

Abstract
One of the most feasible methods of measuring presynaptic calcium levels in presynaptic nerve terminals is optical recording. It is based on using calcium-sensitive fluorescent dyes that change their emission intensity or wavelength depending on the concentration of free calcium in the cell. There are several methods used to stain cells with calcium dyes. Most common are the processes of loading the dyes through a micropipette or pre-incubating with the acetoxymethyl ester forms of the dyes. However, these methods are not quite applicable to neuromuscular junctions (NMJs) due to methodological issues that arise. In this article, we present a method for loading a calcium-sensitive dye through the frog nerve stump of the frog nerve into the nerve endings. Since entry of external calcium into nerve terminals and the subsequent binding to the calcium dye occur within the millisecond time-scale, it is necessary to use a fast imaging system to record these interactions. Here, we describe a protocol for recording the calcium transient with a fast CCD camera.
Keywords: Neurobiology, Issue 125, Neuroscience, optical imaging, neuromuscular junction, presynaptic calcium transient, fluorescence calcium dye, calcium
Introduction
Calcium ions (Ca2+) participate in many neuronal signaling processes, including the initiation, maintenance, and plasticity of mediator release1,2,3,4,5. Upon the arrival of the action potential, extracellular Ca2+ enters the nerve terminal and initiates neurotransmitter release. In some synapses, the calcium current can be measured directly by electrophysiological methods6,7,8. In the case of the neuromuscular junction (NMJ), one cannot use direct patch clamp and two-electrode voltage clamp techniques due to the minute size of the nerve endings.
Recordings of inward Ca2+ currents from the nerve endings in the NMJ can be done by indirect electrophysiological methods9,10. However, these methods require the pretreatment of the synapse by sodium and potassium ion channel blockers. Optical methods do not require the pharmacological separation of ionic currents in the nerve terminal and permit recordings of Ca2+ influx, triggered by action potentials, and the subsequent elevation of Ca2+ ions in the axoplasm11,12,13,14. These methods are based on recordings of changes in the fluorescence of specific Ca2+-sensitive dyes upon the binding of free Ca2+ ions15,16,17,18,19.
Ca2+ indicators can be loaded into the cells through a variety of methods, depending on the purpose of the experiment. Researchers use the bath application of membrane-permeable dye forms20,21, loading via patch pipette22, or microinjection23,24,25. However, all these methods have some limitations in the case of the NMJ due to its peculiarities in synaptic architectonics. For the NMJ, the most convenient and successful method is to load the dye through the nerve stump, a forward-filling method26,27,28,29. This technique can be used for loading various fluorescence dyes into the peripheral nerve endings. This method was successfully used for Drosophila nerve terminals28, the lizard motor nerve28, and frog motor nerve terminals17,26,27,30. Depending on the object under study, methodical details can vary. A glass micro-pipette can be employed for small nerves from larvae28. Several researchers have described a method27,28 in which a freshly cut end of nerve innervating a muscle is immersed into a well pre-filled with a dye. The preparation is then left for several hours to soak the dye. The dye is soaked up by the axons and transported to the nerve terminals. In this paper, we describe a method of loading a fluorescence indicator into frog motor nerve terminals through the nerve stump. Our protocol uses a plastic pipette tip for the incubation of the tissue with a dye. We also describe how to acquire and analyze Ca2+ fluorescence transients.
Protocol
Experiments were performed on isolated nerve-muscle preparations of the musculus cutaneous pectoris from the Rana ridibunda frog. The size of animals of both genders was about 5-9 cm. The experimental procedures were performed in accordance with the guidelines for the use of laboratory animals of the Kazan Federal University and the Kazan Medical University, in compliance with the NIH Guide for the Care and Use of Laboratory Animals. The experimental protocol met the requirements of the European Communities Council Directive 86/609/EEC and was approved by the Ethical Committee of the Kazan Medical University.
1. Preparation of the Solutions
- Preparation of Ringer's solution.
