over 400 time points, encompassing multiple neurons and their axonal and dendritic projections (Fig. 4A) [28]. Calcium waves propagated along dendrites (Fig. 4B), and dissipated asymmetrically in space and time (Fig. 4C). Because of the large field of view, individual firing events, evident as spikes in the intensity time traces, could be observed at sites distant from one another and could provide insight into dendritic multiplexing (Fig. 4D and Visualization 7). Indeed, this is true even in thick, overlapping regions, which is particularly important for quantifying signal propagation between synapses, and is not possible with 2D imaging modalities (Figs. 4E and F, Visualization 7).
Fig. 4.
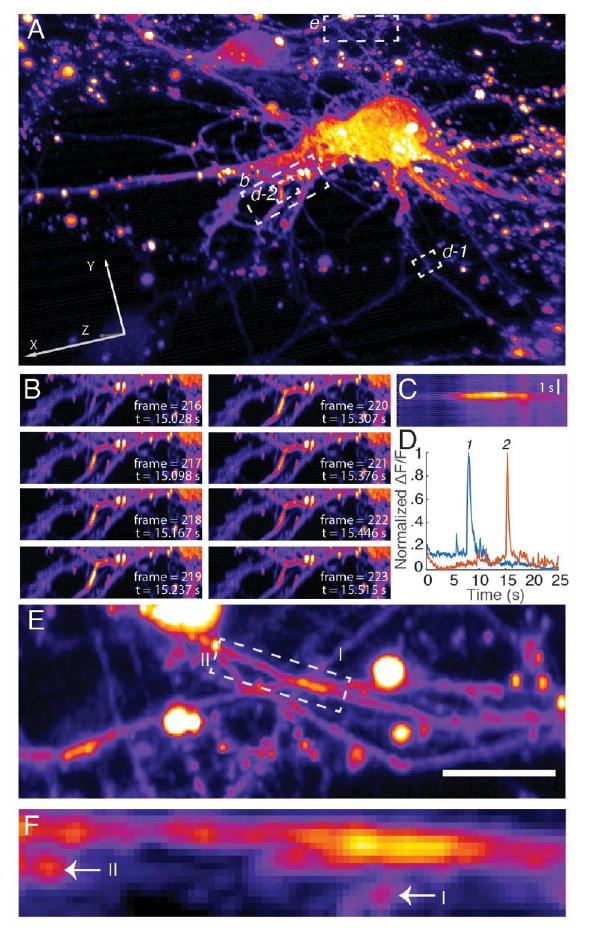
Calcium signaling in primary cortical neurons visualized with pLSFM. (A) 3D rendering of primary cortical neurons, labeled with GCaMP-6f. Action potentials were volumetrically imaged at 14.37 Hz for 500 time points. (B) Propagation of a calcium wave through a dendritic arbor. Sub-region from A. (C) Kymograph of action potential propagating along dendrite shown in B. (D) Temporal analysis of calcium bursts from regions specified in A. (E) Dense, overlapping region of axonal projections from region specified in A. Image shown as a maximum intensity projection parallel to the coverslip. (F). Cross-section normal to the coverslip of boxed region in E, shown as a maximum intensity projection over a depth of 3.2 μm. Arrows point at crossing dendrites that are also labeled in E. Scale bar 10 μm.
3. DISCUSSION
We have demonstrated a 3D parallelized imaging scheme that achieves for every image plane a performance that is comparable to a conventional single plane microscope. This has the important consequence that 3D imaging can be performed more delicately (i.e., with lower excitation intensities, thereby decreasing photodamage), with higher sensitivity, and with increased volumetric acquisition rates. Such improvements are necessary to image rapid subcellular processes with dimly labeled constituents, throughout the entire 3D volume of a cell. Our initial experiments revealed rapid vesicular dynamics that were previously unobservable in 3D, and point towards exciting new research opportunities. For example, compared to other 3D imaging modalities, Rab5a-vesicles were ~10 and 20-fold faster than EB3 comets and mitochondrial translocation events observed by Lattice LSFM [4] and structured illumination microscopy [29], respectively.