- Prepare Ringer's solution: 113.0 mM NaCl, 2.5 mM KCl, 3.0 mM NaHCO3, and 1.8 mM CaCl2. Adjust the pH to 7.2-7.4.
- Prepare Ringer's solution with a low Ca2+ and high Mg2+ content: 113.0 mM NaCl, 2.5 mM KCl, 3.0 mM NaHCO3, 6.0 mM MgCl2, 0.9 mM CaCl2. Adjust the pH to 7.2-7.4.
- Preparation of the dye-loading solution.
- Prepare the water-based solution containing HEPES-Na at 10 mM (pH 7.2-7.4).
- Add 14 µL of the HEPES solution to a vial with the dye30. NOTE: the Ca2+ indicator dye comes in a 500-µL vial with 500 µg of powder.
- Vortex and spin down to mix thoroughly.
- Dilute the solution to bring the final concentration of the Ca2+ indicator down to 30 mM. Avoid exposure to light and store at -20 °C.
2. Dye-loading Procedure
- Dissect the cutaneous pectoris muscle with a piece of the pectoralis proprius nerve. NOTE: The dissection procedure is available in a free download of the paper by Blioch et al., 196831.
- For the dissection procedure, use two fine forceps and corneal scissors (see the Table of Materials). Transfer the dissected tissue into a silicon elastomer-coated Petri dish pre-filled with Ringer's solution and fix the tissue with fine stainless-steel pins such that it is slightly stretched in the dish.
- Re-fill the Petri dish with a fresh aliquot of the Ringer's solution. Remove the connective tissues. Do not damage the nerve.
Prepare the filling pipette: using a razor blade, cut out a ~2 mm-long piece of the conical part of a standard plastic 10 µL pipette tip.
Prepare a piece of modeling clay to mount the filling pipette on the Petri dish.
Connect the back of the filling pipette to a plastic syringe via silicone tubing and plastic connecting adapters made from pipette tips.
Before the dye loading procedure, remove the Ringer's solution from the Petri dish using a plastic pipette. Dry the muscle-nerve preparation using a fine syringe; this will prevent the dilution of the Ca2+ dye upon the subsequent loading of the filling pipette.
Remove the Ca2+ indicator vial from the freezer and allow it to thaw at room temperature in a dark place.
Under stereomicroscope control with low magnification (10×), detect the junction between the muscle and the nerve. With fine tweezers and scissors, cut the pectoralis proprius nerve close to the muscle surface (see step 2.1). Leave a nerve stump about 2 mm long.
Fix the filling pipette attached to tubing and the syringe on the Petri dish using modeling clay.
Move the tip of the pipette close to the nerve stump.
Without pinching it, gently aspirate the nerve stump into the tip of the filling pipette.
Remove the suction tubing from the blunt end of the filling pipette.
Carefully remove the excess solution from the filling pipette using a syringe with a long needle (see the Table of Materials). Do not pinch the nerve stump.
Vertically elevate the tip of the filling pipette slightly, keeping the nerve stump aspirated in the tip.
Insulate the aspirated part of the nerve stump from the outside of the filling pipette tip using petroleum jelly.
Dry up the nerve stump insulated in the filling pipette if necessary: gently aspirate the excess of solution from the filling pipette using a syringe with a long needle.
Draw 0.5 µL of the dye-loading solution (see step 1) using a pipette with a long pipette tip.
Gently insert the pipette tip with loading solution into the filling pipette. Eject the mixture directly onto the nerve stump.
Seal the open end of the filling pipette with petroleum jelly.
Add a small aliquot of Ringer's solution to the Petri dish to keep the preparation wet.
Incubate the preparation at room temperature under dark and wet conditions for 5 h.
Remove the filling pipette with loading solution, rinse the preparation with the Ringer's solution, and keep it overnight in the refrigerator at 8 °C.
3. Preparing the Tissue for Microscopy
Mount the preparation into the silicon elastomer-coated chamber and fix it with steel micro-needles such that it is slightly stretched.
Rinse the tissue with an aliquot of fresh Ringer's solution.
Use a suction electrode to stimulate the nerve; construction of the electrode is available from the free download of the paper by Kazakov et al., 201532. Position the electrode tip close to the cut end of the nerve and aspirate the nerve stump into the electrode orifice.
Mount the preparation chamber on the microscope stage. Place the temperature probe and the inlet and outlet firings in the chamber.
Connect the power cord to the Peltier element.
To superfuse the preparation, use a simple gravity-driven system. To remove the excess solution, turn on the perfusion suction pump.
Switch on the thermo-controller unit.
Set the temperature control to 20 °C.
Mount the ultraviolet protection shield.
Connect the stimulating wire electrode to the electric stimulator and observe the muscular contractions under the microscope with a 4x objective lens.
Fill up the perfusion system with the Ringer's solution with low-Ca2+ and high-Mg2+ content. NOTE: This solution is used to prevent muscular contractions. A decrease in the concentration of external calcium and an elevation of external magnesium result in the reduction of the amplitude of Ca2+ transients.However, based on previous experience, 0.9 mM CaCl2 and 6 mM MgCl2 are still sufficient to reliably resolve the amplitude of Ca2+ transients. It is worth mentioning that there exist some other ways to diminish muscular contractions without reducing the Ca2+ concentration.For example, the use of d-tubocurarine or alpha-bungarotoxin, specific blockers of nicotinic acetylcholine receptors, would completely or partially block muscle twitches17,27,28,30.However, the addition of these toxins can also affect presynaptic calcium entry33. To avoid this, µ-conotoxin GIIIA can be used27.
Switch on the pump and start the superfusion of the preparation with the Ringer's solution with low Ca2+ and high Mg2+.
Switch to the 40× objective lens on the microscope.
Switch on the monochromator (see the Table of Materials).
Select an emission wavelength of 488 nm and a continuous mode of illumination in the monochromator control software.
Under high magnification in fluorescence mode, ensure that the nerve terminals were loaded with the dye.
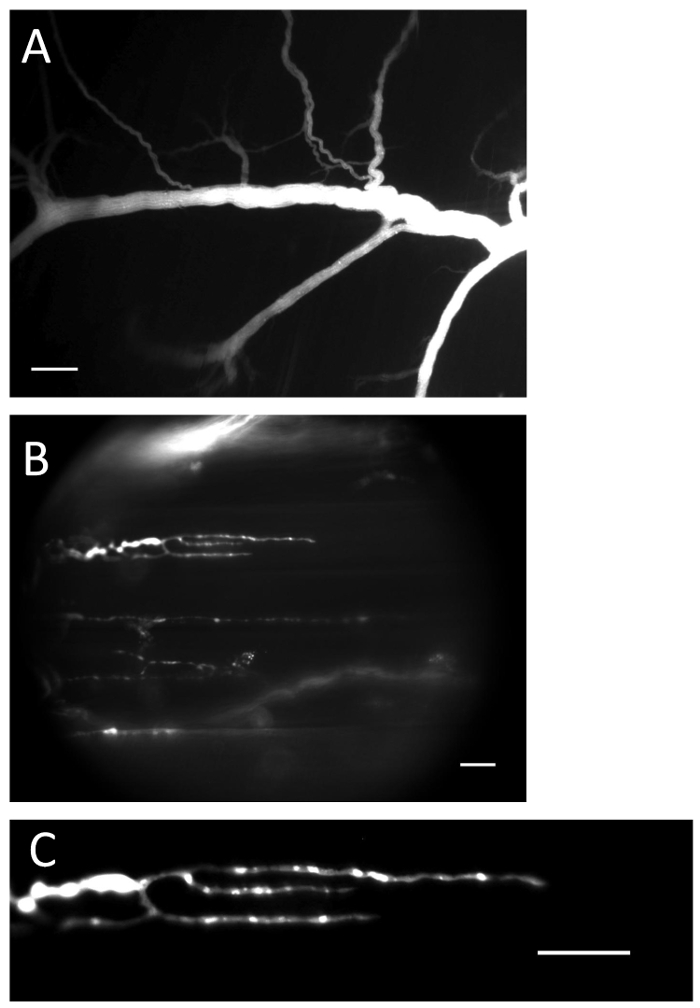
Figure 1: Nerve and terminals with loaded Ca2+ indicator. (A) Nerve filled with Ca2+ indicator after the loading procedure. Scale bar = 200 µm. (B) Nerve endings filled with Ca2+ indicator. Scale bar = 20 µm. (C) Ca2+-indicator fluorescence is clearly visible in the nerve ending. Scale bar = 20 µm. Please click here to view a larger version of this figure.