In principle, pLSFM can be performed with a greater number of light-sheets, allowing further improvements in parallelization. However, as light-sheets are positioned further away from the nominal focal planes of the excitation and detection objectives, aberration correction becomes increasingly important. Here, we observed a loss of peak intensity for cameras 1 and 3, which were oppositely positioned ~25 microns away from the detection objective’s nominal focal plane, by ~25%. To minimize the effect that these aberrations had on image resolution, we deconvolved the data from each camera with their corresponding point-spread functions. Since the aberrations are stationary, they can be corrected optically, which we performed as proof of principle with a deformable mirror (See Fig. S3). In future implementations, compensation of spherical aberrations could be incorporated directly into the relay optics (using similar principles as a correction collar on an objective). This is in direct comparison to previous parallelization methods, where ~66% of the remaining light is irrevocably lost to adjacent image planes as image blur (not accounting for additional losses by diffractive elements) [17].
The arrangement of the three light-sheets and camera planes in pLSFM was static and best suited for an image volume spanning 100 microns in the scan direction. While we found this arrangement well-suited for a wide range of adherent cell types, more flexibility in the spacing of the light-sheets could be beneficial. On the illumination side, light-sheets could be generated holographically with a spatial light modulator, which would allow effortless changes to the number of light-sheets, and their respective positioning. However, to maximize the detection light efficiency, refractive optics should be used to separate the different imaging planes. To accommodate variable beam positioning, the pick-up mirrors, as well as the remaining optical path would need to be mechanically adjusted in an automated fashion.
As described here, pLSFM is optimized for imaging adherent, mammalian cells in a shallow volume adjacent to a coverslip. Mechanical scanning of the coverslip, which we used to develop and demonstrate the governing principles of this method, can introduce vibrations into the sample at higher scanning frequencies. For example, frame-to-frame vibrations on the order of 20-50 nm were observed in the 7.26 Hz Rab5a data(Fig. S8). Furthermore, sample scanning becomes rate-limiting at volumetric image acquisition rates >14 Hz for piezoelectric actuators. At such fast acquisition rates, the piezo actuator response is expected to become nonlinear and distort the image volumes in the scan direction. In principle, greater scan rates could be achieved with more-advanced piezo control systems [30], but agitation of the sample and excitation of vibrational eigenmodes in the sample stage could become limiting. As such, we envisage replacing the mechanical cell movement with an optical scan of the light-sheets along the coverslip, thus keeping cells stationary [6]. To improve spatial resolution, our parallelization approach could be advantageously combined with lattice LSFM [4]. In fact, parallelization could help speed up the structured illumination mode in lattice LSFM, which so far has not been widely adopted for imaging rapid cellular dynamics due to the higher number of raw images needed for reconstruction.
pLSFM could also be adapted to image larger 3D environments, including synthetic hydrogels and reconstituted extracellular matrices, by scanning staggered light-sheets in their propagation direction with selective fluorescence detection [31]. In such a scheme, some out of focus cross talk from adjacent image planes will occur, which could be suppressed with two-photon excitation [32, 33].
In summary, pLSFM provides parallelized 3D fluorescence microscopy without increasing the illumination burden, or reducing the detected photon flux. As an important consequence, pLSFM greatly improves imaging speed without a concomitant increase in photobleaching or phototoxicity. Furthermore, the high duty-cycle of illumination at each plane (in contrast to digitally scanned methods), provides excellent sensitivity. Thus, pLSFM offers an unparalleled combination of gentle illumination, resolution, speed, and sensitivity. Together, these attributes make pLSFM the ideal choice for imaging fast timescale subcellular events in a complete, three-dimensional perspective.
4. METHODS
A. Microscope Layout
An optically pumped semiconductor laser (Coherent, Obis 488 LX) is spatially filtered with a 30-micron pinhole and telescopically expanded (ThorLabs, BE05M-A) to a beam size of 9 mm (1/e2). Laser intensity is controlled with the manufacturer-provided software (Coherent Connection) and shuttering is achieved by direct digital modulation of the laser driver after preconditioning of the FPGA-derived trigger with a scaling amplifier (Stanford Research Systems, SIM983 and SIM900). The laser light is serially directed through two non-polarizing beam-splitters (ThorLabs, BS019 and BS013), resulting in 3 beams of approximately equal power (30:35:35). Each individual beam is truncated with a variable aperture slit (ThorLabs, VA100C), focused to a 1D Gaussian beam with a cylindrical achromatic doublet (ThorLabs, ACY254-150-A), reflected with a knife-edge right-angle prism mirror (ThorLabs, MRAK25-P01), and imaged into sample space with an infinity-corrected tube lens (ThorLabs, ITL200) and a 40x water-dipping objective (Nikon Instruments, NA = 0.8). Each variable slit was adjusted that all light-sheets have the same beam width, as verified by measuring the axial FWHM of fluorescent nanospheres. Truncation of the beam with the variable slit apertures and the small waist size in the sample plane make the beam differ from a true Gaussian beam. Yet the light sheets share some characteristics of Gaussian beams (Fig. S7). Fine adjustments of the beam intensity of each light-sheet was achieved with neutral density filters. All three beams intersected with the sample, mounted at 49 degrees, in a custom stainless steel environment chamber. Gross sample positioning is achieved with a 3D stage (ThorLabs, PT3). However, we recommend using a stage with lockable positioning as this improves long-term stability and decreases the amplitude of external vibrations (data not shown).
Fluorescence was collected at 90 degrees with an identical 40x water-dipping objective and imaged with an infinity-corrected tube lens (ThorLabs, ITL200), creating a series of staggered image planes. The first two focal planes are reflected with knife-edge right-angle prism mirrors and relayed to a sCMOS camera (Hamamatsu, Flash 4.0) with a pair of 150 mm focal length achromatic doublets (ThorLabs, AC254-150A). The third focal plane is transmitted and relayed to a sCMOS camera (Hamamatsu, Flash 4.0) with a pair of 200 mm focal length achromatic doublets (ThorLabs, AC2545-200A).
Sample scanning is performed with a 100-micron piezo stage (Physik Instrumente, P-621.1CD) with a step size of 600 nm. Given the 49-degree rotation of the stage relative to the optical axis, this resulted in an axial step size of 393 nm. Despite the high-bandwidth of the piezo (unloaded resonant frequency of 800 Hz), the fly back time between Z-stacks is rate-limiting. To decrease the bandwidth demands of the piezo, we implemented a bidirectional scan mode where images are acquired in both the forward and reverse scan directions. At very fast acquisition rates, the response of the piezo becomes slightly phase delayed relative to the input triangular waveform. Since this phase delay is deterministic, we phase delayed the camera trigger pulse train by an equivalent amount such that image acquisition remained well synchronized with the actual piezo scan. The duration of these delays depended upon the camera integration time, and were 2, 2.5, 2.7, 2.8, 3.1, 3.0, and 4.5 ms, for camera integration times of 20, 10, 7.5, 5, 2, 1, and 0.5 ms, respectively.
B. Distributed acquisition system
The distributed data acquisition system consists of a master computer and two remote nodes communicating via a dedicated 10G switch (NETGEAR, XS708E). To accelerate data transfer between the nodes and the master computer, each computer is configured such that their 10G Ethernet cards operate in a static link aggregation mode with two Ethernet cables. The nodes and the master computer each house a FireBird PCI Express Camera Link, allowing maximum frame rate imaging with Hamamatsu Flash 4.0 cameras. The master computer also houses the field programmable gate array (National Instruments, PCIe-7852R) for analog and digital control of the microscope, controlled by custom LabView (National Instruments) software. The distributed acquisition system is compatible with four remote nodes, allowing simultaneous imaging with 5 cameras.
The remote nodes are 64-bit Dell Precision 5810 machines operating Windows 8.1 and equipped with an Intel Xeon Processor E5-1620 (4C, 3.5GHz), 64 Gb DDR4 RAM, an NVIDIA Quadro NVS 310 512 MB Video Card, a 500 GB Serial-ATA (7,200 RPM) hard drive, and an Intel X540-T2 10GbE NIC Dual Port network card. The master node is a 64-bit Dell Precision 5810 machine operating Windows 8.1 and equipped with an Intel Xeon Processor E5-1650 (6C, 3.5GHz), 128 GB 2133 MHz DDR4 RAM, an NVIDIA Quadro K420 1 GB video card, 4x 512 Gb Solid State Drives operating in a software-based RAID 0 configuration via an Integrated Intel AHCI chipset SATA controller, and an Intel X540-T2 10GbE NIC Dual Port network card.
C. Image Deconvolution
For image deconvolution, PSFs of isolated 100 nm fluorescent beads (Polysciences Inc., Fluoresbrite Plain YG) were acquired for each camera in both the forward and reverse scans. These PSFs were then individually applied to deconvolve each image volume, depending upon the scan trajectory, using a Richardson-Lucy algorithm with 10 iterations implemented in Matlab. The PSF was kept in its raw form (skewed) and the deconvolution was performed on the raw data (before de-skewing and stitching).