Allow the preparation to equilibrate for at least 30 min in the low-Ca2+ and high-Mg2+ solution.
4. Video Capture with the Digital CCD Camera
Note: The details of capturing fluorescence signals are specific for each microscope and camera type, but the key consideration is the image capturing speed.
Use 1 kHz as the minimum capture frequency for recordings of single Ca2+ transients in the NMJ. NOTE: Fast digital CCD cameras are necessary for fluorescence imaging (see the Table of Materials). The data acquisition system and software (see the Table of Materials) were used here for the synchronization of the camera, monochromator, and stimulator. In brief, this protocol permits the generation of synchronization pulses on digital outputs of the data acquisition system to open the shutter, capture the video signal, and initiate stimulation. All temporal parameters can be set in the protocols and/or on the apparatuses. A typical protocol is a series of 500 frames acquired at 1 kHz (80 x 80 pixels). Illumination with excitation light can bleach out the Ca2+ indicator and photodamage the cell tissue. Thus, avoid long exposures to excitation light. In this protocol, the shutter is open only for the time needed to capture the video. Acquire twenty series per specific nerve terminal. The aim here is to monitor the same sites in the control group and after drug delivery.
Under the 4X objective lens of a microscope, use the bright-field regime to visualize the muscle and nerve branches.
Switch to the 40X objective lens and, using the epifluorescence regime and an excitation wavelength of 488 nm, search for dye-loaded nerve endings. Identify a nerve-ending region of interest.
On the trinocular tube of the microscope, select the light path exchange levels: 100% light to camera.
Start the acquisition software for the CCD camera.
Under "Live" mode, find the ROI and adjust the focus.
Select the menu "Change" settings.
Use the "Basic Configuration" at 1,000 frames per second (fps), with a resolution of 80 x 80.
Set the number of input frames to 500.
Enter the name of the experiment.
Choose "External Trigger."
Set the pre-trigger time to 10 ms.
Set the number of repeats to 20.
In the monochromator control software, select an emission wavelength of 488 nm and "external trigger illumination" mode.
Run the data acquisition software.
Load the stimulation protocol.
Before recording the video, capture the dark frame using the video acquisition software.
Run the stimulation protocol.
Select the ROI and check the recorded signal.
5. Data Analysis
NoOTE: For data analysis, use the CCD camera software and ImageJ; the data is represented as a curve in a spreadsheet program. In the CCD camera software, average 20 repeats and export the results to an ImageJ support file. In ImageJ, select the ROI and background. Subtract the background from the ROI. Represent the data as a ratio: (ΔF/F0 -1) x 100%, where's ΔF is the intensity of fluorescence during stimulation and F0 is the intensity of fluorescence at rest.
In the acquisition software for the CCD camera, click on File > Average Files. Select the files and average them.
Save the averaged file as a .fit file by clicking "Save as Fit file."
Run the ImageJ software. Perform the following steps:
Click Image > adjust > brightness/contrast.
Click Image > stacks > tools > stack sorter.
Click Analysis > tools > ROI manager.
Drag and drop the averaged .fit file into the ImageJ window.
Zoom in on the window for a better view.
By moving the cursor, select the last frame and delete it (this is the dark frame)
Select a rectangular ROI over the area believed to be the background. Add it to the ROI manager
Measure the background by clicking More > Multi Measure. Note the MEAN. Copy the data, export it to a spreadsheet program, and calculate the average value of the threshold for a ratio.