D. 3D Image Registration and Stitching
To register and stitch the full image volume, the relative volume positions, orientations, and scale are calibrated using dense arrays of surface-immobilized 200 nm beads (Polysciences Inc., Fluoresbrite Plain YG) imaged with the full 100 μm scan range of the sample piezo. This results in ~20 μm of overlap between the image volumes acquired with cameras 1 and 2, and cameras 2 and 3. Subsequently, cameras 1 and 3 are registered to camera 2 with Matlab (MathWorks). To ensure convergence of the estimators, the registration approach uses three steps: a) It computes the axial distance between the central volume and volumes 1 and 3, b) the lateral and scaling components of the affine transform, and c) the rotational component of the affine transform. Each volume is then intensity scaled to the same median intensity, and the affine transforms are combined with a deskewing transform, and applied in a single image transformation to cameras 1 and 3. For camera 2, only the deskewing transform is applied. In regions with overlap between the image volumes, the average intensity was kept. These transforms were then applied to deconvolved cellular data, which were imaged with a 40 μm scan range and ~4 μm of overlap. The Matlab scripts used for data processing can be obtained by contacting the corresponding author. Unless otherwise stated, no photobleaching correction was used.
E. Image Analysis and Evaluation of Relative Detection Efficiency of Cameras
Action potentials were identified by summing all voxels over a small 3D region of interest on undeconvolved, but deskewed, data. Action potentials in Visualization 7 were highlighted by performing a temporal FFT on the data. To this end, the low frequency components from 0-0.1Hz were set to zero in the temporal frequency domain. In addition, the highest frequency component in the FFT (the Nyquist frequency) was set to zero, which removed some spurious pixel artefacts that arose in the backwards FFT and damped oscillations. Since the frequency step size is small (0.036Hz), this causes some minor low pass filtering of the data (highest detectable frequency was reduced from 7.19 Hz to 7.15 Hz). This high-pass filtered data, which highlights rapidly occurring events such as action potentials, was pseudocolored and superimposed with data that was not high pass filtered. Importantly, for the analysis of the data shown in Fig. 4, no such Fourier filtering was applied, but instead the raw data was used. The temporal filtering was solely employed to aid the viewer in the Visualization 7 by better highlighting the high-frequency events.
The light loss due to spherical aberrations on cameras 1 and 3 was measured relative to camera 2 by illuminating a solution of fluorescein with a single focused beam originating from the excitation objective. The detection objective was translated until it was centered and in focus on each camera, and the fluorescence intensities were compared. To find the nominal focal plane (which must coincide with camera 2), all three cameras were translated axially and the Z-position of the detection objective was adjusted accordingly. This was repeated until losses due to spherical aberrations on camera 1 and 3 were equivalent. The observed intensity losses for camera 1 and 3 of ~25% are in good agreement with those reported below in Section 4J regarding adaptive optics.
Rab5a-associated vesicles were automatically detected and tracked with a 3D version of uTrack, as previously reported [6, 31]. Brownian and directed motion are modeled with multiple Kalman filters. The actively transport Rab5 are identified by high diffusion coefficient (>500 μm2/s) and lifetime (> 2.75 s). The highest measured velocity amounted to 10.58 μm/s. We manually inspected the particle movement of the corresponding track in the raw data and confirmed the consistency of the track as well as the reported velocity.
F. Animal Care
Rat primary neurons were obtained in accordance to protocols approved by the University of Texas Southwestern Medical Center Institutional Animal Care and Use Committee (IACUC).
G. Cell Culture and Labeling
Male and female embryonic day 18 (E18) primary cortical neurons were prepared from timed pregnant Sprague Dawley rats (Charles River Laboratories, Wilmington, MA) as previously reported [34]. Embryonic cortices were harvested, neurons dissociated, and plated on 5 mm coverslips coated with poly-D-lysine at a density of ~7,000 neurons per coverslip. Neurons were cultured in completed Neurobasal medium (Gibco 21103049) supplemented with 2% B27 (Gibco 17504044), 1 mM glutamine (Gibco 25030081), and penicillin streptomycin (Gibco 15140148) at 37°C in a 5% CO2 environment. Neurons were infected with purified and concentrated lentivirus encoding GCaMP6f after 4 days in vitro.
Retinal pigment epithelial cells (hTERT RPE-1, ATCC CRL-4000) were cultured in DMEM:F12 (Gibco 11330-032) supplemented with 10% Fetal Bovine Serum (FBS, Equitech-Bio, Inc.) and 1% Antibiotic-Antimycotic (Gibco 15240062). Chinese Hamster Ovary cells (ChoK1), kindly provided by Dr. R. Alan Horwitz (Allen Institute for Cell Science), were cultured in DMEM (Gibco 11995-065) supplemented with 10% FBS, 1% Antibiotic-Antimycotic, and 1% Non-Essential Amino Acids (Gibco 11140050). MDA-MB-231 breast cancer cells, kindly provided by Dr. Sean Morrison (UT Southwestern Medical Center), were cultured in DMEM supplemented with 10% FBS. U2OS osteosarcoma cells, kindly provided by Dr. Dick McIntosh (University of Colorado) were cultured in McCoy’s 5A media (Sigma-Aldrich, M4892-10X1L) supplemented with 2.2 g/L sodium bicarbonate, 10% FBS and 1% Antibiotic-Antimycotic. Cells were not authenticated, but were routinely tested for mycoplasma contamination.
Transient transfections were performed with Lipofectamine 2000 (ThermoFisher, 11668027) and Opti-MEM reduced serum medium (Gibco 31985088). Stable transduction was performed with a modified pLVX lentiviral system (Clontech) where expression is downregulated by truncating the CMV promoter. All mNeonGreen constructs are under license from Allele Biotechnology.
H. Photobleaching Measurements
To quantify the rate of photobleaching, confluent U2OS cells stably expressing mNeonGreen-EB3 from a truncated CMV promoter were volumetrically imaged. For multiplane imaging with three cameras, 61 images were acquired per camera, with an integration time of 15 ms and a total laser power of 0.57 mW in the pupil plane (~0.19mW per light-sheet). The final stitched data had ~5% overlap between adjacent image volumes. For single plane imaging, 166 images were acquired on a single camera with an integration time of 15 ms and a laser power of 0.19 mW in the pupil plane. Thus, both modes imaged the same volume for 50 Z-stacks, with the same camera integration time, and approximately equal signal-to-noise ratios, but the multiplane volumetric image acquisition rate was 3-fold faster due to the parallelized detection. Data were deskewed into their proper Euclidian reference, and EB3 particles were detected automatically in a parameter-free manner, allowing both the intensity and number of detected particles to be evaluated through time. The experiment was replicated 6 and 8 times, for pLSFM and single-plane SPIM imaging, respectively. Only data that remained in focus throughout the experiment were analyzed. Data were not randomized, but handled identically with the same analytical pipeline.
I. Image Vibration Analysis
To measure and assess the impact of vibrations, we performed frame-to-frame registrations of small 3D regions of interest. Regions were selected in disparate parts of the cellular volume, and tracked throughout the entire image sequence. The measurement was performed using the imregister function in Matlab (Mathworks) configured with a translational motion model. Fig. S8 presents vibrations in a stitching region, as well as the region used to illustrate our tracking results. The 95th percentile of displacement does not exceed 50 nm in either sub-volume. However, the maximum displacement can reach up to 100 nm (Fig. S8B-H). To assess the impact of these vibrations, we measured the minimum inter-particle distance for each particle detected in the cell. Fig. S8E and S8I shows that the maximum vibration measured is 5-10x smaller than the mean inter-particle distance, hence creating very little ambiguities in the reconstruction of trajectories.
J. Adaptive Optics
To correct the spherical aberrations present in image planes 1 and 3, a modified microscope was built that included a deformable mirror (Mirao52e, Imagine Optics) conjugate to the back-pupil plane of the detection objective, but dispensed with the pick-off mirrors in the detection path (see Figure S3A). The illumination was changed from a light-sheet to a collimated beam to decrease sample position-dependent fluctuations in fluorescence intensity. The detection objective was equipped with a piezoelectric positioner (P-726.1CD, Physik Instrumente) for Z-stepping, and the sCMOS camera could be translated along the optical axis to refocus at planes away from the nominal focal plane. Fluorescence from the intermediate image plane was relayed to the deformable mirror and camera with a pair of 350 mm focal length lenses. An interaction matrix for the mirror was established using a Shack-Hartmann sensor (Haso 4, Imagine Optics) and a collimated 632 nm laser beam, which was injected in the space between the detection objective and the tube lens, and propagated along the optical axis defined by the detection objective. A flip mirror allowed switching between the Shack-Hartmann sensor and the sCMOS camera. Using the Shack-Hartmann sensor, a flat mirror shape was optimized using an adaptive optics feedback loop.