Subtract the threshold from the stacks by clicking Process > Main > Subtract. Enter the averaged value of the threshold.
Select a rectangular ROI around a nerve terminal. Add it to the ROI manager.
Measure by clicking More > Multi Measure. Note the MEAN. Copy and export it to a spreadsheet program.
Average the offset of the signals. NOTE: Use the first several dozen points demonstrating base dye fluorescence without stimulation; this is the fluorescence at rest.
Divide the signals by the fluorescence at rest.
Subtract "1" and multiply by 100%.
Plot the signal and calculate the amplitude of the Ca2+ transient.
Representative Results
After the dye loading and upon motor nerve stimulation, an increase in the amplitude of the fluorescent signal (Ca2+ transient) can be detected in the nerve terminals (see Figure 2). Parameters of Ca2+ transients are presented in Table 1. Quantitatively, the parameters of the Ca2+ transients measured in our study are close to the data obtained by other scientists at synapses of cold-blooded animals15,34. The parameters of the Ca2+ transients depend on the rate of binding of Ca2+ with the dye and the subsequent dissociation. The rate of entry of Ca2+ into the nerve ending, interaction with the dye, and diffusion in the cytoplasm all affect the rise time of the Ca2+ transient. The decay time of the fluorescent signal depends on the affinity of the dye, the speed of the Ca2+ interaction with intracellular buffers, and the removal by ion pumps35. The amplitude analysis of Ca2+ transients can be used to study the influence of various substances on the calcium entry that participates in neurotransmitter release33.
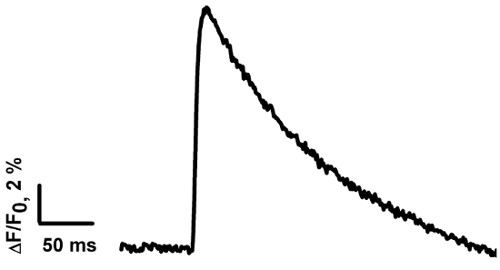
Figure 2: The averaged Ca2+transient measured in the frog NMJ. The Ca2+ transient was calculated based on the average of signals from 13 frog NMJs. Please click here to view a larger version of this figure.
| Peak ΔF/F (%) | Rise time 20%-80% (ms) | τ (ms) |
| 12,6 ±1,1 (n = 13) | 4.6 ±0.5 (n = 13) | 115.3 ±8.3 (n = 13) |
Table 1: The averaged parameters of theCa2+ transient. The data are presented as the mean ± SE; n is the number of measurements in distinct NMJs. The peak ΔF/F is the averaged amplitude of ΔF/F.
Discussion
In this paper, we presented the method of performing Ca2+-sensitive dye loading into frog nerve endings through the nerve stump. By the end of the loading procedure, all terminals in the proximal part of the nerve have significant levels of fluorescence. It has been estimated that the intra-terminal concentration of the probe varies between 40 and 150 µM17.
The incubation procedure is conducted in two steps: at room temperature and then at a lower temperature in a refrigerator. It is important to control the time of tissue incubation with the dye at room temperature. Depending on the actual length of the nerve stump, the specific dye, and the temperature, the incubation time may vary. If overexposed, terminals in the proximal parts close to the nerve stump can be overloaded. However, in the middle part of the nerve, it is still possible to find terminals that are satisfactorily loaded. During the long incubation in the refrigerator, the dye is evenly distributed over the nerve endings.