The sample consisted of 200 nm fluorescent nanospheres immobilized on a coverslip, which was mounted parallel to the focal plane of the detection objective. First the nominal focal plane was found by translating camera and objective along the optical axis (the beads remained stationary) until spherical aberrations were minimal and the bead intensity was maximal. From this position, the objective was offset by ±25 microns (in respect to the Z-axis shown in Figure S3) and the camera was translated to bring the beads back into focus. These two settings recreated the spherical aberrations encountered by image planes 1 and 3. We then applied different amounts of spherical aberration to the mirror to maximize the intensity for these two offset image planes. The intensity was measured in a 3D stack, and the peak intensity of each setting for 20 beads was compared to imaging at the nominal focal plane with a flat mirror shape. For an offset of +25 microns (corresponding to image plane 3), compensation of spherical aberrations improved the bead intensity from 78% to 98% compared to the nominal focal plane. For an offset of −25 microns (corresponding to image plane 1), compensation of spherical aberrations improved the bead intensity from 76% to 90% compared to the nominal focal plane.
We assume that the difference in compensation efficiency comes from the divergence and convergence of the fluorescent light for the offset focal planes. As such, for an implementation of adaptive optics in pLSFM system, the magnification between the objective pupil and the mirror would need to be matched for each beam path individually.
Supplementary Material
Acknowledgments
The authors would like to thank Gaudenz Danuser of UT Southwestern Medical Center for his generous support, and Brenda Shields and Kimberley Ritola of Janelia Farms for helpful advice on culturing neurons. The LabView software developed by Coleman Technologies is built upon a core set of functions licensed from the Howard Hughes Medical Institute’s Janelia Farm Research Campus.
Funding. Cancer Prevention Research Institute of Texas R1255 (to Gaudenz Danuser) and RR160057 (to Reto Fiolka), Human Frontier Science Program LT000954/2015 (to Philippe Roudot), National Institutes of Health F32GM117793 (to Kevin Dean), K25CA204526 (to Erik Welf), R01 NS093382 and R37 HL06376 (to Joachim Herz), Bright Focus Foundation (to Joachim Herz), and the Consortium for Frontotemporal Dementia Research (to Joachim Herz).
Footnotes
See Supplement 1 for supporting content.
References
- 1.Liu YJ, Le Berre M, Lautenschlaeger F, Maiuri P, Callan-Jones A, Heuzé M, Takaki T, Voituriez R, Piel M. Confinement and Low Adhesion Induce Fast Amoeboid Migration of Slow Mesenchymal Cells. Cell. 2015;160:659–672. doi: 10.1016/j.cell.2015.01.007. [DOI] [PubMed] [Google Scholar]
- 2.Dean KM, Palmer AE. Advances in fluorescence labeling strategies for dynamic cellular imaging. Nature Chemical Biology. 2014;10:512–523. doi: 10.1038/nchembio.1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Danuser G. Reply to “Acquisition Frame Rate Affects Microtubule Plus-End Tracking Analysis”. Nature Methods. 2014;11:220–220. doi: 10.1038/nmeth.2860. [DOI] [PubMed] [Google Scholar]
- 4.Chen BC, Legant WR, Wang K, Shao L, Milkie DE, Davidson MW, Janetopoulos C, Wu XS, Hammer JA, Liu Z, English BP, Mimori-Kiyosue Y, Romero DP, Ritter AT, Lippincott-Schwartz J, Fritz-Laylin L, Mullins RD, Mitchell DM, Bembenek JN, Reymann AC, Bohme R, Grill SW, Wang JT, Seydoux G, Tulu US, Kiehart DP, Betzig E. Lattice light-sheet microscopy: Imaging molecules to embryos at high spatiotemporal resolution. Science. 2014;346:1257998–1257998. doi: 10.1126/science.1257998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cianfrocco MA, DeSantis ME, Leschziner AE, Reck-Peterson SL. Mechanism and Regulation of Cytoplasmic Dynein. Annual Review of Cell and Developmental Biology. 2015;31:83–108. doi: 10.1146/annurev-cellbio-100814-125438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dean Kevin M, Roudot P, Reis Carlos R, Welf Erik S, Mettlen M, Fiolka R. Diagonally Scanned Light-Sheet Microscopy for Fast Volumetric Imaging of Adherent Cells. Biophysical Journal. 2016;110:1456–1465. doi: 10.1016/j.bpj.2016.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tomer R, Lovett-Barron M, Kauvar I, Andalman A, Burns Vanessa M, Sankaran S, Grosenick L, Broxton M, Yang S, Deisseroth K. SPED Light Sheet Microscopy: Fast Mapping of Biological System Structure and Function. Cell. 2015;163:1796–1806. doi: 10.1016/j.cell.2015.11.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quirin S, Vladimirov N, Yang C-T, Peterka DS, Yuste R, Ahrens M. Calcium imaging of neural circuits with extended depth-of-field light-sheet microscopy. Optics Letters. 2016;41:855. doi: 10.1364/OL.41.000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar S, Wilding D, Sikkel MB, Lyon AR, MacLeod KT, Dunsby C. High-speed 2D and 3D fluorescence microscopy of cardiac myocytes. Optics Express. 2011;19:13839. doi: 10.1364/OE.19.013839. [DOI] [PubMed] [Google Scholar]
- 10.Sikkel MB, Kumar S, Maioli V, Rowlands C, Gordon F, Harding SE, Lyon AR, MacLeod KT, Dunsby C. High speed sCMOS-based oblique plane microscopy applied to the study of calcium dynamics in cardiac myocytes. Journal of Biophotonics. 2016;9:311–323. doi: 10.1002/jbio.201500193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bouchard MB, Voleti V, Mendes CS, Lacefield C, Grueber WB, Mann RS, Bruno RM, Hillman EMC. Swept confocally-aligned planar excitation (SCAPE) microscopy for high-speed volumetric imaging of behaving organisms. Nature Photonics. 2015;9:113–119. doi: 10.1038/nphoton.2014.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fahrbach FO, Voigt FF, Schmid B, Helmchen F, Huisken J. Rapid 3D light-sheet microscopy with a tunable lens. Optics Express. 2013;21:21010. doi: 10.1364/OE.21.021010. [DOI] [PubMed] [Google Scholar]
- 13.Kong L, Tang J, Little JP, Yu Y, Lämmermann T, Lin CP, Germain RN, Cui M. Continuous volumetric imaging via an optical phase-locked ultrasound lens. Nature Methods. 2015;12:759–762. doi: 10.1038/nmeth.3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Botcherby EJ, Smith CW, Kohl MM, Debarre D, Booth MJ, Juskaitis R, Paulsen O, Wilson T. Aberration-free three-dimensional multiphoton imaging of neuronal activity at kHz rates. Proceedings of the National Academy of Sciences. 2012;109:2919–2924. doi: 10.1073/pnas.1111662109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Winter PW, Shroff H. Faster fluorescence microscopy: advances in high speed biological imaging. Current Opinion in Chemical Biology. 2014;20:46–53. doi: 10.1016/j.cbpa.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geissbuehler S, Sharipov A, Godinat A, Bocchio NL, Sandoz PA, Huss A, Jensen NA, Jakobs S, Enderlein J, Gisou van der Goot F, Dubikovskaya EA, Lasser T, Leutenegger M. Live-cell multiplane three-dimensional super-resolution optical fluctuation imaging. Nature Communications. 2014;5:5830. doi: 10.1038/ncomms6830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abrahamsson S, Chen J, Hajj B, Stallinga S, Katsov AY, Wisniewski J, Mizuguchi G, Soule P, Mueller F, Darzacq CD, Darzacq X, Wu C, Bargmann CI, Agard DA, Dahan M, Gustafsson MGL. Fast multicolor 3D imaging using aberration-corrected multifocus microscopy. Nature Methods. 2012;10:60–63. doi: 10.1038/nmeth.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hajj B, Wisniewski J, El Beheiry M, Chen J, Revyakin A, Wu C, Dahan M. Whole-cell, multicolor superresolution imaging using volumetric multifocus microscopy. Proceedings of the National Academy of Sciences. 