Our own observations33,35, as well as the data of other researchers30, prove the lack of any appreciable influence of the loading procedure on the amplitude of the postsynaptic response or on the frequency of miniature end-plate potentials. Good longevity was documented in the loaded preparations. There are some important points we would like to draw attention to. It is very essential to place the nerve stump into the dye-loading solution withinseveral minutes after excision to enable the dye to enter into the axons of the cut nerve; delays can cause ineffective loading, presumably due to the resealing of nerve axons27,36. Some investigators immerse the nerve stump in 100 mM EDTA (a Ca2+- and Mg2+-chelator) immediately after the excision of the nerve to prevent the cut axons from resealing. The buffer is removed after 1-2 min and replaced with a dye-loading solution37. The use of a petroleum-jelly well instead of plastic tubing for the loading procedure permits the use of a shorter nerve stump. While using this approach, the nerve is cut after it is immersed in the HEPES solution with dye, and the axons do not reseal because of the lack of divalent ions in the dye solution27,28.
In our study, we used the water-soluble salt form of the Ca2+ indicator instead of dextran. The dextran conjugates diffuse in the axon more slowly than the salt forms. However, the use of the dextran conjugate reduces dye compartmentalization and handling by the nerve and NMJs. Calcium Green 1-3,000 MW dextran conjugate has a good diffusion rate and demonstrates reduced compartmentalization38.
It is very important to avoid a long period of fluorescent illumination of the tissue, because this affects its health and survival. We use Nomarski optics in the visible-light channel to search for nerve terminals. During the recording, we limit the illuminated field using a diaphragm.
It is noteworthy that this loading method is suitable only for preparations that can withstand long incubations. To reduce the dye-loading time when studies are being conducted on more fragile tissues (e.g., synapses of warm-blooded animals), it is necessary to downscale the nerve stump length and to use micropipettes for loading29,39.
This loading technique is well suited for imaging changes in cytosolic Ca2+, with fluorescent indicators under both single-nerve stimulation and rhythmic synaptic activity17,27,35. The analysis of the Ca2+-transient amplitude can be used to study the influence of different substances on the calcium entry that participates in neurotransmitter release33.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This research was performed under the Russian Government's Program for Competitive Growth of the Kazan Federal University and a grant from the Russian Foundation for Basic Research (16-04-01051; 16-34-00817; 15-04-02983). We thank four anonymous reviewers for providing helpful comments on earlier drafts of the manuscript. We express our gratitude to Yuliya Aratskaya for the voice recording. We are grateful to Dr. Victor Ilyin for the many helpful comments and the help with the finale editing of the manuscript.
References
- Llinás R, Sugimori M, Silver RB. Microdomains of high calcium concentration in a presynaptic terminal. Science. 1992;256:677–679. doi: 10.1126/science.1350109. [DOI] [PubMed] [Google Scholar]
- Augustine GJ. How does calcium trigger neurotransmitter release? Curr Opin Neurobiol. 2001;11:320–326. doi: 10.1016/s0959-4388(00)00214-2. [DOI] [PubMed] [Google Scholar]
- Burnashev N, Rozov A. Presynaptic Ca2+ dynamics, Ca2+ buffers and synaptic efficacy. Cell Calcium. 2005;37:489–495. doi: 10.1016/j.ceca.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Neher E. Presynaptic calcium and control of vesicle fusion. Curr Opin Neurobiol. 2005;15:266–274. doi: 10.1016/j.conb.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Pang ZP, Südhof TC. Cell biology of Ca2+-triggered exocytosis. Curr Opin Cell Biol. 2010;22:496–505. doi: 10.1016/j.ceb.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG, Sakmann B. Calcium influx and transmitter release in a fast CNS synapse. Nature. 1996;383:431–434. doi: 10.1038/383431a0. [DOI] [PubMed] [Google Scholar]