2014;111:17480–17485. doi: 10.1073/pnas.1412396111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abrahamsson S, Ilic R, Wisniewski J, Mehl B, Yu L, Chen L, Davanco M, Oudjedi L, Fiche J-B, Hajj B, Jin X, Pulupa J, Cho C, Mir M, El Beheiry M, Darzacq X, Nollmann M, Dahan M, Wu C, Lionnet T, Liddle JA, Bargmann CI. Multifocus microscopy with precise color multi-phase diffractive optics applied in functional neuronal imaging. Biomedical Optics Express. 2016;7:855. doi: 10.1364/BOE.7.000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma Q, Khademhosseinieh B, Huang E, Qian H, Bakowski MA, Troemel ER, Liu Z. Three-dimensional fluorescent microscopy via simultaneous illumination and detection at multiple planes. Scientific Reports. 2016;6:31445. doi: 10.1038/srep31445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huisken J, Stainier DYR. Selective plane illumination microscopy techniques in developmental biology. Development. 2009;136:1963–1975. doi: 10.1242/dev.022426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wäldchen S, Lehmann J, Klein T, van de Linde S, Sauer M. Light-induced cell damage in live-cell super-resolution microscopy. Scientific Reports. 2015;5:15348. doi: 10.1038/srep15348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jaqaman K, Loerke D, Mettlen M, Kuwata H, Grinstein S, Schmid SL, Danuser G. Robust single-particle tracking in live-cell time-lapse sequences. Nature Methods. 2008;5:695–702. doi: 10.1038/nmeth.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vale RD. The Molecular Motor Toolbox for Intracellular Transport. Cell. 2003;112:467–480. doi: 10.1016/s0092-8674(03)00111-9. [DOI] [PubMed] [Google Scholar]
- 25.Qiu W, Derr ND, Goodman BS, Villa E, Wu D, Shih W, Reck-Peterson SL. Dynein achieves processive motion using both stochastic and coordinated stepping. Nature Structural & Molecular Biology. 2012;19:193–200. doi: 10.1038/nsmb.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Delprato AM, Flores-Rodriguez N, Rogers SS, Kenwright DA, Waigh TA, Woodman PG, Allan VJ. Roles of Dynein and Dynactin in Early Endosome Dynamics Revealed Using Automated Tracking and Global Analysis. PLoS ONE. 2011;6:e24479. doi: 10.1371/journal.pone.0024479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chhetri RK, Amat F, Wan Y, Höckendorf B, Lemon WC, Keller PJ. Whole-animal functional and developmental imaging with isotropic spatial resolution. Nature Methods. 2015;12:1171–1178. doi: 10.1038/nmeth.3632. [DOI] [PubMed] [Google Scholar]
- 28.Chen T-W, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, Looger LL, Svoboda K, Kim DS. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295–300. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.York AG, Chandris P, Nogare DD, Head J, Wawrzusin P, Fischer RS, Chitnis A, Shroff H. Instant super-resolution imaging in live cells and embryos via analog image processing. Nature Methods. 2013;10:1122–1126. doi: 10.1038/nmeth.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schitter G, Menold P, Knapp HF, Allgöwer F, Stemmer A. High performance feedback for fast scanning atomic force microscopes. Review of Scientific Instruments. 2001;72:3320. [Google Scholar]
- 31.Dean Kevin M, Roudot P, Welf Erik S, Danuser G, Fiolka R. Deconvolution-free Subcellular Imaging with Axially Swept Light Sheet Microscopy. Biophysical Journal. 2015;108:2807–2815. doi: 10.1016/j.bpj.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Truong TV, Supatto W, Koos DS, Choi JM, Fraser SE. Deep and fast live imaging with two-photon scanned light-sheet microscopy. Nat Meth. 2011;8:757–760. doi: 10.1038/nmeth.1652. [DOI] [PubMed] [Google Scholar]
- 33.Welf Erik S, Driscoll Meghan K, Dean Kevin M, Schäfer C, Chu J, Davidson Michael W, Lin Michael Z, Danuser G, Fiolka R. Quantitative Multiscale Cell Imaging in Controlled 3D Microenvironments. Developmental Cell. 2016;36:462–475. doi: 10.1016/j.devcel.2016.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bock HH, Herz J. Reelin Activates Src Family Tyrosine Kinases in Neurons. Current Biology. 2003;13:18–26. doi: 10.1016/s0960-9822(02)01403-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.