- Borst JG, Sakmann B. Calcium current during a single action potential in a large presynaptic terminal of the rat brainstem. J Physiol. 1998;506:143–157. doi: 10.1111/j.1469-7793.1998.143bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazejian B, DiGregorio DA, Vergara JL, Poage RE, Meriney SD, Grinnell AD. Direct measurements of presynaptic calcium and calcium-activated potassium currents regulating neurotransmitter release at cultured Xenopus nerve-muscle synapses. J Neurosci. 1997;17:2990–3001. doi: 10.1523/JNEUROSCI.17-09-02990.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molgó J, Mallart A. Effects of Anemonia sulcatatoxin II on presynaptic currents and evoked transmitter release at neuromuscular junctions of the mouse. Pflugers Arch. 1985;405(4):349–353. doi: 10.1007/BF00595687. [DOI] [PubMed] [Google Scholar]
- Slutsky I, Rashkovan G, Parnas H, Parnas I. Ca2+-independent feedback inhibition of acetylcholine release in frog neuromuscular junction. J Neurosci. 2002;22(9):3426–3433. doi: 10.1523/JNEUROSCI.22-09-03426.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugland RP. Indicators for Ca2+, Mg2+, Zn2+ and other metal ions. In: Gregory J, editor. Handbook of fluorescent probes and research products. Eugene,Oregon: MolecularProbes; 2002. pp. 771–826. [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260(6):3440–3450. [PubMed] [Google Scholar]
- Tsien RY. Fluorescent indicators of ion concentrations. Methods Cell Biol. 1989;30:127–156. doi: 10.1016/s0091-679x(08)60978-4. [DOI] [PubMed] [Google Scholar]
- Adams SR. How calcium indicators work. Cold Spring Harb Protoc. 2010;2010(3) doi: 10.1101/pdb.top70. [DOI] [PubMed] [Google Scholar]
- DiGregorio DA, Vergara JL. Localized detection of action potential-induced presynaptic calcium transients at a Xenopus neuromuscular junction. J Physiol. 1997;505:585–592. doi: 10.1111/j.1469-7793.1997.585ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG. Optical measurement of presynaptic calcium currents. Biophys J. 1998;74:1549–1563. doi: 10.1016/S0006-3495(98)77867-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S. Ca2+ dynamics at the frog motor nerve terminal. Pflug Arch Eur J Phisiol. 2000;440:351–365. doi: 10.1007/s004240000278. [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Oertner TG, Svoboda K. The life cycle of Ca2+ ions in dendritic spines. Neuron. 2002;33:439–452. doi: 10.1016/s0896-6273(02)00573-1. [DOI] [PubMed] [Google Scholar]
- Luo F, Dittrich M, Stiles JR, Meriney SD. Single-pixel optical fluctuation analysis of calcium channel function in active zones of motor nerve terminals. J Neurosci. 2011;31:11268–11281. doi: 10.1523/JNEUROSCI.1394-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG. Monitoring presynaptic calcium dynamics with membrane-permeant indicators. In: Yuste R, Konnerth A, editors. Imaging in neuroscience and development: a laboratory manual. New York: Cold Spring Harbor Laboratory Press; 2005. pp. 307–314. [Google Scholar]
- Macleod GT. Topical Application of Indicators for Calcium Imaging at the Drosophila Larval Neuromuscular Junction. Cold Spring Harb Protoc. 2012;2012(7):786–790. doi: 10.1101/pdb.prot070086. [DOI] [PubMed] [Google Scholar]
- Eilers J, Konnerth A. Dye loading with Patch Pipettes. Cold Spring Harb Protoc. 2009;2009(4):277–281. doi: 10.1101/pdb.prot5201. [DOI] [PubMed] [Google Scholar]
- Coleman WL, Bill CA, Simsek-Duran F, Lonart G, Samigullin D, Bykhovskaia M. Synapsin II and calcium regulate vesicle docking and the cross-talk between vesicle pools at the mouse motor terminals. J Physiol. 2008;586(19):4649–4673. doi: 10.1113/jphysiol.2008.154666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macleod GT. Direct Injection of Indicators for Calcium Imaging at the Drosophila Larval Neuromuscular Junction. Cold Spring Harb Protoc. 2012;2012(7):797–801. doi: 10.1101/pdb.prot070102. [DOI] [PubMed] [Google Scholar]
- Talbot JD, David G, Barrett EF, Barrett JN. Calcium dependence of damage to mouse motor nerve terminals following oxygen/glucose deprivation. Exp Neurol. 2012;234(1):95–104. doi: 10.1016/j.expneurol.2011.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YY, Zucker RS. Release of LHRH is linearly related to the time integral of presynaptic Ca2+ elevation above a threshold level in bullfrog sympathetic ganglia. Neuron. 1993;10:465–473. doi: 10.1016/0896-6273(93)90334-n. [DOI] [PubMed] [Google Scholar]
- Tsang CW, Elrick DB, Charlton MP. alpha-Latrotoxin releases calcium in frog motor nerve terminals. J Neurosci. 2000;20(23):8685–8692. doi: 10.1523/JNEUROSCI.20-23-08685.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman Z, Malik P, Wu TY, Ochoa C, Watsa N, Lindgren C. Endocannabinoids mediate muscarine-induced synaptic depression at the vertebrate neuromuscular junction. Eur J Neurosci. 2007;25(6):1619–1630. doi: 10.1111/j.1460-9568.2007.05422.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macleod GT. Forward-filling of dextran-conjugated indicators for calcium imaging at the drosophila larval neuromuscular junction. Cold Spring Harb Protoc. 2012;2012(7):3440–3450. doi: 10.1101/pdb.prot070094. [DOI] [PubMed] [Google Scholar]
- Wu LG, Betz WJ. Nerve activity but not intracellular calcium determines the time course of endocytosis at the frog neuromuscular junction. Neuron. 1996;17(4):769–779. doi: 10.1016/s0896-6273(00)80208-1. [DOI] [PubMed] [Google Scholar]
- Blioch ZL, Glagoleva IM, Liberman EA, Nenashev VA. A study of the mechanism of quantal transmitter release at a chemical synapse. J Physiol. 1968. pp. 11–35. [DOI] [PMC free article] [PubMed]
- Kazakov A, Aleksandrov M, Zhilyakov NV, Khaziev EF, Samigullin DV. A simple suction electrode for electrical stimulation of biological objects. Mezhdunarodnyj nauchno-issledovatel'skij zhurnal. 2015;40(9):13–16. In Russian. [Google Scholar]
- Khaziev E. Acetylcholine-induced inhibition of presynaptic calcium signals and transmitter release in the frog neuromuscular junction. Front Physiol. 2016;7(621):1–10. doi: 10.3389/fphys.2016.00621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bélair EL, Vallée J, Robitaille R. Long-term in vivo modulation of synaptic efficacy at the neuromuscular junction of Rana pipiens frogs. J Physiol. 2005;569(1):163–178. doi: 10.1113/jphysiol.2005.094805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samigullin D, Fatikhov N, Khaziev E, Skorinkin A, Nikolsky E, Bukharaeva E. Estimation of presynaptic calcium currents and endogenous calcium buffers at the frog neuromuscular junction with two different calcium fluorescent dyes. Front Synaptic Neurosci. 2015;6(29):1–10. doi: 10.3389/fnsyn.2014.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angleson JK, Betz WJ. Intraterminal Ca2+ and spontaneous transmitter release at the frog neuromuscular junction. J Neurophysiol. 2001;85(1):287–294. doi: 10.1152/jn.2001.85.1.287. [DOI] [PubMed] [Google Scholar]
- Shahrezaei V, Cao A, Delaney KR. Ca2+ from one or two channels controls fusion of a single vesicle at the frog neuromuscular junction. J Neurosci. 2006;26(51):13240–132499. doi: 10.1523/JNEUROSCI.1418-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troncone LR, et al. Promiscuous and reversible blocker of presynaptic calcium channels in frog and crayfish neuromuscular junctions from Phoneutria nigriventer spider venom. J Neurophysiol. 2003;90(5):3529–3537. doi: 10.1152/jn.00155.2003. [DOI] [PubMed] [Google Scholar]
- Samigullin DV, Khaziev EF, Zhilyakov NV, Sudakov IA, Bukharaeva EA, Nikolsky EE. Calcium transient registration in response to single stimulation and during train of pulses in mouse neuromuscular junction. BioNanoSci. 2016;7(1):162–166. [Google Scholar]